
Abstract
A better understanding of the structure and extent of genetic variability in a breeding population of a crop is essential for translating genetic diversity to genetic gain. We assessed the nature and pattern of genetic variability and differentiation in a panel of 100 winged-yam (Dioscorea alata) accessions using 24 phenotypic traits and 6,918 single nucleotide polymorphism (SNP) markers. Multivariate analysis for phenotypic variability indicated that all phenotypic traits assessed were useful in discriminating the yam clones and cultivars. Cluster analysis based on phenotypic data distinguished two significant groups, while a corresponding analysis with SNP markers indicated three genetic groups. However, joint analysis for the phenotypic and genotypic data provided three clusters that could be useful for the identification of heterotic groups in the D. alata breeding program. Our analysis for phenotypic and molecular level diversity provided valuable information about overall diversity and variation in economically important traits useful for establishing crossing panels with contrasting traits of interest. The selection and hybridization of parental lines from the different heterotic groups identified would facilitate maximizing diversity and exploiting population heterosis in the D. alata breeding program.
Similar content being viewed by others


Introduction
Yam (Dioscorea spp.) is an important tuber crop supporting the livelihood of millions of people in the tropics and subtropics1,2. Globally, the average annual yam production is about 60 million tons with a gross value of ~14 billion US dollars. West Africa is the major yam growing area accounting for 94% of global yam production3. There are over 600 Dioscorea species of which ten are cultivated and are of economic importance4. The winged yam (Dioscorea alata), which is also called water yam or greater yam, is widely distributed in Africa and other tropical areas of the world5. Dioscorea alata is known for its vigorous plants with high tuber yield and plays a vital role in food security and income generation6. Different ecotypes are currently grown and used7.
Assessing genetic diversity present within a plant species is essential to identify genes controlling useful biological functions that can then be rationally used to develop new varieties8. The level and extent of genetic diversity in yam crop in general9,10,11,12 and winged yam specifically13,14 have been assessed using phenotypic attributes and low throughput molecular markers. Norman et al.13 successfully characterized 52 accessions of D. alata from Sierra Leone using 28 morphological descriptors. Anokye et al.14 dissected phenotypic diversity in a collection of D. alata from Ghana and Nigeria using morphological descriptors and grouped the accessions into distinct clusters independent of geographic origin. In addition to the phenotypic diversity, DNA-based marker systems using low throughput marker types4,7,15,16 have been applied to assess the genetic diversity of various D. alata populations. Dioscorea alata accessions from diverse geographical areas of West and central Africa and Puerto Rico were clustered into three genetic groups using AFLP markers irrespective of geographical origin16. Arnau et al.7 profiled 348 accessions using 24 microsatellite markers and reported wide genetic variation and significant structuring associated with geographic origin, ploidy levels, and morpho-agronomic characteristics.
Recent advances in sequencing techniques and their cost reduction has provided new insight into population structure and genetic variability in crops. Reference genomes are now available for both D. rotundata and D. alata (https://yambase.org/jbrowse_yambase/?data=data/json/) as well as a high-density genetic map of winged yam2. The availability of genomic resources in D. alata enables genome-wide study on genetic diversity with large datasets of high-density SNP markers from the next-generation sequencing platforms. Diversity Array Technology (DArT) characterized by high call rates and scoring reproducibility is among the sequencing techniques that are successfully applied in exploring genetic diversity and population genetic parameters of diploid root crops including cassava17 or polyploids such as potato18 but has not yet been applied to yam.
Previous diversity studies in yam have generally been conducted using either morphological or low throughput molecular markers. However, joint analysis of morphological and molecular information has the potential to provide in depth insight into population structure and genetic variability and it has been successfully applied in other crops4,19,20,21,22. The usefulness of this approach for genetic variability analysis in yam was demonstrated by Sartie et al.4. Employing a combination of data from 32 microsatellite markers and 24 phenotypic traits, D. alata and D. dumetorum which hitherto were categorized as sub-groups were clearly separated into distinct genetic groups4.
The objective of this study was to assess the genetic structure and diversity in a panel of D. alata clones and cultivars using phenotypic traits and SNP markers. These results will serve as a basis for a protocol to evaluate the genetic diversity and make an informed choice of parental lines to design new varieties in D. alata.
Results
Diversity and differentiation based on phenotypic traits
We performed principal component analysis to identify the most discriminative variables among the accessions. The first seven principal components cumulatively explained 76% of the total phenotypic variation (Table 1). The first principal component explained 25% of the total variation with the main contribution for this observed variation coming from eight variables. Principal component two accounted for 18% of the total variation with traits such as the number of medium and small tubers, number of stems, total tuber number per plant, the weight of medium and small tubers and length of medium and small tubers contributing much to this factor. Three variables, including tuber texture, tuber appearance, and tuber hairiness, were the qualitative traits that contributed much to the principal component three. The width of medium tubers contributed most to the variation explained by the fourth principal component. Two variables, senescence class, and tuber surface cracks contributed much to the variation explained by principal component 5. Yam anthracnose disease severity and tuber shape contributed most to the principal components 6 and 7, respectively. Based on the contribution of each of the measured traits to the most informative principal components, all the 24 variables were found to be relevant in discriminating the water yam accessions.
Correlation analysis revealed high and positive correlation between tuber yield components such as total tuber number (big, medium and small), the weight of the tuber (big, medium and small) and the length (big, medium and small) while negative correlations were observed between the tuber yield components and the two disease variables (Supplementary Fig. S1).
Assessment of phenotypic diversity using morphological attributes grouped the accessions into two distinct clusters (Fig. 1). Cluster one recorded the highest number of accessions (68), all of which were breeding lines. Accessions in cluster one had long tubers, high tuber yield and moderate tolerance to the yam mosaic virus (YMV) and to yam anthracnose diseases (YAD). Cluster two had 32 accessions, including the three farmer’s varieties (TDa291, TDa297, and TDa92_2). Accessions in this cluster were poor in tuber yield (small tubers) and susceptible to YAD and YMV.

Hierarchical cluster dendrogram based on ‘Gower’ genetic similarity matrix of 24 phenotypic traits showing the grouping pattern of the D. alata accessions evaluated.
Genetic diversity parameters and population structure with SNP markers
Minor allele frequency varied from 0.05 to 0.5, with an average of 0.25. Expected heterozygosity varied from 0.10 to 0.50, with an average of 0.34. The observed heterozygosity ranged from 0.03 to 0.69, with an average of 0.38 across the SNP markers. Average polymorphism information content was 0.36. The SNP markers were unequally distributed across the 21 chromosomes, with the highest number observed on chromosome 5 and the lowest observed on chromosome 11 (Supplementary Fig. S2). Chromosome 5 presented the highest SNP density followed by chromosomes 4 and 8.
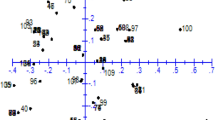
Using the Bayesian information criterion (BIC) and the discriminant analysis of principal components (DAPC), a maximum of k = 3 was obtained (Supplementary Fig. S3) which corresponded to the number of clusters under the DAPC. Estimation of the cluster membership revealed that cluster two had the highest number of accessions (42) followed by cluster one with 33 accessions and cluster three with the smallest number of accessions (27) (Fig. 2). The genetic similarity among and within the three DAPC clusters showed variation for the fixation index (FST) with the highest FST (0.13) obtained between accessions in cluster one and cluster three, while the lowest value was found between accessions of DAPC clusters one and two (FST = 0.06). Assessment of the genetic diversity among accessions within each cluster revealed very low FST variability of −0.012, −0.011 and −0.014 for clusters 1, 2 and 3, respectively (Supplementary Fig. S4).

Discriminant analysis of principal components (DAPC) using 6918 SNP markers. The axes represent the first two Linear Discriminants (LD). Each colour represents a cluster, and each dot represents an individual. Numbers represent the different subpopulations identified by DAPC analysis.
The genetic distance of 0.03 was observed between the intentional duplicate DNA samples of TDa1000512a and TDa1000512, while 0.02 was obtained between TDa1100242a and TDa1100242 (Supplementary Table S1). Hence, a threshold of 0 ≤ x ≤ 0.03 (where x is the genetic distance between two accessions) was set, and accessions with genetic distances lower than this threshold were considered as duplicates or closely related. The genetic distance varied from 0.02 to 0.83, and the highest value was observed between TDa291 and TDa0000005.
Based on the dissimilarity matrix generated from the SNP markers, the cophenetic coefficient correlation for the ward.D2 was 0.70, and the accessions categorized into three major clusters (Fig. 3). A total of 26 accessions were grouped into cluster one with genetic distance ranging from 0.02 to 0.75 (Supplementary Table S2). In this cluster, clones TDa0500056, TDa0200012, TDa0700015, and TDa98011174 shared most of alleles and hence they can be considered as closely related as shown through the hierarchical cluster. In the same cluster, TDa1401249 and TDa1401359 were found to have the lowest genetic distance (GD = 0.02) and can also be classified as duplicates or very closely related. Cluster two included 40 accessions, of which 38 are breeding lines and two landraces (TDa291 and TDa297). In this cluster, the lowest genetic distance (GD = 0.03) was observed between the TDa1000512a and TDa10000512 (intentional duplicate) while the highest genetic distance was observed between TDa291 (landrace) and TDa0000005 (breeding line). In cluster three, the genetic distance varied from 0.02 (TDa0100039 and TDa1100432) to 0.62 (TDa1100280 and TDa1100302). Apart from the intentional duplicate DNA TDa1100242 and TDa1100242a accessions like TDa1100432, TDa0100039 and TDa1400432 were found with very minimal genetic distances, hence can be considered as closely related.

Hierarchical cluster dendrogram based on ward.D2 method showing the genetic relationships among the yam accessions based on a Jaccard genetic similarity matrix obtained from the 6918 SNP markers. In each group, accessions with high genetic similarity (near 0) were grouped in the same branch.
Population structure analysis performed at K = 3 revealed high genetic diversity with a large proportion of admixture (individuals with multiple groups affiliation) (Fig. 4). Out of the 100 accessions, TDa0900026, TDa0200012, TDa0000005, TDa1100204, TDa0900128, TDa0700015, TDa9801174, TDa0500056, TDa0900146 were identified to be without admix and can be considered as pure lines. Also, the intentional duplicate genotype DNA TDa1000512 and TDa1000512a were found to have the same admix pattern (Fig. 4).

Admixture plot showing clustering of the D. alata accessions into three clusters based on the molecular data using Bayesian-based clustering analysis. A vertical bar represents each accession. The coloured sections in a bar indicate membership coefficients of the accessions in the different clusters. Identified subgroups are: cluster 1 (red), cluster 2 (black), and cluster 3 (blue).
Combined analysis of phenotypic and genotypic data
Hierarchical cluster generated from the phenotypic information was compared to that originating from the genotypic data, and very few clones (2%) were identified to be clustered into the same position across the two hierarchical clusters (Fig. 5). We noticed grouping patterns and membership changes between phenotypic and molecular information. Forty percent of the total accessions evaluated maintained their groups both across the phenotypic and molecular clustering.

Comparison of hierarchical cluster dendrograms from phenotypic (A) and the genotypic (B) data. The black lines in between the two dendrograms represent mismatched accessions while the purple lines are accessions in the same position from phenotypic to the genotypic cluster.
Genetic diversity assessment using the combined phenotypic and molecular information revealed the presence of three well defined genetic groups in the current set of materials (Supplementary Figs. S5 and S6). Cluster one is composed of 35 accessions (Fig. 6), all of which are breeding lines. Accessions in this group mainly originated from bi-parental crossing between TDa0500015 (female parent) and TDa0200012 (male parent) (Supplementary Table S3). Thirty-seven breeding lines and three landraces were grouped in cluster two. A total of 25 accessions were grouped in the third cluster. Seventy-two percent of the clones in this cluster originated from open pollination with TDa00000194 as the female plant.

Hierarchical cluster based on the combined phenotypic and molecular data using Ward.D2 method.
Discriminant analysis carried out on the three clusters emanating from the combined matrix showed a variation of both phenotypic traits and genotypic parameters (Table 2, Supplementary Fig. S6). Accessions categorized into cluster one have good tuber yield traits such as many tubers (long and large) without cracks (Table 2). Accessions grouped into cluster two expressed an average tuber yield but with rough tuber skin characteristics. The 25 accessions identified in cluster three were mainly characterized by less preferred tuber yield attributes such as small tuber weight, size, and length (Table 2). Concerning the genotypic parameters, polymorphic information content (PIC) varied considerably across the three groups while slight variation was observed for the rest of the parameters such as observed heterozygosity, expected heterozygosity (HE), minor allele frequency and major allele frequency (Table 2).
Dissimilarity matrix generated from the phenotypic data was compared with that obtained from the molecular data, and very low correlation (r = 0.097) was observed between the two matrices (Supplementary Fig. S7). High correlation values were detected between the joint matrix of combined distances and the genotypic and phenotypic information, with a Mantel test showing high (r = 0.84) and moderate (r = 0.53) values for phenotypic and genotypic data, respectively (Supplementary Fig. S7).
Discussion
This study dissected the level and extent of genetic diversity in a panel of 100 D. alata clones and varieties routinely utilized in yam breeding program at IITA using both phenotypic and molecular markers. The results of our multivariate analysis with phenotypic traits indicated substantial diversity in the current set of D. alata accessions with all the measured traits having relevance for characterization of yam germplasm. However, a subset of traits including number of tubers, tuber weight, tuber length and width, stem diameter, number of stems and yam anthracnose disease had very high contributions to the principal components and could be used to efficiently assess diversity in D. alata collections. Several authors have also reported the importance of phenotypic traits in unravelling the diversity and differentiation in yam genotypes13,14. Phenotypic characterization is very relevant to define germplasm accessions or breeding lines and serves as meaningful criteria for selecting materials with desirable traits for breeding endeavors.
Various types of molecular markers ranging from dominant to codominant are available to estimate and assess the level of genetic diversity. This study also used 6918 SNP markers from Diversity Array Technology (DArT) platform to elucidate the genetic diversity and population structure of the D. alata accessions. The usefulness of the DArT technology for genetic diversity analysis has been well established for many crops23,24,25. The three complementary approaches implemented here, namely DAPC, hierarchical cluster, and admixture ancestry to define the optimal number of groups all showed the presence of three genetic groups among the 100 winged yam accessions. The high agreement in the membership of the hierarchical clusters and DAPC groups found in this study was also observed by Wang et al.26 and Fatokun et al.27 in their study of genetic diversity and population structure of rice and cowpea accessions, respectively. The observed genetic differentiation among the studied materials was further confirmed by the high fixation index (Fst) value among accessions between clusters and low FST value among accessions within clusters. This indicated high connectivity among accessions within clusters and low connectivity among accessions between clusters as also reported in Cowpea27 and Cassava28 using DAPC analysis with SNP markers from genotyping by sequencing. The high cophenetic coefficient correlation (>0.70) observed here for the hierarchical cluster constructed based on the molecular data indicates the reliability of this approach to summarize the information of dissimilarity matrices21.
The high proportion of admixture observed in our study is an indication of gene flow in the population of D. alata. Many of the accessions in our study are breeding lines emanating from the hybridization of genotypes of diverse genetic backgrounds. Such hybridization has enhanced the genetic diversity as rightly asserted by Arnau et al.7. The few accessions identified with no admixture can be utilized to develop bi-parental crosses for QTL identification while focusing on the traits of interest in the breeding program.
The low correlation observed between the genotypic and phenotypic distance matrices observed in this study was also reported in sweet potatoes21, Cupuassu22, Jatropha29 and yellow passion fruit30, and in agreement with the inconsistencies observed between the clusters generated from the phenotypic and the molecular diversity analyses. The negligible association observed between the phenotypic and genotypic data could be because the variation in phenotypic traits may be as a result of one or few mutations and therefore often not concordant with the overall genetic distance between two accessions. Consequently, to assess the amount (and differentiation among e.g. geographical areas or other relevant entities) of total anonymous variability, DNA markers are often more efficient since they rely on numerous mutations along the entire genome. This low correlation between phenotypic and molecular diversity matrices could also be because the variation often detected by molecular markers is commonly of the non-adaptive type and hence not liable to natural and or artificial selection, unlike phenotypic characters which are usually subject to natural and artificial selection31. Additionally, molecular markers have been assumed to be selectively neutral, whereas the portions of the genome associated with phenotypic expression are subject to selection under environmental influence32,33. Several authors, therefore, suggest that the best way to identify divergence among accessions is the combined use of molecular and phenotypic data as these tools are complementary4,20,31,32. The low correlation between morphological and molecular data in the D. alata accessions generally suggests that the two data types are appropriate for a combined use, which can deepen understanding and discriminate the genotypes better due to the non-overlapping information.
The genetic diversity and differentiation in the population were also assessed using a joint dissimilarity matrix of the phenotypic and molecular marker data resulting in three distinct clusters. The high and moderate correlation observed between the combined dissimilarity matrix and the genotypic and phenotypic dissimilarity matrices, respectively, is an indication that genetic diversity analysis based on the joint matrix is a very valuable tool to cumulatively and reliably allocate genotypic and phenotypic information. The usefulness of the combined dissimilarity matrix of morphological and molecular data in deciphering the genetic variability and differentiation among accessions in this study corroborate the findings of Sartie et al.4, Andrade et al.21, Alves et al.22, Reis et al.30 and Alves et al.32. By analyzing morphological and molecular data together, Alves et al.32 found that the average dissimilarity among physic nut accessions improved by 156% and 64% with respect to the original mean phenotypic and molecular dissimilarities, respectively. Similarly, Martins et al.34 and Bosettiet al.35 observed a marked increase in the sampling intensity of the genetic diversity by combining phenotypic and molecular data. This could be as a result of the combination of non-overlapping information into one data set32.
In conclusion, our analysis of the diversity in the D. alata clones and cultivars using genotypic, phenotypic, and combined genotypic/phenotypic distances revealed ample genetic variation in the population. This diversity can be explored for D. alata improvement and genetic resource conservation. As a result of the inconsistencies and reshuffling of cluster membership observed across the phenotypic and genotypic characterizations, the combined matrix could be employed for genetic diversity analysis if one desires to use both the phenotypic and genotypic information as this approach maintains high correlation with both matrices and gives a cumulative overview of the total diversity in the population. Future analysis to estimate the combining ability of the accessions in the three clusters identified by the combined distance will enhance the partitioning of the accessions into distinct heterotic groups. The selection and hybridization of parental lines from the three genetic groups identified by the combined morphological and molecular marker analysis is expected to maximize genetic diversity and heterosis in yam breeding program.
Methods
Plant material
A panel of 100 winged yam accessions obtained from the IITA yam breeding program was used for this study. These accessions comprised of 97 breeding lines emanating from full-sib and half-sib breeding and three landrace cultivars (Supplementary Table S3).
Phenotypic evaluation
The accessions were planted in three locations in Nigeria: Ibadan (N7°40′19.62, E3°91′73.13) and Ikenne (N6°51′00.873, E3°41′48.528) in the south-west, and Ubiaja (N6°39′48.772″, E6°20′29.533) in the south-central part of the country. The experiment was laid out in a simple lattice design with three plants per genotype. Twenty-four phenotypic variables were recorded according to the yam crop ontology36. The different variables, collection period, and methods of assessment are summarized in Supplementary Table S4.
Genotyping
One gram of young, healthy and fully expanded leaves from fully grown plants in the field were collected per genotype. The leaf samples were immediately put in Ziploc bags and kept on ice before transferring to −80 °C for freeze drying. Deoxyribonucleic acid (DNA) was extracted from the leaf samples using the CTAB procedure with slight modification37. DNA quality was estimated for each sample based on electrophoresis in an agarose gel.
DArT genotyping was performed using the methodology described by Sansaloni et al.38 with slight modification. For the sequencing-based DArT genotyping, complexity reduction methods optimised for yam at DArT was used: PstI_ad/TaqI/HpaII_ad with TaqI restriction enzyme used to eliminate a subset of PstI –HpaII. PstI-site specific adapter was tagged with 102 different barcodes enabling encoding a plate of DNA samples to run within a single lane on an Illumina GAIIx. After the sequencing, FASTQ files generated by DArT were aligned against the yam genome reference available on (https://drive.google.com/drive/folders/0B54dLJoyUZ5SUDY5TzV2eGFWaWs) and hapmap file was generated by DArT.
Multivariate analysis of phenotypic data and hierarchical cluster construction
Analysis of variance was performed to determine differences among the accessions for the various traits across the three locations using the SAS version 9.439 according to the model:
where: Y = trait, µ = grand mean, E = environment effect (location), B(E) = Block effect in environment, G = genotype effect, GE = genotype by environment interaction, e = error.
The LSmeans from the genotype by environment analysis was used for principal component analysis in the FactorMiner and missMDA R packages40. The optimal number of factors to be retained was determined using the dimdesc function in R, and the contribution of each trait was determined using the principle of Peres-Nero et al.41. A dissimilarity matrix was generated using the Gower method implemented in cluster and graphics R packages42. Count variables (such as number of tubers, numbers of stems) were transformed using log ratio function in R. The final hierarchical cluster was then performed using the ward.D2 method in cluster R package42. Correlation among the different phenotypic variables was performed using the R software and result was displayed as heatmap.
Analysis of molecular data
To evaluate the sequencing error and reliability of the Diversity Array Technology for yam, two intentionally duplicated DNA samples were included. The Duplicated DNA was used to define the threshold for duplicate identification in the sample. The HapMap file received from the DArT platform was converted into a variant call format (VCF). A total of 22,140 SNP markers were identified from the raw data and after filtering with VCFtools43 for MAF (0.05), no missing data, depth (>5), Genotype Quality (GQ = 20), maximum and minimum allele = 2 and no indels, a total of 6,918 SNP markers were retained for further analysis.
Summary statistics including major and minor allele frequencies, heterozygosity both for SNP markers and genotypes were assessed using TASSEL software version 5.2.5144 and the plink command–freq and –hardy45. A population structure analysis was performed based on Admixture46 through the Bayesian Information Criterion (BIC). The optimal number of clusters was inferred using k-means analysis after varying the possible number of clusters from 2 to 40. A discriminant analysis of principal components (DAPC) was carried out on the identified clusters using the first 40 principal components in the adegenet R package47. Membership probabilities of the individuals for the different groups were estimated using a cut-off value of 40% suggested through the DAPC. Pairwise population differentiation statistics (FST) was calculated using VCFtools43 to estimate the genetic distance and the relationships among the different DAPC groups formed. A pairwise genetic distance matrix was calculated using the Jaccard method implemented in the phylentropy R package48. A Ward’s minimum variance hierarchical cluster dendrogram was then generated from the genetic distance matrix using the analyses of phylogenetics and evolution (ape) R package49,50.
Joint analysis of phenotypic and molecular data
Genetic groups were defined using a combination of the phenotypic and genotypic dissimilarity matrices. This joint matrix was generated by the summation of the genotypic and phenotypic dissimilarity matrices. A Mantel test was performed for analysis of correspondence between the dissimilarity matrices obtained from the phenotypic, genotypic and joint distance matrices using the Monte-Carlo method with 9999 permutations for estimation of significance. The hierarchical clusters generated from the phenotypic and genotypic data sets were compared using the viridis R package51 and the similarity of the two dendrograms assessed using the tanglegram function implemented in the dendextend R package52.
References
Asiedu, R. & Sartie, A. Crops that feed the world 1. Yams: Yams for income and food security. Food Security 2(4), 305–315, https://doi.org/10.1007/s12571-010-0085-0 (2010).
Cormier, F. et al. A reference high‐density genetic map of greater yam (Dioscorea alata L.). Theor. Appl. Genet. 132, 1733–1744, https://doi.org/10.1007/s00122-019-03311-6 (2019).
FAO. Food and Agriculture Organization of the United Nations Statistics database, FAOSTAT. http://www.fao.org/faostat/en/#data/QC (2018).
Sartie, A., Asiedu, R. & Franco, J. Genetic and phenotypic diversity in a germplasm working collection of cultivated tropical yams (Dioscorea spp.). Genet Resour Crop Evol. 59, 1753–65, https://doi.org/10.1007/s10722-012-9797-7 (2012).
Lebot, V. Tropical root and tuber crops: cassava, sweet potato, yams and aroids. Crop production science in horticulture, CAB books, CABI, Wallingford, UK 17 (2009).
Bhattacharjee, R. et al. An EST-SSR based genetic linkage map and identification of QTLs for anthracnose disease resistance in water yam (Dioscorea alata L.). PLoS ONE 13, e0197717, https://doi.org/10.1371/journal.pone.0197717 (2018).
Arnau, G. et al. Understanding the genetic diversity and population structure of yam (Dioscorea alata L.) using microsatellite markers. PLoS ONE 12, e0174150, https://doi.org/10.1371/journal.pone.0174150 (2017).
Onda, Y. & Mochida, K. Exploring genetic diversity in plants using high-throughput sequencing techniques. Curr Genomic 17, 358–367 (2016).
Islam, M. D., Chowdhury, R. U., Afroz, R., Rahman, S. & Haque, M. D. Characterization and maintenance of yam (Dioscorea spp.) germplasm. Bangladesh. J. Agril. Res. 36, 605–621 (2011).
Zannou, A., Struik, P., Richards, P. & Zoundjihékpon, J. Yam (Dioscorea spp.) responses to the environmental variability in the Guinea Sudan zone of Benin. Afr. J. Agric. Res. 10, 4913–4925 (2015).
Loko, Y. L., Adjatin, A., Dansi, A., Vodouhe, R. & Sanni, A. Participatory evaluation of Guinea yam (Dioscorea-cayenensis Lam.–D. rotundata Poir. complex) landraces from Benin and agro-morphological characterization of cultivars tolerant to drought, high soil moisture and chips storage insects. Genet. Resour. Crop Evol 62, 1181–1192, https://doi.org/10.1007/s10722-015-0221-y (2015).
Sheikh, N. & Kumar, Y. J. Morphological characterization of Meghalayan Dioscorea spp. (yam). North East India. Agr. Sci. Tech 19, 487–497 (2017).
Norman, P. E., Tongoona, P. & Shanahan, P. E. Diversity of the morphological traits of yam (Dioscorea spp.) genotypes from Sierra Leone. J. appl. biosci 45, 3045–3058 (2011).
Anokye, M., Tetteh, J. P. & Otoo, E. Morphological characterization of some water yam (Dioscorea alata L.) germplasm in Ghana. J. Agr. Sci. Tech. 4, 518–532 (2014).
Asemota, H. N., Ramser, J., Lopéz-peralta, C., Weising, K. & Kahl, G. Genetic variation and cultivar identification of Jamaican yam germplasm by Random Amplified Polymorphic DNA analysis. Euphytica 92, 341–35 (1996).
Egesi, C. N., Asiedu, R., Ude, G., Ogunyemi, S. & Egunjobi, J. K. AFLP marker diversity in water yam (Dioscorea alata L.). Plant Genet. Resour 4, 181–187 (2006).
Xia, L. et al. DArT for high-throughput genotyping of cassava (Manihot esculenta) and its wild relatives. Theor Appl Genet 110, 1092–1098, https://doi.org/10.1007/s00122-005-1937-4 (2005).
Rungis, D. E. et al. Assessment of genetic diversity and relatedness in the Latvian potato genetic resources collection by DArT genotyping. Plant Genet Resour-C. 15(1), 72–78, https://doi.org/10.1017/S1479262115000398 (2017).
Najaphy, A., Parchin, R. A. & Farshadfar, E. Comparison of phenotypic and molecular characterizations of some important wheat cultivars and advanced breeding lines. Aust. J. Crop Sci. 6, 326–332 (2012).
da Silva, M. J. et al. Phenotypic and molecular characterization of sweet sorghum accessions for bioenergy production. PloS ONE 12, e0183504 (2017).
Andrade, E. K. V. et al. Genetic dissimilarity among sweet potato genotypes using morphological and molecular descriptors. Acta Sci. Agron 39, 447–455, https://doi.org/10.4025/actasciagron.v39i4.32847 (2017).
Alves, R. M., de Sousa, S. C. R., de Albuquerque, P. S. B. & dos Santos, V. S. Phenotypic and genotypic characterization and compatibility among genotypes to select elite clones of cupuassu. Acta Amaz. 47, 175–184, https://doi.org/10.1590/1809-4392201602104 (2017).
Wenzl, P. et al. Diversity Arrays Technology (DArT) for whole-genome profiling of barley. Proc. Natl. Acad. Sci. USA 101, 9915–9920, https://doi.org/10.1073/pnas.0401076101 (2004).
Roorkiwal, M. et al. Exploring germplasm diversity to understand the domestication process in Cicer spp. Using SNP and DArT Markers. PLoS ONE 9, e102016, https://doi.org/10.1371/journal.pone.0102016 (2014).
Boakyewaa, A. G. et al. Genetic diversity and population structure of early-maturing tropical maize inbred lines using SNP markers. PLoS ONE 14, e0214810, https://doi.org/10.1371/journal.pone.0214810 (2019).
Wang, C.-H. et al. Genetic diversity and classification of Oryza sativa with emphasis on Chinese rice germplasm. Heredity (Edinb) 112, 489–496, https://doi.org/10.1038/hdy.2013.130 (2014).
Fatokun, C. et al. Genetic diversity and population structure of a mini-core subset from the world cowpea (Vigna unguiculata (L.) Walp.) germplasm collection. Sci. Rep. 8, 1–10, https://doi.org/10.1038/s41598-018-34555-9 (2018).
Rabbi, I. Y. et al. Tracking crop varieties using genotyping-by-sequencing markers: a case study using cassava (Manihot esculenta Crantz). BMC Genetics 16, 1–11, https://doi.org/10.1186/s12863-015-0273-1 (2015).
Sunil, N. et al. Correlating the phenotypic and molecular diversity in Jatropha curcas L. Biomass and bioenergy 35(3), 1085–1096 (2011).
Reis, R. V. D., Viana, A. P., Oliveira, E. J. D. & Silva, M. G. D. M. Phenotypic and molecular selection of yellow passion fruit progenies in the second cycle of recurrent selection. Crop Breed. Appl. Biotechnol. 12, 17–24 (2012).
Singh, S. P., Nodari, R., Gepts, P. & Singh, S. P. Genetic diversity in cultivated common bean: I. allozymes. Crop Sci 31, 19–23, https://doi.org/10.2135/cropsci1991.0011183X003100010004x (1991).
Alves, A. A. et al. Joint analysis of phenotypic and molecular diversity provides new insights on the genetic variability of the Brazilian physic nut germplasm bank. Genet. Mol. Biol. 36, 371–381, https://doi.org/10.1590/S1415-47572013005000033 (2013).
Collard, B. C., Jahufer, M. Z. Z., Brouwer, J. B. & Pang, E. C. K. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts. Euphytica 142(1–2), 169–196 (2005).
Martins, F. A., Carneiro, P. C. S., Silva, D. J. H. D., Cruz, C. D. & Carneiro, J. E. D. S. Integration of data in studies of genetic diversity of tomato. Pesquisa Agropecuária Brasileira 46(11), 1496–1502 (2011).
Bosetti, F., Zucchi, M. I. & Pinheiro, J. B. Molecular and morphological diversity in Japanese rice germplasm. Plant Genetic Resources 9(2), 229–232 (2011).
Asfaw, A. Standard operating protocol for yam variety performance evaluation trial. IITA, Ibadan, Nigeria, 27 (2016).
Dellaporta, S. L., Wood, J. & Hicks, J. B. A plant DNA minipreparation: version II. Plant Mol. Biol. Rep. 1, 19–21 (1983).
Sansaloni, C. et al. Diversity Arrays Technology (DArT) and next-generation sequencing combined: genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proceedings 5(7), P54, https://doi.org/10.1186/1753-6561-5-s7-p54 (2011).
SAS. Statistical Analysis System Institute, SAS/STATS User’s Guide. SAS Institute Inc., Cary, NC, USA (2012).
Le, S., Josse, J. & Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 25, 1–18, http://www.jstatsoft.org/v25/i01/ (2008).
Peres-Neto, P. R., Jackson, D. A. & Somers, K. M. Giving meaningful interpretation to ordination axes: assessing loading significance in principal component analysis. Ecology 84, 2347–2363 (2003).
Maechler, M. et al. Methods for cluster analysis. Much extended the original from Rousseeuw, P., Struyf, A. & Hubert, H. based on Kaufman and Rousseeuw 1990 “Finding Groups in Data”. https://svn.r-project.org/R-packages/trunk/cluster (2019).
Danecek, P. 1000 genomes project analysis group. The variant call format and VCFtools. Bioinformatics (Oxford, England) 27, 2156–2158, https://doi.org/10.1093/bioinformatics/btr330 (2011).
Bradbury, P. J. et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635, https://doi.org/10.1093/bioinformatics/btm308 (2007).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575, https://doi.org/10.1086/519795 (2007).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664, https://doi.org/10.1101/gr.094052.109 (2009).
Jombart, T., Devillard, S. & Balloux, F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics 11, 1–15, https://doi.org/10.1186/1471-2156-11-94 (2010).
Drost, H. G. Philentropy: Information theory and distance quantification with R. J. open source software 3, 1–4, https://doi.org/10.21105/joss.00765 (2018).
Paradis, E., Claude, J. & Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290, https://doi.org/10.1093/bioinformatics/btg412 (2004).
R Core Team. R: A language and environment for statistical computing. R foundation for statistical computing, Vienna, Austria, https://www.R-project.org/ (2018).
Garnier, S. Viridis: Default color maps from ‘matplotlib’. R package version 0.5.1, https://CRAN.R-project.org/package=viridis (2018).
Tal, G. Dendextend: an R package for visualizing, adjusting, and comparing trees of hierarchical clustering. Bioinformatics 31, 3718–3720, https://doi.org/10.1093/bioinformatics/btv428 (2015).
Acknowledgements
We acknowledge funding from IITA through a research grant (OPP1052998) received from the Bill and Melinda Gates Foundation.
Author information
Authors and Affiliations
Contributions
Asrat Asfaw., Patrick Adebola and Robert Asiedu designed the research presented here, Asrat Asfaw., Paterne Agre., Flora Asibe., Alex Edemodu., Kwabena Darkwa executed the field research, Paterne Agre managed the genotyping, Asrat Asfaw., Patrick Adebola., Guillaume Bauchet designed the analysis, Paterne Agre conducted the analyses. Paterne Agre., Kawabena Darkwa and Asrat Asfaw wrote the first draft; all authors read and edited the subsequent versions of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Agre, P., Asibe, F., Darkwa, K. et al. Phenotypic and molecular assessment of genetic structure and diversity in a panel of winged yam (Dioscorea alata) clones and cultivars. Sci Rep 9, 18221 (2019). https://doi.org/10.1038/s41598-019-54761-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-54761-3
- Springer Nature Limited
This article is cited by
-
Genomic prediction for agronomic traits in a diverse Flax (Linum usitatissimum L.) germplasm collection
Scientific Reports (2024)
-
Assessment of genetic diversity among sago palms (Metroxylon sagu Rottb.) in Bengkulu, Indonesia using simple sequence repeats
Genetic Resources and Crop Evolution (2024)
-
Exploration and morphological characterization of local yams from the Tanimbar Islands, Maluku, Indonesia
Genetic Resources and Crop Evolution (2024)
-
Genetic Diversity of Cowpea Parental Lines Assembled for Breeding in Uganda
Plant Molecular Biology Reporter (2023)
-
Phenotypic, chemical component and molecular assessment of genetic diversity and population structure of Morinda officinalis germplasm
BMC Genomics (2022)