
Abstract
Distance decay principles predict that species with larger geographic ranges would have greater intraspecific genetic diversity than more restricted species. However, invasive pest species may not follow this prediction, with confounding implications for tracking phenomena including original ranges, invasion pathways and source populations. We sequenced an 815 base-pair section of the COI gene for 441 specimens of Bactrocera correcta, 214 B. zonata and 372 Zeugodacus cucurbitae; three invasive pest fruit fly species with overlapping hostplants. For each species, we explored how many individuals would need to be included in a study to sample the majority of their haplotype diversity. We also tested for phylogeographic signal and used demographic estimators as a proxy for invasion potency. We find contrasting patterns of haplotype diversity amongst the species, where B. zonata has the highest diversity but most haplotypes were represented by singletons; B. correcta has ~7 dominant haplotypes more evenly distributed; Z. cucurbitae has a single dominant haplotype with closely related singletons in a ‘star-shape’ surrounding it. We discuss how these differing patterns relate to their invasion histories. None of the species showed meaningful phylogeographic patterns, possibly due to gene-flow between areas across their distributions, obscuring or eliminating substructure.
Similar content being viewed by others


Introduction
A fundamental assumption of species-level research is that sampling specimens from across their range accurately represents intraspecific genetic diversity1,2,3,4. This tenet is based on population-level processes such as genetic drift and selection, which follow the distance decay principle5 resulting in regional differences that increase with geographic distance as the probability of gene-flow between more distant populations decreases. These divergences may be augmented in heterogenous landscapes or under strong environmental gradients6. Indeed, studies using various, predominantly native, taxa over large geographic ranges typically find that species with broader distributions, on average, have more intraspecific diversity than species with more restricted distributions (e.g.3,4,7). However, depending on the life-history characters and evolutionary history, intraspecific genetic diversity can be regionally heterogeneous and may vary widely between taxa (e.g.6,8).
We tested distance decay assumptions with three of the worst agricultural pests in Southeast Asia, since this has implications for their control, quarantine and the reconstruction of invasion patterns based on demographic history. When intraspecific variation is correlated with geography it becomes possible to trace the geographic origins of invasive species and provides a framework to prevent future incursions and better manage invasive populations9,10,11. Moreover, populations can be adapted to their local environments, which influences the chance of a successful future invasion12 or can even reinvigorate invasions by enabling access to new hosts or through biocontrol resistance13,14. If, on the other hand, no intraspecific variation is detected in the invasive range, it suggests a single small initial invasive population or multiple invasions from the same source. In this situation, finding the source of the invasion can be important in predicting infestation potential and to locate natural enemies for control. This kind of information would be important for understanding the invasion threat posed by other serious pests, an increasingly common problem as globalization intensifies.
When using molecular methods for species identification, commonly using the cytochrome c oxidase subunit I (COI) gene, most automated species delimitation algorithms, such as BIN, CROP, jMOTU, GMYC and ABGD (for comparisons see15,16,17), rely, at least in part, on there being a diagnosable difference between intra- and interspecific variation and then averaging these values within phylogenetic groupings, in order to ultimately detect differences between species. Commonly, large DNA barcoding initiatives have focused on sequencing as many species as possible, with the implicit assumption that genetic diversity has been adequately sampled to reflect species identity2,18,19,20,21,22. Few studies have focused on haplotype accumulation, and those that have studied it found that even with sample sizes >50, generally only a small portion of the actual diversity was sampled (e.g.3,23). Little attention has been paid to what qualifies as sufficient sampling for reliable delimitation, identification or population assignment, with potentially significant ramifications for monitoring pest species in the context of control and quarantine.
Larvae of the globally distributed fruit fly family Tephritidae feed on fruit or flowers and roughly 250 of the ca. 5,000 species have become agricultural pests, some of which have greatly expanded their distributions due to human activity24,25,26,27. The recent demographic and geographic changes of pests in this group make them ideal models for testing distance decay predictions, the results of which would be directly relevant to other invasive pest systems. The billions in USD of economic damage that tephritid fruit flies inflict across the planet28,29,30 has sparked multiple initiatives to build COI DNA barcode reference libraries in order to detect and identify problematic species11,31,32,33,34,35. Three invasive polyphagous pests; peach fruit fly - Bactrocera zonata Saunders, guava fruit fly - Bactrocera correcta Bezzi and melon fly - Zeugodacus cucurbitae Coquillett have overlapping host ranges that include over fifty species of commercially grown fruit, as well as partly overlapping distribution ranges24,36. Zeugodacus cucurbitae is the most invasive of the three, and has spread out of South-East Asia across Africa and much of the Pacific37. Bactrocera zonata is likely native to the Indian subcontinent and is spreading westward into northern Africa. The extent of the invasive versus the native ranges of B. correcta have not been studied in much detail, but likely include parts of South-East Asia. Plant protection agencies, including the United States Department of Agriculture and the European Plant Protection Agency have dedicated programs to exclude the introduction and establishment of these insects38,39. However, none of these species have hitherto been studied molecularly throughout their range.
We performed high-density sampling of Zeugodacus cucurbitae and Bactrocera correcta and B. zonata, whereby we attempted to include samples from across their ranges. Using an 815 base-pair section of the COI gene (3′-P region) we explored with rarefaction curves how many specimens hypothetically needed to be sampled to obtain all the haplotypes. We additionally tested whether the species could be identified reliably using COI and looked for a phylogeographic signal in this gene that may be useful in quarantine efforts and for reconstructing invasion pathways. Finally, we evaluated the efficacy of demographic estimators to infer historic and current population expansion as a proxy for pest severity and future invasion threat.
Material & Methods
Taxon sampling
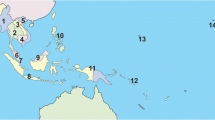
Based on morphology and a seven-gene molecular phylogeny with 178 Dacini species40, B. correcta and B. zonata are sister species, and Z. cucurbitae is closely related to Z. tau Walker, Z. synnephes Hendel and Z. choristus May. Zeugodacus cucurbitae is widespread and is documented to be expanding its distribution, which currently includes much of subtropical Africa, South-East Asia and Pacific Islands, including Guam and Hawaii24,37 (Fig. 1C). Bactrocera zonata is present in northern Africa and Saudi Arabia, the Indian subcontinent and has been reported from Thailand and Vietnam (Fig. 1A). Partly overlapping with this distribution, B. correcta can be found throughout most of mainland South-East Asia (Fig. 1B)41. Samples were collected between 2005 and 2017 (Fig. 1, BOLD, https://doi.org/10.5883/DS-ZOCOCU). Bactrocera correcta and B. zonata were collected with the male attractant methyl eugenol, and Z. cucurbitae with cue lure42. Bucket traps with one of these lures and a 1 × 1 cm dichlorvos strip as a killing agent were maintained for three to five days along trails in natural areas or near agricultural land. Specimens were preserved in 95% ethanol and stored at −20 °C. We selected 441 B. correcta, 214 B. zonata and 372 specimens of Z. cucurbitae for DNA extraction. All identifications were done based on morphology.

Distribution and sampling points of the three species; (A) Bactrocera zonata, (B) Bactrocera correcta and (C) Zeugodacus cucurbitae. Green areas indicate countries for which the species has been reported and we have sampled, brown areas indicate countries for which the species has been reported but we were unable to sample. Red circles indicate sampling localities. The map insert in C shows the Hawaiian Islands. Note that the actual distributions of the species do not always follow political boundaries, the flies do not occur in northern China, or the northern Islands of Japan.
DNA extraction, amplification, and sequencing
One to three legs of each specimen were typically used for total genomic DNA extraction, voucher specimens are deposited in the University of Hawaii Insect Museum (UHIM). Some specimens were additionally used for genomic studies (e.g.43), such flies were fully ground up to obtain higher concentration DNA extracts. DNA was extracted using the Qiagen (Valencia, CA) DNeasy animal blood and tissue kit, following manufacturer’s protocols. A 780 base-pair (bp) section of the cytochrome c oxidase subunit I 3′P region (COI-3P) of the mitochondrial DNA was amplified using the forward primer HCO-2198rc (5′-3′ GCT CAA CAA ATC ATA AAG ATA TTG G)44 and reverse primer Pat-k508 (5′-3′ TCC AAT GCA CTA ATC TGC CAT ATT A)45. The PCR thermal conditions were 2 min. at 94 °C, 40 cycles of (94 °C for 30 sec., 53 °C for 30 sec. and 70 °C for 60 sec.) with a final extension at 70 °C for 10 min. Bidirectional Sanger sequencing was outsourced to Eurofins (Louisville, Kentucky, USA). We combined chromatograms of the forward and reverse reads in Geneious R10, trimmed primer regions, and created alignments for each species. The alignments contained no insertions or deletions, and all sequences were checked for stop-codons, which can indicate the presence of nuclear pseudogenes. The sequences are deposited both in BOLD (https://doi.org/10.5883/DS-ZOCOCU) and NCBI Genbank (accession numbers MT257264 - MT258375).
Phylogenetic and population genetic analyses
The final datasets used for analyses included 428 B. correcta, 756 bp aligned, 214 B. zonata, 752 bp aligned and 372 Z. cucurbitae, 764 bp aligned (https://doi.org/10.5883/DS-ZOCOCU). Some papers have studied the COI diversity of B. correcta regionally46,47, but because of only partially matching COI target regions (i.e. COI-5P versus COI-3P) we have decided to not include their data in our study. Also, Kunprom et al.46 reported a strongly divergent COI lineage within B. correcta, matching earlier results based on the COI-3P region47. They attributed this anomaly to Wolbachia bacterial infection, but upon comparison of the published COI-3P sequences with our COI reference dataset (data not shown) we conclude this lineage to match a cluster that includes B. nigrotibialis, B. nigrifacia and B. nigrofemoralis, which share some of the same hosts as B. correcta and can be morphologically similar36,41. For maximum likelihood tree inference of the relationships of the target species and their closest relatives (based on40), we used unique haplotypes only and added B. nigrotibialis (Perkins), Zeugodacus tau (Walker), Z. synnephes (Hendel) and Z. choristus (May) to the alignments. Maximum likelihood tree inference was done using RaxML v8 with 50 searches for the best tree and subsequent multiparametric bootstrapping with the extended majority rule parameter to estimate statistical support values. We calculated haplotype networks using the TCS statistical parsimony algorithm48, employed in the software “TCS” v.1.2149. The raw output of TCS was visualized in the web implementation of tcsBU50 and optimized for publication in Adobe Illustrator CC. Haplotype rarefaction curves were estimated in R using the HaploAccum() function from the package SpideR with 1,000 permutations, and we used the chaoHaplo() function from the same package to obtain the non-parametric Chao 1 estimator of total haplotype diversity. The “tools and recipes for evolutionary genetics and genomics” (EggLib51); python library was used to calculate the descriptive statistics for nucleotide diversity π, Tajima’s D, Watterson’s estimator of θ, and Fu’s Fs.
Results
COI Phylogeny
We successfully obtained COI sequences of 428 Bactrocera correcta, 214 B. zonata and 372 Zeugodacus cucurbitae. The COI maximum likelihood trees (Fig. 2) based on unique haplotypes, with added outgroup sister taxa, show that each of the study species can reliably be separated from its closest sister species using a monophyly criterion, and that the intraspecific variation does not overlap with the variation between species. All haplotypes are unique to each respective species. The intraspecific variation ranges from 1.32% in Z. cucurbitae to 3.06% in B. zonata (Table 1), and this variation is distributed across the COI region (Supplemental Information Table 1).

COI maximum likelihood trees based on unique haplotypes showing a monophyletic origin for (A) Zeugodacus cucurbitae and its closest congeners and (B) Bactrocera zonata and (B) correcta and their closest sister species. Scale bar indicates substitutions per site; the taxa are ‘collapsed’ into a triangle for each species, where the horizontal width indicates the maximum intraspecific variation. Bootstrap support values are indicated on the respective branches.
Haplotype networks
We find contrasting patterns of haplotype diversity between the three species (Fig. 3), where B. zonata has the highest diversity and most haplotypes are represented by singletons, B. correcta has ~7 dominantly represented haplotypes and Z. cucurbitae has a single dominant haplotype with closely related singeltons in a ‘star-shape’ surrounding it. None of the species displays a biogeographic signal, where certain haplotypes or parts of the haplotype network can be correlated with a biogeographic region. For B. zonata, many of the haplotypes are unique to a region, but also unique in the network and further sampling will likely show that these are shared between regions.

TCS COI haplotype networks for (A) Bactrocera zonata, (B) Bactrocera correcta and (C) Zeugodacus cucurbitae and the legend for all three in (D) Each circle represents a haplotype, circle size indicates the occurrence of each haplotype, but note that the scale differs between (A–C) The colors refer to the sampling locality, with shades of the same color roughly following biogeographical regions; white circles connecting sampled haplotypes indicate hypothetical (unsampled) haplotypes.
Haplotype rarefaction
The haplotype rarefaction curves with randomly sampled diversity do not reach an asymptote for any of the species (Fig. 4), indicating that we have only sampled part of the actual diversity. Despite their strongly different haplotype networks (Fig. 3), Bactrocera correcta and Zeugodacus cucurbitae have largely overlapping haplotype rarefaction curves and confidence intervals, although the Chao 1 estimated total diversity for the former is 1,110 haplotypes and only 270 for the latter. The angle on the curve for Bactrocera zonata is close to a 1:1 ratio where every new sample represents a new haplotype, indicating a much higher diversity for this species, which is underlined by a Chao 1 estimated total haplotype diversity of 1,890.

Haplotype rarefaction curves and the Chao 1 estimated total haplotypes for each species, shaded areas indicate 95% confidence interval from 1,000 permutations. The 1:1 line indicates the maximum where each new sample represents a new haplotype.
Population genetic statistics
From the lack of geographic partitioning in the haplotype networks (Fig. 3), we assumed the different sampling points for each species to be part of a panmictic population and estimated descriptors of their genetics (Table 1). The number of observed haplotypes ranged from 70 to 184. With Chao 1 total estimated haplotypes per species ranging from 270 to 1,890 (Fig. 4.), we sampled 7.2% (in B. correcta) –25.9% (in Z. cucurbitae) of the total estimated diversity. Watterson’s estimator of θ and nucleotide diversity π estimators show that B. zonata is the most diverse species, followed by B. correcta, and Z. cucurbitae shows little diversity, matching the results from the haplotype network (Fig. 3). Wattersons’s estimator of θ, calculated through the formula θw = 4Neµ can also be used as an estimate of effective population size (Ne), and shows that Z. cucurbitae has the smallest effective population size, followed by B. correcta and B. zonata. Negative values of Fu’s Fs are evidence for an excess number of alleles relative to the expected. Fu’s Fs is negative for all three species, but the largest negative is in B. zonata. An excess of low frequency polymorphisms relative to expectation is expressed by negative values of Tajima’s D, and are commonly interpreted as demographic estimators for population expansion or selection. We found negative values of Tajima’s D for all three species, but most strongly in Z. cucurbitae, followed by B. zonata and B. correcta.
Discussion
Distance decay COI diversity
We examined the role of the distance-decay model in the mitochondrial gene COI as a tool to understand the degree of spatially heterogeneity in genetic diversity and whether important pest species have experienced recent range expansions. In general, intraspecific genetic diversity is a result of demographic history, mutation and selection52. The different genetic patterns observed can reveal much about the forces, such as rapid expansion or significant bottlenecks, directly affecting species of interest. Considering the prevalence of horticulture in human history, it is difficult to assess what should be considered the native range of the three species. The native range can sometimes be assessed a posteriori, because it will commonly have the highest genetic diversity (e.g.53), but a priori assumptions rely on historical records of observations and these may be inaccurate due to, e.g., faulty taxonomy54. For our study, we therefore attempted to include as many samples from throughout the current ranges of the species as possible, and we did not detect variation in haplotype diversity between regions that would enable the distinction between native and non-native ranges. Despite the fact that we did not detect regional variation, all three species in our study show markedly different COI diversity patterns. This might be expected to influence their invasion success if genetic diversity is a boon for invasive species. The differences may be a result of the differing topology of the landscape of their range, ecological interactions and/or species-specific variation in the mutation rate (μ) of COI. Zeugodacus cucurbitae has the widest distribution in our study, yet, conversely, it shows a lack of haplotype diversity. This could be due to very recent expansion in Z. cucurbitae from a very genetically limited source population that has adapted to the agricultural environments and since spread across the globe. The pattern of a single adapted population spreading rapidly due to humans has also been observed in other organisms (e.g.13,55,56). Regardless of the underlying reasons for the contrasting patterns found in this study, the diverging haplotype patterns suggest that distance decay based principles likely do not apply to these pest species, at least not anymore. As a consequence, the management strategies undertaken for each pest should be based on the absence or presence of intraspecific diversity in regional populations observed. However, it should be kept in mind that different markers may reveal different intraspecific patterns, depending on their resolution and evolutionary history10. These results also pertain to species of conservation concern, since understanding current and past population retractions as measured by genetic diversity across their range has important implications for the evaluation and identification of declining taxa.
Invasion history and potential
Inherent to the host preferences of these insects, there is a fairly detailed record of their range expansion24,25,57. The intraspecific mtDNA diversity reveals additional aspects of the invasive history and, possibly potential by assessing the demographic history and biogeographic distribution of diversity. The low levels of variation and star-like shape of the mtDNA genealogy of Z. cucurbitae are consistent with a sudden population expansion. Large negative values of Fu’s Fs and Tajima’s D-tests further indicate a population demographic expansion58,59. In stark contrast with these results, we found a new haplotype for almost every additional sample of B. zonata that we included. The demographic estimators for the latter do not suggest that there has been rapid population expansion, at least not from a small source population, and in the regions covered by our sampling. A study of the COI variation of 1,600 specimens of two other prominent pests of the tribe, B. dorsalis and B. carambolae, also showed high levels of haplotype variation and a large global distribution with multiple recently invaded continents60. Overall, the results of Tajima’s D estimator seem congruent with the historical record of invasions. Zeugodacus cucurbitae has the largest negative value for Tajima’s D and is the most invasive and widespread of the three pests, B. zonata is the second and B. correcta third. However, it is worth noting that the melon, gourd and squash hosts of Z. cucurbitae are also the most widely grown and transported. The demographic estimators can reveal if there have been recent changes in population sizes, but the values depend on the original population structure in the ‘native’ range and only partly reflect the species’ pest ‘potential’. The pest potential, which can be regarded as the combined factors of the likelihood of reaching new areas and the possibility of becoming established24, is likely mostly determined extrinsically, by human actions – such as the growing of host plants and transporting of fruit. It appears that depending on these circumstances, any of the species in the tribe can develop into pests, with serious implications for quarantine which currently focuses on the handful of already known pest species.
Undetected(?) biogeographic patterns
Although COI has been shown to reveal geographic patterning in some species (e.g.61,62,63), we did not detect any biogeographic structure in our three pest species. The effects of historical climatic change were not as severe in Southeast Asia as in temperate regions, with more continuous possibilities for gene-flow throughout the region which may have resulted in less strongly pronounced biogeographic patterns64,65. However, a high resolution (1,097 SNPs) study of the melon fly Z. cucurbitae revealed geographic structuring with six to ten clusters that were only partly detected using microsatellite data43, and are not detected in our current study using COI. Despite being a widespread species occurring from Africa across Asia into the Pacific, we only found a single dominant haplotype. Our sampling is spread over the full range of Z. cucurbitae, including different invasive populations in Africa. The results from the demographic estimators and the star-shape of the haplotype network therefore all suggest that Z. cucurbitae was restricted to a small geographic area and began to expand rapidly relatively recently– an expansion that is still ongoing as more of Africa is invaded37. There are, however, some alternative explanations that we cannot exclude based on our present data, such as the presence of Wolbachia bacteria that could have influenced the mitochondrial diversity66. Nonetheless, the SNP data supports biogeographic patterning. The differences between mtDNA versus nuclear DNA are partly due to the differences in mutation rates and inheritance, but also to the fact that the method to derive nuclear DNA SNP’s results in a much larger data set of independent sites with a greater ability to resolve small differences between populations. As more SNP data becomes available for more species, it will be interesting to test at which time-scales, or under which evolutionary circumstances, the resolution between mtDNA and SNP data are similar.
In contrast to the data available for Z. cucurbitae, the biogeographic structures of B. zonata and B. correcta have not been studied with high-resolution (i.e. genomic) methods, which may similarly reveal patterning that we did not detect using COI. The peach fruit fly B. zonata is mostly found on the Indian subcontinent and is expanding westward into Africa. Of the three species included in our study, our sampling of B. zonata was most limited, with high density sampling in Bangladesh but lacking specimens from the distribution west of Bangladesh. Nonetheless, it had the highest haplotype diversity. Despite the peach reference in its common name, it was likely first introduced into Africa with guava in 199325. The area sampled by us likely is part of the original native range. The hosts of B. zonata and B. correcta largely overlap and similar biogeographical and population patterns might have been expected. Based on their current distribution, overlapping host range, phylogenetic relation and similar morphology, the most parsimonious conclusion is that B. correcta and B. zonata speciated through vicariance. Bactrocera correcta is now found mostly in eastern Asia, and B. zonata is mostly found in the Indian subcontinent with recent westward expansion37. The two species only overlap in Sri Lanka, India, Nepal, Bangladesh and Thailand. These vicariant distributions, despite the likely possibilities for human-mediated dispersal (fruit trade) into either range, may indicate strong competition or selection pressures that limit their avenues for invading regions already occupied by the other species. Fragments of the COI barcode region (COI-5P) of B. zonata have been studied in India67, where they also concluded that B. zonata has high intraspecific variability, but showed no biogeographic patterning. The third species in our study, the guava fruit fly Bactrocera correcta is mostly found in mainland Southeast Asia. Our sampling covers a large part of the distribution, only lacking Malaysia and India, and we likely included most of its native distribution and COI haplotype diversity. A regional study of the COI-5P diversity of B. correcta in Thailand concluded that there was much genetic homogeneity, likely because host plants are spatially and temporally continuously available46. However, we found samples from Thailand to belong to at least nine different haplotypes, mostly those dominantly represented in our dataset. All three species pose large invasion risks and biogeographical analysis of intercepted specimens can help to diagnose pathways and block them (e.g.60,68). However, from the lack of biogeographic patterning we conclude that COI data cannot be used to trace the origins of introductions or intercepted flies of Z. cucurbitae, B. correcta and B. zonata.
COI-based identification
Percentage-wise, the ranges for intra- and interspecific COI variation we find in our study species are similar to those found in other groups of life, such as moths, beetles, or birds, which commonly show <3% intraspecific and >2% interspecific variation4,16,20,69,70. Even though the estimated total number of haplotypes between the three species varies seven-fold, the maximum intraspecific diversity is at most ~3% and all species can be identified reliably using COI. The high mutation rates in COI, although predominantly in the third-codon position and synonyms are generally known across all animal taxa71, and have made the marker a useful part of a broader dataset for species delimitation and recognition, and to some extent population-level studies. Contrasting patterns of COI evolution between higher groups has been observed before71,72, which has been linked to the functional properties of the gene, but here we show that large differences can exist between closely related (sister) species as well, or species within the same tribe. Although we only studied three species, the patterns of haplotype diversity are starkly contrasting with very low haplotype diversity in Z. cucurbitae to very high in B. zonata. The estimates of the proportion of the diversity that we sampled range from 7.2–25.9%. All three of these species are pests associated with crops and would be expected to have high effective population sizes (Ne), given the potentially high population density in agricultural systems. The statistic Watterson’s θ is dependent on Ne and genetic diversity µ, and it is possible that only µ varies significantly between these three pests. Mitochondrial genetic diversity, in contrast to nuclear genetic diversity, may not be linked to Ne but only to µ and selection52,73. In Bactrocera dorsalis and B. carambolae, high mutation rates were postulated to cause hyper-diversity in their mitochondrial genomes60. However, another explanation for this pattern may have to do with the demographic and invasion history of each species. Despite the common application of COI sequence data for large groups of life across the planet2,19,20,74, there are many open-ended questions on how this molecule functions, and to what extent there is active selection71,75. There have been few studies thus far on the intraspecific variation of COI using large sample sizes, but as DNA sequencing continues to become cheaper (e.g.76) there will hopefully be larger sample sizes for more groups and we can evaluate to what extent general rules apply and how many samples are required to cover the haplotype diversity, to ensure reliable species identification.
Conclusion
Our results indicate that COI was effective for species level identification across the ranges of these three invasive pest species, even in the presence of considerable intraspecific heterogeneity among species. The intraspecific heterogeneity compared between two closely and one more distantly related species with a non-overlapping host range suggests strongly different demographic histories between all three. Generalized distance decay predictions may not be applicable for pest species, because recently expanded distributions due to globalization may obscure historic biogeographic patterns. As a result, we could not use COI intraspecific variation to identify source populations or invasion pathways to augment pest management. Future studies based on high-resolution genomic data are likely to uncover additional patterns, which may be suitable for identifying invasion pathways. We found that simple demographic estimators based on COI haplotype diversity can be used to infer the historical expansion speed and complement otherwise documented distribution data, but that it cannot predict the virulence of a pest because its spread is more likely dependent on extrinsic factors, such as globalization.
Data availability
All data used in this paper is available through BOLD, https://doi.org/10.5883/DS-ZOCOCU as well as Genbank accession numbers MT257264 - MT258375.
References
Fernandez-Triana, J., Shaw, M., Cardinal, S. & Mason, P. Contributions to the study of the Holarctic fauna of Microgastrinae (Hymenoptera, Braconidae). I. Introduction and first results of transatlantic comparisons. J. Hymenopt. Res. 37, 61–76 (2014).
Steinke, D., Zemlak, T. S., Boutillier, J. A. & Hebert, P. D. N. DNA barcoding of Pacific Canada’s fishes. Mar. Biol. 156, 2641–2647 (2009).
Bergsten, J. et al. The effect of geographical scale of sampling on DNA barcoding. Syst. Biol. 61, 851–869 (2012).
Huemer, P., Hebert, P. D. N., Fiedler, K. & PLOS, O. N. E. Large geographic distance versus small DNA barcode divergence: Insights from a comparison of European to South Siberian Lepidoptera. 13, 1–19 (2018).
Nekola, J. C. & White, P. S. Special Paper: The distance decay of similarity in biogeography and ecology. J. Biogeogr. 26, 867–878 (1999).
Alonso-Blanco, C. et al. 1,135 Genomes Reveal the Global Pattern of Polymorphism in Arabidopsis thaliana. Cell 166, 481–491 (2016).
Lukhtanov, V. A., Sourakov, A., Zakharov, E. V. & Hebert, P. D. N. DNA barcoding Central Asian butterflies: increasing geographical dimension does not significantly reduce the success of species identification. Mol. Ecol. Resour. 9, 1302–1310 (2009).
Domyan, E. T. & Shapiro, M. D. Pigeonetics takes flight: Evolution, development, and genetics of intraspecific variation. Dev. Biol. 427, 241–250 (2017).
Zhang, W., Fan, X., Zhu, S., Zhao, H. & Fu, L. Species-Specific Identification from Incomplete Sampling: Applying DNA Barcodes to Monitoring Invasive Solanum Plants. PLoS One 8 (2013).
Dupuis, J. R. et al. Population genomics and comparisons of selective signatures in two invasions of melon fly, Bactrocera cucurbitae (Diptera: Tephritidae). Biol. Invasions 20, 1211–1228 (2018).
Armstrong, K. F. & Ball, S. L. DNA barcodes for biosecurity: Invasive species identification. Philos. Trans. R. Soc. B Biol. Sci. 360, 1813–1823 (2005).
Violle, C., Reich, P. B., Pacala, S. W., Enquist, B. J. & Kattge, J. The emergence and promise of functional biogeography. Proc. Natl. Acad. Sci. 111, 13690–13696 (2014).
Reil, J. B. et al. Transpacific coalescent pathways of coconut rhinoceros beetle biotypes: Resistance to biological control catalyses resurgence of an old pest. Mol. Ecol. 1–16 (2018).
Mimura, M. et al. Understanding and monitoring the consequences of human impacts on intraspecific variation. Evol. Appl. 10, 121–139 (2017).
Ratnasingham, S. & Hebert, P. D. N. A DNA-Based Registry for All Animal Species: The Barcode Index Number (BIN) System. PLoS One 8 (2013).
Pentinsaari, M., Hebert, P. D. N. & Mutanen, M. Barcoding beetles: A regional survey of 1872 species reveals high identification success and unusually deep interspecific divergences. PLoS One 9, pone0108651 (2014).
Kekkonen, M., Mutanen, M., Kaila, L., Nieminen, M. & Hebert, P. D. N. Delineating species with DNA barcodes: A case of Taxon dependent method performance in moths. PLoS One 10, pone0122481 (2015).
Funk, D. J. & Omland, K. E. Species-level paraphyly and polyphyly: Frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 34, 397–423 (2003).
Mutanen, M. et al. Species-Level Para- and Polyphyly in DNA Barcode Gene Trees: Strong Operational Bias in European Lepidoptera. Syst. Biol. 65, 1024–1040 (2016).
Kerr, K. C. R. R. et al. Comprehensive DNA barcode coverage of North American birds. Mol. Ecol. Notes 7, 535–543 (2007).
Gibbs, J. DNA barcoding a nightmare taxon: assessing barcode index numbers and barcode gaps for sweat bees. Genome 31, 1–11 (2017).
Ward, R. D., Zemlak, T. S., Innes, B. H., Last, P. R. & Hebert, P. D. N. DNA barcoding Australia’s fish species. Philos. Trans. R. Soc. B Biol. Sci. 360, 1847–1857 (2005).
Phillips, J. D., Gwiazdowski, R. A., Ashlock, D. & Hanner, R. An exploration of sufficient sampling effort to describe intraspecific DNA barcode haplotype diversity: examples from the ray-finned fishes (Chordata: Actinopterygii). DNA Barcodes 3, 66–73 (2015).
Vargas, R. I., Pinero, J. C. & Leblanc, L. An overview of pest species of Bactrocera fruit flies (Diptera: Tephritidae) and the integration of biopesticides with other biological approaches for their management with a focus on the pacific region. Insects 6, 297–318 (2015).
Meyer, M. de, Mohamed, S. & White, I. M. Invasive Fruit Fly pests in Africa. (2007).
White, I. M. & Elson-Harris, M. M. Fruit Flies of Economic Significance. (CABI Publishing, 1992).
Norrbom, A. L., Carroll, L. E., Thompson, F. C., White, I. M. & Freidberg, A. Systematic Database of Names. in Fruit fly expert identification system and systematic information database (ed. Thompson, F. C.) 65–251 (Backhuys Publishers, 1998).
Siebert, J. B. & Cooper, T. Embargo on California produce would cause revenue, job loss. Calif. Agric. 49, 7–12 (1995).
Badii, K. B., Billah, M. K., Afreh Nuamah, K., Obeng Ofori, D. & Nyarko, G. Review of the pest status, economic impact and management of fruit-infesting flies (Diptera: Tephritidae) in. Africa. African J. Agric. Res. 10, 1488–1498 (2015).
CNAS. Economic Impacts of Losing the Fruit Fly Trapping Program. CNAS Issue Br. (2015).
Smit, J., Reijnen, B. & Stokvis, F. Half of the European fruit fly species barcoded (Diptera, Tephritidae); a feasibility test for molecular identification. Zookeys 365, 279–305 (2013).
Aketarawong, N., Isasawin, S., Sojikul, P. & Thanaphum, S. Gene flow and genetic structure of Bactrocera carambolae (Diptera, Tephritidae) among geographical differences and sister species, B. dorsalis, inferred from microsatellite DNA data. Zookeys 540, 239–272 (2015).
Barr, N. B. et al. Identifying Anastrepha (Diptera: Tephritidae) Species Using DNA Barcodes. J. Econ. Entomol. XX(X), 1–17 (2017).
Jiang, F., Jin, Q., Liang, L., Zhang, A. B. & Li, Z. H. Existence of species complex largely reduced barcoding success for invasive species of Tephritidae: A case study in Bactrocera spp. Mol. Ecol. Resour. 14, 1114–1128 (2014).
Virgilio, M., Jordaens, K., Breman, F. C., Backeljau, T. & de Meyer, M. Identifying insects with incomplete DNA barcode libraries, African fruit flies (Diptera: Tephritidae) as a test case. PLoS One 7 (2012).
Allwood, A. J. et al. Host plant records for fruit flies (Diptera: Tephritidae). Raffles Bull. Zool. Suppl. 7, 1–92 (1999).
De Meyer, M. et al. A review of the current knowledge on Zeugodacus cucurbitae (Coquillett) (Diptera, Tephritidae) in Africa, with a list of species included in Zeugodacus. Zookeys 2015, 539–557 (2015).
EPPO. European and Mediterranean Plant Protection Organization. (2018).
CABI. Invasive Species Compendium. (2018). Available at: https://www.cabi.org/isc/. (Accessed: 11th October 2018).
San Jose, M. et al. Incongruence between molecules and morphology: a seven-gene phylogeny of Dacini fruit flies paves the way for reclassification (Diptera: Tephritidae). Mol. Phylogenet. Evol. 121, 139–149 (2018).
Drew, R. A. I. & Romig, M. C. Tropical fruit flies of South-East Asia. (CABI, 2013).
Metcalf, R. L., Mitchell, W. C. & Metcalf, E. R. Olfactory receptors in the melon fly Dacus cucurbitae and the oriental fruit fly Dacus dorsalis. Proc. Natl. Acad. Sci. USA 80, 3143–3147 (1983).
Dupuis, J. R. et al. HiMAP: Robust phylogenomics from highly multiplexed amplicon sequencing. Mol. Ecol. Resour. 18, 1000–1019 (2018).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial Cytochrome C Oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3, 294–299 (1994).
Simon, C. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 87, 651–701 (1994).
Kunprom, C., Sopaladawan, P. N. & Pramual, P. Population genetics and demographic history of guava fruit fly Bactrocera correcta (Diptera: Tephritidae) in northeastern Thailand. Eur. J. Entomol. 112, 227–234 (2015).
Jamnongluk, W., Baimai, V. & Kittayapong, P. Molecular evolution of tephritid fruit flies in the genus Bactrocera based on the cytochrome oxidase I gene. Genetica 119, 19–25 (2003).
Templeton, A. R., Crandall, K. A. & Sing, C. F. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 132, 619–633 (1992).
Clement, M., Posada, D. & Crandall, K. A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659 (2000).
Santos, M. D. et al. TcsBU: A tool to extend TCS network layout and visualization. Bioinformatics 32, 627–628 (2015).
De Mita, S. & Siol, M. EggLib: processing, analysis and simulation tools for population genetics and genomics. BMC Genet. 13, 1–12 (2012).
Bazin, E., Glémin, S. & Galtier, N. Population size does not influence mitochondrial genetic diversity in animals. Science. 312, 570–572 (2006).
Lees, D. C. et al. Tracking origins of invasive herbivores through herbaria and archival DNA: the case of the horse-chestnut leaf miner. Front. Ecol. Environ. 9, 322–328 (2011).
Schutze, M. K. et al. Synonymization of key pest species within the Bactrocera dorsalis species complex (Diptera: Tephritidae): taxonomic changes based on a review of 20 years of integrative morphological, molecular, cytogenetic, behavioural and chemoecological data. Syst. Entomol. 40, 456–471 (2015).
Branch, G. M. & Nina Steffani, C. Can we predict the effects of alien species? A case-history of the invasion of South Africa by Mytilus galloprovincialis (Lamarck). J. Exp. Mar. Bio. Ecol. 300, 189–215 (2004).
Rubinoff, D., Holland, B. S., Shibata, A., Messing, R. H. & Wright, M. G. Rapid Invasion Despite Lack of Genetic Variation in the Erythrina Gall Wasp (Quadrastichus erythrinae Kim). Pacific Sci. 64, 23–31 (2010).
Meyer, M. D. et al. Notes on the frugivorous fruit fly (Diptera: Tephritidae) fauna of western Africa, with description of a new Dacus species. Eur. J. Taxon. 50, 1–17 (2013).
Schmidt, D. & Pool, J. The effect of population history on the distribution of the Tajima’s D statistic. 1–8 (2002).
Pramual, P., Kongim, B. & Nanork, P. Phylogeography of Simulium siamense Takaoka and Suzuki complex (Diptera: Simuliidae) in Thailand. Entomol. Sci. 14, 428–436 (2011).
San Jose, M. et al. Tracking the Origins of Fly Invasions; Using Mitochondrial Haplotype Diversity to Identify Potential Source Populations in Two Genetically Intertwined Fruit Fly Species (Bactrocera carambolae and Bactrocera dorsalis [Diptera: Tephritidae]). J. Econ. Entomol. 1–13 (2018).
O’Loughlin, S. M. et al. Complex population history of two Anopheles dirus mosquito species in Southeast Asia suggests the influence of Pleistocene climate change rather than human-mediated effects. J. Evol. Biol. 21, 1555–1569 (2008).
Morgan, K. et al. Inter-specific gene flow dynamics during the Pleistocene-dated speciation of forest-dependent mosquitoes in Southeast Asia. Mol. Ecol. 19, 2269–2285 (2010).
Valade, R. et al. Mitochondrial and microsatellite DNA markers reveal a Balkan origin for the highly invasive horse-chestnut leaf miner Cameraria ohridella (Lepidoptera, Gracillariidae). Mol Ecol 18, 3458–3470 (2009).
Penny, D. A 40,000 year palynological record from north-east Thailand; implications for biogeography and palaeo-environmental reconstruction. Palaeogeogr. Palaeoclimatol. Palaeoecol. 171, 97–128 (2001).
Cannon, C. H. & Manos, P. S. Phylogeography of the Souteast Asian stone oak (Lithocarpus). J. Biogeogr. 30, 211–226 (2003).
Smith, M. A. et al. Wolbachia and DNA barcoding insects: patterns, potential, and problems. PLoS One 7, e36514 (2012).
Choudhary, J. S. et al. Genetic analysis of Bactrocera zonata (Diptera: Tephritidae) populations from India based on cox1 and nad1 gene sequences. Mitochondrial DNA Part A DNA Mapping, Seq. Anal. 0, 1–10 (2017).
Shi, W., Kerdelhué, C. & Ye, H. Genetic structure and inferences on potential source areas for Bactrocera dorsalis (Hendel) based on mitochondrial and microsatellite markers. PLoS One 7 (2012).
Schmidt, S., Schmid-Egger, C., Morinière, J., Haszprunar, G. & Hebert, P. D. N. DNA barcoding largely supports 250 years of classical taxonomy: Identifications for Central European bees (Hymenoptera, Apoidea partim). Mol. Ecol. Resour. 15, 985–1000 (2015).
Rivera, J. & Currie, D. C. Identification of Nearctic black flies using DNA barcodes (Diptera: Simuliidae). Mol. Ecol. Resour. 9, 224–236 (2009).
Pentinsaari, M., Salmela, H., Mutanen, M. & Roslin, T. Molecular evolution of a widely-adopted taxonomic marker (COI) across the animal tree of life. Sci. Rep. 6, 1–12 (2016).
Meier, R., Shiyang, K., Vaidya, G. & Ng, P. K. L. DNA Barcoding and Taxonomy in Diptera: A Tale of High Intraspecific Variability and Low Identification Success Author (s): Rudolf Meier, Kwong Shiyang, Gaurav Vaidya and Peter K. L. Ng Published by: Oxford Universi. Syst. Biol. 55, 715–728 (2006).
Wang, J. Estimation of effective population sizes from data on genetic markers. Philos. Trans. R. Soc. B Biol. Sci. 360, 1395–1409 (2005).
Huemer, P., Mutanen, M., Sefc, K. M. & Hebert, P. D. N. Testing DNA barcode performance in 1000 species of European Lepidoptera: Large geographic distances have small genetic impacts. PLoS One 9, 1–21 (2014).
Rubinoff, D., Cameron, S. & Will, K. A genomic perspective on the shortcomings of mitochondrial DNA for ‘barcoding’ identification. J. Hered. 97, 581–594 (2006).
Hebert, P. D. N. et al. A Sequel to Sanger: Amplicon sequencing that scales. BMC Genomics 19 (2018).
Acknowledgements
We greatly appreciate help from Dan Nitta, Julian Dupuis and Kimberly Morris with the molecular work for efficiently processing large numbers of samples. We would also like to thank Scott Geib for allowing us kind use of his laboratory resources. We further thank Ken Bloem for providing comments that improved the manuscript. Funding for this project was provided by the United States Department of Agriculture (USDA) Farm Bill Section 10007 Plant Pest and Disease Management and Disaster Prevention Program in support of suggestion “Genomic approaches to fruit fly exclusion and pathway analysis”: 3.0497-FY17. These funds were managed as cooperative agreements between USDA Animal and Plant Health Inspection Service and the University of Hawaii’s College of Tropical Agriculture and Human Resources (8130-0565-CA) and the University of Idaho’s College of Agriculture and Life Sciences (8130-0665-CA). This material was made possible, in part, by a Cooperative Agreement from the United States Department of Agriculture’s Animal and Plant Health Inspection Service (APHIS). It may not necessarily express APHIS’ views. Additional funding was provided by the USDA Cooperative State Research, Education and Extension (CSREES) project HAW00942-H administered by the College of Tropical Agriculture and Human Resources, University of Hawaii.
Author information
Authors and Affiliations
Contributions
C.D., M.S.J. and D.R. conceived the project idea. M.S.J., L.L. and D.R. collected specimens, C.D. and M.S.J. performed experiments and generated sequence data. C.D., M.S.J., N.B., L.L. and D.R. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Doorenweerd, C., San Jose, M., Barr, N. et al. Highly variable COI haplotype diversity between three species of invasive pest fruit fly reflects remarkably incongruent demographic histories. Sci Rep 10, 6887 (2020). https://doi.org/10.1038/s41598-020-63973-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-63973-x
- Springer Nature Limited
This article is cited by
-
A comprehensive DNA barcoding of Indian freshwater fishes of the Indus River system, Beas
Scientific Reports (2024)
-
Coconut rhinoceros beetle in Solomon Islands: a tale of two invasions
Biological Invasions (2023)
-
Phylogenetic analysis of Chirocephalus (Branchiopoda: Anostraca), with the description of a new species from Algeria
Biologia (2023)
-
Wing phenotypic plasticity, quantitative genetics, modularity, and phylogenetic signal analysis revealed the niche partitioning in two fruit fly species, Bactrocera dorsalis and Zeugodacus cucurbitae
International Journal of Tropical Insect Science (2022)
-
Genetic and endosymbiotic diversity of Greek populations of Philaenus spumarius, Philaenus signatus and Neophilaenus campestris, vectors of Xylella fastidiosa
Scientific Reports (2021)