
Abstract
Whole body lean mass (WBLM) is a heritable trait predicting sarcopenia. To identify genomic locus underlying WBLM, we performed a genome-wide association study of fat-adjusted WBLM in the Framingham Heart Study (FHS, N = 6,004), and replicated in the Kansas City Osteoporosis Study (KCOS, N = 2,207). We identified a novel locus 3p27.1 that was associated with WBLM (lead SNP rs3732593 P = 7.19 × 10−8) in the discovery FHS sample, and the lead SNP was successfully replicated in the KCOS sample (one-sided P = 0.04). Bioinformatics analysis found that this SNP and its adjacent SNPs had the function of regulating enhancer activity in skeletal muscle myoblasts cells, further confirming the regulation of WBLM by this locus. Our finding provides new insight into the genetics of WBLM and enhance our understanding of sarcopenia.
Similar content being viewed by others

Introduction
Sarcopenia is defined as a progressive, and generalized loss of skeletal muscle mass, strength and function1. Sarcopenia plays a vital role in the frailty process, also being a key player in its incubation period and it causes serious consequences through frailty, such as decreased function, disability and eventual death2,3. According to Khosla et al.4, the age- and sex-adjusted prevalence of sarcopenia varied from 6 to 15% among subjects 65 years of age or over. The muscular tissue, as characterized by Lean Body Mass (LBM), is frequently used to predict sarcopenia. LBM can be measured accurately by dual energy X-ray absorptiometry (DXA).
Previous studies showed that LBM is under genetic control, with heritability over 50%5,6. Previous studies have identified dozens of genomic loci associated with LBM7,8,9,10,11. Among them, Zillikens et al.12 identified five loci in/near HSD17B11, VCAN, ADAMTSL3, IRS1, and FTO that were associated with lean body mass. Medina-Gomez et al.13 conducted a bivariate genome-wide association meta-analysis of pediatric musculoskeletal traits and revealed pleiotropic effects at the SREBF1/TOM1L2 locus. However, to date, the identified loci only explain a small proportion of the variation observed for a particular phenotype, and the majority of hidden heritability is yet to be identified.
In this study, we performed a GWAS of WBLM using the Framingham Heart Study (FHS) as discovery sample and the Kansas City Osteoporosis Study (KCOS) as replication sample. In addition, we conducted a serious of bioinformatic analysis to explore the functional relevance of the identified variants.
Materials and Methods
All the methods were conducted in accordance with the guidelines and regulations of the Soochow university institutional review board (for the Framingham heart study sample) and the University of Missouri Kansas City institutional review board (for the Kansas City osteoporosis study). The Institutional Review Boards of University of Missouri Kansas City and the Soochow university approved the study. All participants signed informed consent before participating.
Discovery sample
The Framingham heart study (FHS) was accessed through the database of genotype and phenotype (dbGAP) portal. The FHS sample is a longitudinal and prospective cohort consisting of over 16,000 individuals spanning three generations of European ancestry. All participants underwent dual-energy X-ray absorptiometry (DXA, Lunar Corp., Madison, WI, USA) scan during different examinations. More details about the FHS sample have been described elsewhere14.
A subset sample of the FHS cohort was genotyped by the Affymetrix high-throughput 500 K genotyping array plus a supplemental 50 K genotyping array. These two genotype sets were merged together to form a single dataset of ~550,000 SNPs.
After checking the availability of both genotypes and phenotypes, we identified 6,004 Caucasian subjects aged 23–93 years, of which 637, 2,222, and 3,145 were from the original, offspring, and third generations, respectively.
Replication sample
The replication sample was the Kansas City Osteoporosis Study (KCOS). The KCOS is a cross-sectional study of osteoporosis with 2,286 unrelated European ancestry participants living in and around Kansas City, Missouri, USA. Participants are normal healthy subjects defined by a complete set of exclusion criteria, as described elsewhere15. The KCOS cohort was genotyped by the Affymetrix SNP 6.0 genotyping array. WBLM was measured by a DXA bone densitometer (QDR 4500 W, Hologic Inc., Bedford, MA, USA) according to the manufacture protocol.
After checking the availability of both genotypes and phenotypes, we identified 2,207 individuals aged 18–92 years.
Phenotype modeling
In both discovery and replication samples, the stepwise linear regression model implemented in the R function stepAIC was used to screen the significance of covariates, including whole body fat mass, gender, age, age^2, height, height^2 and the first five principle components derived from genome-wide genotype data. Raw WBLM was adjusted by significant covariates, and the residuals were normalized by inverse quantiles of standard normal distribution.
Genotype quality control
We followed strict genotype quality control (QC) procedure at both individual and SNP levels. At the individual level, genetic sex was inferred from genotype data on X-chromosome with PLINK16 and was compared with the self-reported sex. Individuals of inconsistent sex were removed. At the SNP level, SNP that violates the Hardy-Weinberg equilibrium (P < 1.0 × 10−5) were removed. Population outliers were monitored by genotype-derived principle components, and if present, outliers were removed. In the FHS sample, SNPs with the Mendel error were set to missing value.
Genotype imputation
Both discovery and replication samples were imputed using sequencing data generated by the 1000 genomes project17. Specifically, phased variants of 240 individuals of European ancestry were downloaded from the project website. Haplotypes of bi-allelic variants, including SNPs and bi-allelic insertions/deletions (INDELs), were extracted to form reference panels for imputation. As a QC procedure, variants with zero or one copy of minor alleles were removed.
Before imputation, it is necessary to check for the consistency of allele strandedness between the test sample and the reference sample. The chi-square test was used to examine the consistency. SNPs that failed the consistency test (p < 1.0 × 10−6) were transformed into the reverse strand in the test sample. SNPs that again failed the consistency test were removed. Imputation was performed with FISH18, a fast and accurate diploid genotype imputation algorithm.
The imputation certainty was measured by the imputation score r2, which was defined as the correlation between imputed dosage and the best imputed genotype. Variants of low imputation score (r2 < 0.3) or of low frequency (MAF < 0.05) were excluded from subsequent analyses.
Association testing
In the FHS sample, an additive mode of inheritance was applied to test genetic association between normalized phenotype residuals with genotyped and imputed genotypes. A mixed linear regression model was applied to account for genetic relatedness within each pedigree in the FHS sample19. Association test was examined within the variance-components framework.
Association was examined in the KCOS sample by a linear regression model with MACH2QTL20.
Functional annotation
Functional annotation of the identified SNPs was performed using the bioinformatical software HaploReg21. HaploReg provides functional information for non-coding SNPs with multiple functional categories, including conservation sites, DNase hypersensitivity sites (DHS), transcription factor binding sites (TFBS), promoter sites, enhancer sites, and others. We annotated lead SNP and its neighbor SNPs with strong LD pattern (r2 > 0.8).
We used the GTEx project dataset22 and the Westra et al.‘s study23 to perform cis-eQTL analysis. The GTEx project collects and analyzes a variety of human tissues from donors of the same dense genotype to assess genetic variation within their genome. We downloaded the summary results of skeletal muscle tissue from the GTEx website (V7) (https://www.gtexportal.org/home/). The Westra et al.‘s study performed eQTL meta-analysis reported in non-transformed peripheral blood samples of 5,311 individuals, with replication in 2,775 individuals23. We obtained cis-eQTL results from the study website (http://www.genenetwork.nl/bloodeqtlbrowser/).
Replication of previously identified loci
We assessed the GWAS Catalog (https://www.ebi.ac.uk/gwas/) web portal to identify loci that were previously reported for lean mass. For each locus, we listed effect size and p-value of the lead SNP in the present study to evaluate the replicability of those previously reported loci.
Ethics approval and consent to participate
The study was approved by the local institutional review board of all agencies. All participants signed informed consent before participating in the study.
Results
Basic characteristics of both discovery and replication samples are listed in Table 1. A total of 6,004 subjects are available in the FHS sample for analysis, including 637, 2,222 and 3145 from the first, offspring and third generation, respectively, 3,479 of whom are women. In the KCOS sample, a total of 2,207 subjects are available for analysis, 1,691 of whom are women.
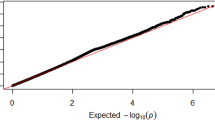
In the FHS sample, after performing genotype imputation and removing variants with low frequency and poor imputation certainty, a total of 6,879,267 variants were available for analysis, of which 88% were SNPs and the rest were bi-allelic deletion/insertion variants (DIVs). Overall genomic control inflation factor was 1.09. The logarithmic quantile–quantile (QQ) plot results is displayed in Fig. 1. The QQ plot shows a significant deviation in the distribution tail, implying the presence of a true association signals.

QQ plot. Results were plotted for fat-adjusted whole body lean mass in the Framingham heart study.
Manhattan plot of association results across the genome is displayed in Fig. 2. At the genome-wide significance (GWS, 5.0 × 10−8) level, a total of 4 SNPs at one single locus 6p21.1 were identified, with the lead SNP rs513688 (P = 7.48 × 10−9). In a previous study, this locus was found to be associated with leg lean mass24 with the same lead SNP, indicating that they arise from one single association signal.

WBLM genome-wide association manhattan plot.
SNPs from one additional locus 3p27.1 showed evidence of association. The lead SNP rs3732583 at this locus nearly reaches the GWS level (P = 7.19 × 10−8). The main association results are listed in Table 2.
In the replication sample, rs3732593 was consistent in effect direction to that in the discovery sample. Further, the replication association is nominal significant (one-sided P = 0.04), further supporting the association at this locus.
Regional plot of the lead SNP rs3732593 for the novel locus 3p27.1 is displayed in Fig. 3.

Regional plot of the associated region 3p27.1.
Functional annotation
We used HaploReg to annotate the lead SNP rs3732593 at 3p27.1 and its neighboring SNPs (LD r2 > 0.8). rs3732593 is located in the intron of MCF2 cell line derived transforming sequence-like 2 (MCF2L2) gene. In the latest GTEx cis-eQTL summary results (version 7), rs3732593 is associated with the expression of ATPase phospholipid transporting 11B (ATP11B, P = 0.01) gene in skeletal muscle tissue. In the Roadmap epigenomic study, it is predicted to have enhancer site activity in skeletal myoblasts cells, as implied by core 15-state model and H3K4me1 histone mark. It has 23 neighboring SNPs in strong LD (r2 > 0.8); all are located in intron of MCF2L2. Three of them (rs6767909, rs6770912 and rs12630205, all within a 12.5 kb window) are associated with the expression of MCF2L2 in the study of Westra et al. in whole blood tissue. They are also associated with the expression of another gene betaGal beta-1,3-N-acetylglucosaminyltransferase 5 (B3GNT5) in the same study.
Replication of previously reported loci
The previous studies identified 22 loci for lean mass traits; none of them are significant at the GWS level in the present study. This is probably because of the limited sample size used in the present study.
At the nominal level (P < 0.05), on the other hand, the present study did replicate a total of 4 previously identified loci. Among them, 3 loci are further consistent in effect direction, including 2q36.3 (lead SNP rs2943656, beta = −0.06, p = 2.0 × 10−3), 16q12.2 (rs9936385, beta=0.06, p = 2.4 × 10−3) and 1p36.12 (rs6684375, beta= 0.05, p = 4.9 × 10−2).
Discussion
In this study, we performed a genome-wide association study of fat adjusted whole body lean mass in 6,004 participants from the FHS, and performed replication in the KCOS. we identified one novel locus 3p27.1 that was associated with WBLM after adjustment by whole body fat mass (lead SNP rs3732593 P = 7.19 × 10−8).
Previous study suggested that the third decade is a turning point in muscle mass and the age of 27 years as the threshold, beyond which skeletal mass began to be inversely related to the age of men and women25. Other studies showed that men’s lean body mass and skeletal muscle mass were significantly higher than women’s4,26. In our study, the age and gender were included as covariates. In addition, age square was included as covariate too, to account for the non-linear influence of age to lean mass (e.g., increase to the peak, then decrease).
Moreover, both fat mass and lean mass are important body composition and they are highly correlated. In this study, because of the consistent correlation between fat and lean mass, we chose whole body fat mass as one of the covariates for adjusting whole body lean mass to eliminate the effects of confounding factors. This allows us to estimate the independent effects of lean mass.
Functional annotations highlighted several candidate genes, including MCF2L2, ATP11B and B3GNT5. Among them, ATP11B is phosphorylated in its intermediate state and drives uphill transport of ions across membranes, which are members of the P-type ATPase. P-type ATPases are a family of ATP-dependent ion transporters. The genes most closely related to ATP11B are ATPase phospholipid transporting 11 A (ATP11A) and the type IIB sarco/endoplasmic reticulum Ca2+ transporter (SERCA1)27. SERCA Ca2+-ATPases is intracellular pumps located in the sarcoplasmic or endoplasmic reticula of muscle cells28,29. It is involved in muscle stimulation and contraction. Mutations in gene SERCA1 cause changes in muscle properties and dysfunction-related diseases. However, the functions of ATP11B gene related to skeletal muscle is still unknown.
MCF2L2 gene encodes a guanine nucleotide exchange factor of the Rho family, which plays an important role in the signaling pathway of the Rho protein. Studies have shown that the Rho-guanine nucleotide exchange factor domain of Obscurin can activate RhoA signaling in skeletal muscle30. As a small GTPase of the rho family, RhoA has been well documented to regulate actin reorganization, regulate transcription and participate in cell cycle control31,32. It plays a key role in the development and maintenance of skeletal muscle.
Meta-analysis of multiple individual studies has now become the mainstream of GWAS analysis. The biggest advantage of meta-analysis is to enhance the statistical power of identifying true genetic associations by expanding the sample size. However, modern meta-analysis introduces heterogeneity problems because each sample study was not designed under the same conditions. Although individual studies comprising smaller sample sizes than typical meta-analyses, they are more uniform in terms of sample and experimental design than the latter. In addition, both the discovery and replication samples in this study represented populations of European ancestry. This eliminates the erroneous correlation signals that may result from mismatched ancestors between samples. Therefore, there is irreplaceable value in individual research.
There were 2 different DXA machines used in the discovery and the replication samples, respectively. However, only one machine was used within each sample, and association analysis was done within each sample. As we did not combine raw phenotypes of the two samples, no systematic bias due to machine type would generate. Within each sample, the same phenotype modeling approach was used. The phenotype being analyzed in both samples follows strictly a standard normal distribution so that the regression coefficient was comparable between both samples.
The identified lead SNP rs3732593 was extremely significant (P = 7.19 × 10−8) in the discovery FHS sample but was only nominally significant (P = 0.04) in the replication sample. Due to the winner’s curse effect, replication p-value is difficult to be as low as that in the discovery sample even for true association33. Instead, a nominally significant replication p-value would suggest a successful replication. In addition to p-value, we also checked the effect direction across the discovery and the replication samples. The consistent effect direction strenghtened our confidence towards true association at this locus.
In conclusion, by performing a genome-wide association study and related functional analysis, we identified a novel locus 3p27.1 that is associated with whole body lean mass. Our findings provide useful insights that enhance our understanding of the genetic pathogenesis of sarcopenia.
Data availability
Summary results are available upon request to the corresponding author.
References
Fielding, R. A. et al. Sarcopenia: an undiagnosed condition in older adults. Current consensus definition: prevalence, etiology, and consequences. International working group on sarcopenia. J. Am. Med. Dir. Assoc. 12, 249–256, https://doi.org/10.1016/j.jamda.2011.01.003 (2011).
Fried, L. P. et al. Frailty in older adults: evidence for a phenotype. J. Gerontol. A Biol. Sci. Med. Sci 56, M146–156, https://doi.org/10.1093/gerona/56.3.m146 (2001).
Raphael, D. et al. Frailty: a public health perspective. Can. J. Public. Health 86, 224–227 (1995).
Melton, L. J. III et al. Epidemiology of sarcopenia. J. Am. Geriatr. Soc. 48, 625–630 (2000).
Arden, N. K. & Spector, T. D. Genetic influences on muscle strength, lean body mass, and bone mineral density: a twin study. J. bone Miner. research: Off. J. Am. Soc. Bone Miner. Res. 12, 2076–2081, https://doi.org/10.1359/jbmr.1997.12.12.2076 (1997).
Nguyen, T. V., Howard, G. M., Kelly, P. J. & Eisman, J. A. Bone mass, lean mass, and fat mass: same genes or same environments? Am. J. Epidemiol. 147, 3–16, https://doi.org/10.1093/oxfordjournals.aje.a009362 (1998).
Guo, Y. F. et al. Suggestion of GLYAT gene underlying variation of bone size and body lean mass as revealed by a bivariate genome-wide association study. Hum. Genet. 132, 189–199, https://doi.org/10.1007/s00439-012-1236-5 (2013).
Sun, L. et al. Bivariate genome-wide association analyses of femoral neck bone geometry and appendicular lean mass. PLoS one 6, e27325, https://doi.org/10.1371/journal.pone.0027325 (2011).
Liu, X. G. et al. Genome-wide association and replication studies identified TRHR as an important gene for lean body mass. Am. J. Hum. Genet. 84, 418–423, https://doi.org/10.1016/j.ajhg.2009.02.004 (2009).
Hai, R. et al. Genome-wide association study of copy number variation identified gremlin1 as a candidate gene for lean body mass. J. Hum. Genet. 57, 33–37, https://doi.org/10.1038/jhg.2011.125 (2012).
Hai, R. et al. Bivariate genome-wide association study suggests that the DARC gene influences lean body mass and age at menarche. Sci. China. Life Sci. 55, 516–520, https://doi.org/10.1007/s11427-012-4327-6 (2012).
Zillikens, M. C. et al. Large meta-analysis of genome-wide association studies identifies five loci for lean body mass. Nat. Commun. 8, 80, https://doi.org/10.1038/s41467-017-00031-7 (2017).
Medina-Gomez, C. et al. Bivariate genome-wide association meta-analysis of pediatric musculoskeletal traits reveals pleiotropic effects at the SREBF1/TOM1L2 locus. Nat. Commun. 8, 121, https://doi.org/10.1038/s41467-017-00108-3 (2017).
Cupples, L. A. et al. The Framingham Heart Study 100K SNP genome-wide association study resource: overview of 17 phenotype working group reports. BMC Med. Genet. 8(Suppl 1), S1, https://doi.org/10.1186/1471-2350-8-S1-S1 (2007).
Deng, H. W. et al. A genomewide linkage scan for quantitative-trait loci for obesity phenotypes. Am. J. Hum. Genet. 70, 1138–1151, https://doi.org/10.1086/339934 (2002).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575, https://doi.org/10.1086/519795 (2007).
Genomes Project, C. et al. An integrated map of genetic variation from 1,092 human genomes. Nat. 491, 56–65, https://doi.org/10.1038/nature11632 (2012).
Zhang, L. et al. FISH: fast and accurate diploid genotype imputation via segmental hidden Markov model. Bioinforma. 30, 1876–1883, https://doi.org/10.1093/bioinformatics/btu143 (2014).
Zhang, L., Li, J., Pei, Y. F., Liu, Y. & Deng, H. W. Tests of association for quantitative traits in nuclear families using principal components to correct for population stratification. Ann. Hum. Genet. 73, 601–613, https://doi.org/10.1111/j.1469-1809.2009.00539.x (2009).
Li, Y., Willer, C. J., Ding, J., Scheet, P. & Abecasis, G. R. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 34, 816–834, https://doi.org/10.1002/gepi.20533 (2010).
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–934, https://doi.org/10.1093/nar/gkr917 (2012).
Consortium, G. T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585, https://doi.org/10.1038/ng.2653 (2013).
Westra, H. J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243, https://doi.org/10.1038/ng.2756 (2013).
Pei, Y. F. et al. Two functional variants at 6p21.1 were associated with lean mass. Skelet. Muscle 9, 28, https://doi.org/10.1186/s13395-019-0212-3 (2019).
Silva, A. M. et al. Ethnicity-related skeletal muscle differences across the lifespan. Am. J. Hum. Biol. 22, 76–82, https://doi.org/10.1002/ajhb.20956 (2010).
Janssen, I., Heymsfield, S. B., Wang, Z. M. & Ross, R. Skeletal muscle mass and distribution in 468 men and women aged 18-88 yr. J. Appl. Physiol. 89, 81–88, https://doi.org/10.1152/jappl.2000.89.1.81 (2000).
Halleck, M. S., Schlegel, R. A. & Williamson, P. L. Reanalysis of ATP11B, a type IV P-type ATPase. J. Biol. Chem. 277, 9736–9740, https://doi.org/10.1074/jbc.M200240200 (2002).
Odermatt, A. et al. Mutations in the gene-encoding SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+ ATPase, are associated with Brody disease. Nat. Genet. 14, 191–194, https://doi.org/10.1038/ng1096-191 (1996).
Odermatt, A. et al. Sarcolipin regulates the activity of SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 273, 12360–12369, https://doi.org/10.1074/jbc.273.20.12360 (1998).
Ford-Speelman, D. L., Roche, J. A., Bowman, A. L. & Bloch, R. J. The rho-guanine nucleotide exchange factor domain of obscurin activates rhoA signaling in skeletal muscle. Mol. Biol. Cell 20, 3905–3917, https://doi.org/10.1091/mbc.E08-10-1029 (2009).
Brown, J. H., Del, R. D. P. & Sussman, M. A. The Rac and Rho hall of fame: a decade of hypertrophic signaling hits. Circulation Res. 98, 730–742, https://doi.org/10.1161/01.RES.0000216039.75913.9e (2006).
Kjoller, L. & Hall, A. Signaling to Rho GTPases. Exp. Cell Res. 253, 166–179, https://doi.org/10.1006/excr.1999.4674 (1999).
Palmer, C. & Pe’er, I. Statistical correction of the Winner’s Curse explains replication variability in quantitative trait genome-wide association studies. PLoS Genet. 13, e1006916, https://doi.org/10.1371/journal.pgen.1006916 (2017).
Acknowledgements
We appreciate all the volunteers who participated into this study. The Framingham Heart Study is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with Boston University (Contract No.N01-HC-25195). This manuscript was not prepared in collaboration with investigators of the Framingham Heart Study and does not necessarily reflect the opinions or views of the Framingham Heart Study, Boston University, or NHLBI. Funding for SHARe Affymetrix genotyping was provided by NHLBI Contract N02-HL-64278. SHARe Illumina genotyping was provided under an agreement between Illumina and Boston University. Funding support for the Framingham Whole Body and Regional Dual X-ray Absorptiometry (DXA) dataset was provided by NIH grants R01 AR/AG 41398. The datasets used for the analyses described in this manuscript were obtained from dbGaP at http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap through dbGaP accession phs000342.v14.p10. LZ and YFP are partially supported by the national natural science foundation of China (31571291, 31771417 and 31501026) and a project of the priority academic program development (PAPD) of Jiangsu higher education institutions. HS and HWD are partially supported by the National Institutes of Health (R01AR059781, P20GM109036, R01MH107354, R01MH104680, R01GM109068, R01AR069055, U19AG055373, R01DK115679), the Edward G. Schlieder Endowment and the Drs. W. C. Tsai and P. T. Kung Professorship in Biostatistics from Tulane University. The numerical calculations in this paper have been done on the supercomputing system of the National Supercomputing Center in Changsha. The funders had no role in study design, data collection and analysis, results interpretation or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
L.Z. designed the study. L.Z., Y.F.P., S.R., Y.X.Z., H.S., Q.T. and H.W.D. collected the data. Y.F.P., L.Z. and L.L. analyzed the data. S.R., Y.X.Z., L.L., Z.X.J. X.H. and Y.L. performed the literature search. S.R., Y.X.Z., L.L., Z.X.J., X.H., Y.L., Y.F.P., H.W.D. and L.Z. interpreted the data. S.R., Y.X.Z. generated the figures. S.R. and Y.X.Z. drafted the early version of the manuscript. L.Z. supervised the study. All authors were involved in writing the paper and had final approval of the submitted and published versions.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ran, S., Zhang, YX., Liu, L. et al. Association of 3p27.1 Variants with Whole Body Lean Mass Identified by a Genome-wide Association Study. Sci Rep 10, 4293 (2020). https://doi.org/10.1038/s41598-020-61272-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-61272-z
- Springer Nature Limited