
Abstract
A significant number of Southeast Asian mammal species described in the 19th and 20th century were subsequently synonymized and are now considered subspecies. Many are affected by rapid habitat loss which creates an urgent need to re-assess the conservation status based on species boundaries established with molecular data. However, such data are lacking and difficult to obtain for many populations and subspecies. We document via a literature survey and empirical study how shotgun sequencing of faecal DNA is a still underutilized but powerful tool for accelerating such evaluations. We obtain 11 mitochondrial genomes for three subspecies in the langur genus Presbytis through shotgun sequencing of faecal DNA (P. femoralis femoralis, P. f. percura, P. siamensis cf. cana). The genomes support the resurrection of all three subspecies to species based on multiple species delimitation algorithms (PTP, ABGD, Objective Clustering) applied to a dataset covering 40 species and 43 subspecies of Asian colobines. For two of the newly recognized species (P. femoralis, P. percura), the results lead to an immediate change in IUCN status to Critically Endangered due to small population sizes and fragmented habitats. We conclude that faecal DNA should be more widely used for clarifying species boundaries in endangered mammals.
Similar content being viewed by others


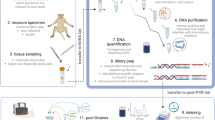
Introduction
Human impacts on the environment have rapidly accelerated species extinction via habitat degradation and climate change and the recent report by the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services (IPBES) predicts that climate change has already affected the distribution of nearly half (47%) of land-mammals1. Protection is urgently needed but is hampered by the lack of data for a large number of mammal species, subspecies, and populations which face extinction2,3,4. A typical example is Asian primates for which 70% of the species are threatened5. Effective population management is needed but it requires a robust understanding of species numbers, boundaries, and distributions based on up-to-date information6,7. Unfortunately, this information is lacking for many rare, globally threatened, and elusive mammalian species. Many lack molecular data and collecting these data is difficult because invasive sampling yielding fresh tissues is usually not feasible.
This leaves only three alternative sources of DNA. The first is museum specimens, but the number of samples in museums tends to be small and many were collected in the 19th or early 20th century thus reflecting historic genetic diversity prior to extensive habitat loss. The second is tissue samples obtained from specimens that died of “natural causes” such as road accidents. The third source of genetic material is non-invasive samples such as hair and faeces. Faecal samples can be collected in good numbers during routine field surveys, which makes faecal samples particularly useful for data-deficient taxa that are in urgent need for re-assessment of species boundaries and distributions using molecular information8. Moreover, it is now straightforward to obtain complete mitochondrial genomes from such samples using shotgun sequencing8,9,10. Faecal samples contain a complex pool of DNA including that of the host. The host DNA is particularly informative because it reflects the current genetic diversity of the species. Faecal samples are nevertheless still an underappreciated source of information and many protocols for field research do not cover the collection of such samples8. Recent review by Srivathsan et al.8 estimates that it is possible to obtain data on host genetics and natural history for nearly 1,000 species of mammals in the next decade if faecal samples were to be collected routinely during field surveys and evaluated using metagenomics. In this study, we document the power of faecal metagenomics for testing the species boundaries in two species of Asian primates that are listed as Data Deficient on the IUCN Red List of Threatened Species.
Asian colobines (langurs and odd-nosed monkeys) are a diverse group of mammals, with 55 recognized species (87 spp.) belonging to seven genera (Nasalis, Presbytis, Pygathrix, Rhinopithecus, Semnopithecus, Simias, Trachypithecus)11; i.e., nearly half of all primate species in Asia are colobines. Unfortunately, many of these species are dependent on habitats that are quickly disappearing. Thus, nine species are already considered Critically Endangered, 23 are Endangered, and nine Vulnerable according to IUCN threat criteria5. This also applies to the genus Presbytis which is one of the most species-rich primate genera12. The 17 recognized species are found in the tropical rainforests of Sundaland, including the Malay Peninsula and the western Indo-Malay Archipelago13,14. Eleven species within Presbytis (chrysomelas, comata, femoralis, frontata, hosei, melalophos, natunae, potenziani, rubicunda, siamensis, and thomasi) were recognized in the last IUCN assessment15. The assessment predated the elevation of six subspecies to species (see Roos et al.11: bicolor, canicrus, mitrata, sabana, siberu, sumatrana) which suggests that many of the Presbytis taxa currently ranked as subspecies are in urgent need for re-assessment with molecular data. Unfortunately, these data are lacking for many subspecies and species which has serious consequences for the proper conservation assessment.
Meyer et al.12 presented the most comprehensive phylogenetic reconstruction of Presbytis which included 13 of the 17 recognized species, but the analysis was based on only two mitochondrial markers (cyt-b and d-loop). This study also covered the banded langur P. femoralis whose relationships had previously already been addressed by several studies. Presbytis femoralis is found on the Malay Peninsula and the island of Sumatra16 (Fig. 1). The species currently consists of three subspecies that were originally described as species because they are distinguishable based on a combination of morphological characters. Yet, many primatologists subsequently considered these characters insufficient for recognizing the three taxa as species (see Table 1).

Distribution of three subspecies of Presbytis femoralis. Image: Ang Yuchen.
The nominal species, Presbytis femoralis, was described by Martin (1838) based on specimens collected by Raffles (1821) from Singapore17,18 (Table 2). Raffles’ banded langur P. f. femoralis occurs in southern Peninsular Malaysia and Singapore. The second subspecies, the East Sumatran banded langur P. f. percura (Fig. 2), occurs only in eastern Sumatra and was described by Lyon (1908) based on specimens collected from near Siak Kecil River, Makapan, Kompei, Pulau Rupat, and Salat Rupat19. The third subspecies, Robinson’s banded langur P. f. robinsoni, was described by Thomas (1910) based on white phenotypic variants collected in Trang, southern Thailand20,21,22. However, typical robinsoni specimens are uniformly dark brown to black, with the inner side of the upper arms, lower abdomen following onto the inside of the thighs to the heel being white. Presbytis f. robinsoni is widespread and ranges from northern Peninsular Malaysia to southern Thailand and Myanmar.

Three subspecies of Presbytis femoralis; clockwise from East Sumatran banded langur P. f. percura (1), Raffles’ banded langur P. f. femoralis (2), to Robinson’s banded langur P. f. robinsoni (3). Photos: Andie Ang.
Presbytis femoralis, consisting of all three subspecies, is listed as Vulnerable in the most recent IUCN Red List assessment, with the nominate subspecies considered Endangered because the known populations are restricted to small and isolated patches of forest. In addition, one population from Singapore showed low genetic variability10,23. Presbytis f. robinsoni is considered Near Threatened, while the least known and least studied subspecies is P. f. percura was considered Data Deficient24. Genetic data suggest that at least P. f. femoralis and P. f. robinsoni are different species25,26 which is also in agreement with the aforementioned morphological characters. However, resolving all subspecies-level boundaries within banded langurs required data for P. f. percura.
For several reasons the subspecies boundaries within P. femoralis remain poorly understood. The main problem is the lack of molecular data for P. f. percura. However, even if the data were available, a comprehensive analysis would still be difficult because the sequence data from three published analyses were not submitted to public sequence repositories (cyt-b, 12 S rDNA, and d-loop)27,28,29. This means that currently the only publicly available molecular data are for P. f. femoralis from the type locality in Singapore (KU899140)10 and P. f. robinsoni from Redang Panjang, Malaysia (DQ355299)30. Note that the genome DQ355299 was initially submitted to Genbank as P. melalophos but it was later clarified by Meyers et al.12 that it is from P. f. robinsoni. Fortunately additional molecular data can be reconstructed based on a table published in Abdul-Latiff et al.28 that lists the variable d-loop sites for several species and subspecies (see Nijman26). Another complication related to resolving species limits within P. femoralis is confusing nomenclatural changes. Abdul-Latiff et al.28 proposed to replace the type species P. femoralis (Martin 1838) with a junior synonym (P. neglectus neglectus Schlegel 1876)31 without considering the detailed information in Low and Lim32 that explains why Martin is the author of the name femoralis and Singapore the type locality of the species. Abdul-Latiff et al.28’s study furthermore violated its own proposed nomenclatural changes by retaining P. f. percura and P. f. robinsoni (see Nijman26).
Here, we solve these problems by providing the first mitogenomes of P. f. percura and thus addressing the taxonomic status of all three subspecies of banded langurs. We also obtain the first mitogenome for the Riau pale-thighed langur P. siamensis cf. cana from Sumatra which helps with resolving subspecies limits within this species. Lastly, we provide an updated dated phylogenetic tree for Asian colobines based on mitochondrial genomes and survey the mammal literature to illustrate that faecal DNA is currently still an underutilized source of genetic information.
Results
Survey of zoological record
In order to investigate to what extent faecal samples have been used in addressing taxonomic problems, we surveyed the literature as captured in Zoological Record. We retrieved 1,852 articles that mentioned faecal samples, but apparently only a subset of 43 articles addressed systematic/taxonomic issues because they were also classified under Systematics/Taxonomy. Inspection of these records revealed only two studies that used faecal DNA for resolving species limits33,34.
Sequence data
Illumina sequencing of faecal metagenomes yielded 60.3–69.7 million sequences for each sample from Sumatra (Presbytis femoralis percura: ESBL1–8, P. siamensis cf. cana: Pres2; Table 3). The data were combined with the Hi-Seq data for six samples from Singapore for P. f. femoralis (BLM1–6) sequenced as part of study by Srivathsan et al.10. All data were quality trimmed using Trimmomatic35 and complete mitochondrial genomes were obtained (P. f. femoralis: 16,548 bp, P. f. percura: 16,548 bp, P. s. cf. cana: 16,558 bp). Human contamination was found to be negligible (0.06% of mitochondrial reads) and did not impact the reconstruction of the mitochondrial genomes (see methods). One sample of P. f. percura ESBL_7 had low average coverage of <5X for the mitochondrial genome and was not analysed further. Data corresponding to the mitochondrial genomes are available as Supplementary Materials 2 and 3.
Species delimitation
Pairwise comparison of cyt-b, hypervariable region HV1 of the d-loop and mitochondrial genomes (CDS + rDNA+d-loop) revealed minimum genetic divergence of 7.1%, 6.1% and 5.3% between P. f. femoralis and P. f. percura. On the other hand, the minimum pairwise distance between either of these taxa with P. f. robinsoni is 6.0% for HV1, 10.3% for cyt-b and 7.6% across the mitochondrial genome. For the two subspecies of P. siamensis, we were only able to compare HV1 sequences. The HV1 sequence of P. s. cf. cana has a 11.1% divergence from P. s. siamensis and 5.1% from P. melalophos (KY117602), while cyt-b and complete mitochondrial genomes show divergence of 2.8% and 2.5% between sequences from P. s. cf. cana and P. mitrata/P. melalophos. Overall, these results suggest that P. s. cf. cana represents a genetically distinct Presbytis lineage.
The high genetic differentiation leads to the recognition of several species based on multiple species delimitation algorithms such as Poisson Tree Processes (PTP)36, Automated Barcode Gap Discovery (ABGD)37 and Objective Clustering38. PTP consistently split P. f. femoralis and P. f. percura into different molecular Operational Taxonomic Units (mOTUs) across the three datasets examined (1. Asian colobine mitogenome, 2. Presbytis mitogenome+cyt-b + HV1, 3. Presbytis HV1-only) (Supplementary Figs. 1–3). ABGD and Objective Clustering (thresholds 2–4%) similarly assigns these two subspecies to different species across the three different datasets and a range of parameters (Table 4). For ABGD, these subspecies would only lump if unusually high priors for intraspecific divergences were used (priors > =0.0215). These parameters are not likely to be appropriate because they also led to the collapse of many recognized Presbytis species into a single mOTU. All three species delimitation methods placed P. f. robinsoni as a distinct species from P. f. femoralis and P. f. percura. Lastly, P. s. cf. cana was placed as a distinct species using PTP and ABGD unless inappropriately high priors for intraspecific divergence are used in ABGD. Objective Clustering based on HV1 also identified P. s. cf. cana as a distinct species. However, the mitogenome and cyt-b datasets lumped P. s. cf. cana with P. melalophos and P. mitrata at 3% and 4% thresholds.
Note that we observed further species-level splitting when species delimitation was based on only HV1 data for the subspecies of P. femoralis. This dataset included sequences for multiple individuals of P. f. femoralis, P. f. robinsoni and P. s. siamensis from the Malay Peninsula (reconstructed in Nijman26, based on Abdul Latiff et al.28). At low prior intraspecific divergences, ABGD split some haplotypes of P. f. femoralis from the Malay Peninsula into separate mOTUs from other haplotypes of the same subspecies from the same region. These, however, consistently grouped together as single mOTU at higher thresholds. Similarly, PTP based analyses of only HV1 data split haplotypes of P. f. robinsoni into multiple mOTUs (Fig. S2) while ABGD consistently placed them as a single species.
Mitochondrial phylogeny of Asian colobines and genus Presbytis
The phylogenetic reconstruction based on mitochondrial genomes of the Asian colobine dataset revealed that P. femoralis is polyphyletic (Fig. 3). The reconstructions based on Maximum Likelihood (ML) and Bayesian Inference (BI) are congruent and reveal that P. f. femoralis and P. f. percura are sister taxa. Divergence time estimates dated the split of P. f. femoralis and P. f. percura at 2.6 Mya (CI: partitioning by codon: 1.96–3.35 Mya, partitioning by gene 1.90–3.37 Mya (Supplementary Fig. S4)). This clade is sister to a clade comprising of P. mitrata, P. comata, P. siamensis cf. cana, and P. melalophos. Presbytis f. robinsoni diverged from these species at 4.5 Mya (CI: partitioning by codon: 3.49–5.48 Mya, partitioning by gene 3.46–5.62). Overall, the mitochondrial phylogeny reveals high support for a clade comprising of Presbytis and Trachypithecus as well as the relationships within a clade consisting of four genera (((Simias + Nasalis)+Pygathrix)+Rhinopithecus). Only the placement of Semnopithecus remains uncertain, as revealed by low support for its relationship to the clade comprising of Nasalis, Simias, Pygathrix and Rhinopithecus on the ML tree. This result is different from Wang et al.39, who found high support for a sister group relationship of Semnopithecus to all remaining genera of Asian colobines. However, a combined analysis of nuclear and mitochondrial data placed Semnopithecus differently thus suggesting our mt-genome phylogeny correctly reflects that the placement of Semnopithecus remains uncertain.

Dated phylogeny of Asian colobine primates based on mitochondrial genomes. The values at nodes represent posterior probability (codon partitioning)/ML bootstrap support for relationships between Asian colobines. Values are omitted if both BI and ML support values are <0.7/70, while * represents support of 1/100. The bars represent the 95% confidence intervals for divergence times estimates. KU899140 is here referred to as BLM5.
With regard to the phylogenetic relationships within Presbytis, our results on Presbytis mitogenome+cyt-b + HV1 dataset (Fig. 4) are largely consistent with the reconstruction by Meyer et al.12. The only differences are as follows: low support for a clade comprising of P. comata, mitrata, melalophos, bicolor, sumatrana, rubicunda and P. siamensis cf. cana but resolution for P. rubicunda, melalophos, mitrata and bicolor which formed a trichotomy in Meyer et al.12. Here, we found P. rubicunda to be sister to P. bicolor. Presbytis f. femoralis and P. f. percura remain sister taxa. Both taxa combined are more closely related to P. mitrata, P. comata and several other taxa of Presbytis than P. f. robinsoni. The split between P. f. femoralis and P. f. percura is again deeper than for most recognized taxa of Presbytis. Divergence estimates based on cyt-b for this taxon set revealed deeper divergence times as compared to mitochondrial genomes, but with overlapping confidence intervals. Presbytis f. femoralis and P. f. percura split 2.93 Mya (2.09–3.78 Mya) (Supplementary Fig. S5), while P. f. robinsoni diverged from the clade comprising of P. femoralis, potenziani, mitrata, melalophos, bicolor, sumatrana and P. siamensis cf. cana at 5.47 Mya (CI: 4.28–6.66 Mya).

ML reconstruction of relationships between Presbytis species based on mitogenome+cyt-b + HV1 dataset. Node values represent bootstrap support, values <70 are excluded, while node support of 100 is represented by *. KU899140 is here referred to as BLM5.
Discussion
The species limits of many Southeast Asian mammal taxa remain unclear which interferes with a conservation assessment at a time when many populations, subspecies, and species face extinction. We here demonstrate how such taxonomic uncertainty can be addressed rapidly through shotgun sequencing of faecal DNA. We document the power of the approach by studying langur species in the genus Presbytis Eschscholtz, 1821 which continue to undergo many taxonomic changes that significantly affect the conservation status of these taxa. At one point all Asian langurs and leaf monkeys in Presbytis, Semnopithecus, and Trachypithecus were included in Presbytis and only five widespread species were recognized (P. aygula, P. melalophos, P. frontata, P. potenziani, P. rubicunda)40,41,42. This has dramatically changed over the last 20 years and currently three genera and 45 species are recognized (17 spp. in Presbytis; eight spp. in Semnopithecus; 20 spp. in Trachypithecus)5,11. Many of these changes in species boundaries were based on genetic data which allowed for the application of explicit species delimitation methods27,39,43. These new data and analyses revealed that many taxa that were initially described as species and later downgraded to subspecies diverged well before the Pleistocene and should be recognized as species; i.e., the morphological characters that were used for the initial species descriptions were appropriate for the delimitation of species and the subsequent lumping was not justified.
Resurrection of Presbytis femoralis, P. percura and P. robinsoni
Based on multiple species delimitation methods, high genetic divergence, placement in the mitochondrial phylogenies, as well as distinct morphological differences, we here resurrect the three species of P. femoralis from their current subspecific status (Table 2). The newly circumscribed Raffles’ banded langur P. femoralis is now only known from southern Peninsular Malaysia (states of Johor and Pahang) and Singapore. The East Sumatran banded langur P. percura only occurs in Riau Province of east-central Sumatra. Lastly, Robinson’s banded langur P. robinsoni has the widest distribution and ranges from northern Peninsular Malaysia (states of Kedah and Perak) through southern Thailand (provinces of Surat Thani, Phetchaburi, and Prachuap Khiri Khan) to southern Myanmar (Tanintharyi Region). These changes to species status mean that Presbytis now comprises 19 species.
A >5% genetic difference and divergence estimates of 2.6–2.9 Mya between P. femoralis and P. percura suggest that these two species diverged prior to the Pleistocene, while several other species of Presbytis originated more recently. These results are particularly intriguing because the changing sea levels during the Pleistocene would have increased connectivity between the land masses of Sumatra and Malay Peninsula. However, the Malacca Straits River flowing northwards with tributaries in what is now Sumatra and the Malay Peninsula44 may have been a substantial barrier between P. femoralis and P. percura, as it would have been significantly wider than the rivers that currently form geographic barriers between some of the Presbytis species in Sumatra. Furthermore, it has been argued that the land bridge between Sumatra and the Malay Peninsula had coarse sandy and/or poorly drained soils which may have limited plant growth in central Sundaland. Unsuitable vegetation may have acted as a dispersal barrier for rainforest plants and animals45. These barriers would have kept the langur populations separate; i.e., it remains unclear whether P. femoralis and P. percura would have formed a hybrid zone if they had encountered each other. Note that it is known that currently recognized primate species in Trachypithecus that radiated ~0.95–1.25 Mya can interbreed43,46, but the genetic divergence between P. femoralis and P. percura is considerably higher.
Conservation status of P. femoralis, P. percura, and P. robinsoni
In the most recent IUCN Red List assessment (unpublished data from a Red List re-assessment in 2015), Presbytis femoralis (comprising femoralis, percura and robinsoni) was listed as Vulnerable (A2cd A3cd A4cd: population size reduction of at least 30% over three generations based on a decline in area of occupancy, extent of occurrence and habitat quality, and actual or potential levels of exploitation). As part of this assessment the status of the three subspecies were also evaluated. Presbytis f. femoralis was considered Endangered (A2cd A3cd A4cd), P. f. percura Data Deficient, and P. f. robinsoni Near Threatened. With their resurrection to species rank, the conservation status of each of the taxa requires re-assessment. Presbytis femoralis has a small global population size which continues to decline mainly due to habitat loss. There are 60 individuals (48 mature individuals) in the Singapore population of P. femoralis47. There are no precise population estimates available for the conspecifics in the Malaysian states of Johor and Pahang, but it is believed that only a few hundred individuals remain (see Abdul-Latiff et al.28); i.e., the overall population of P. femoralis could well be <250 mature individuals. Furthermore, the extensive habitat loss especially to industrial-scale oil palm plantations in southern Peninsular Malaysia is unlikely to cease in the near future (see Shevade et al.48; Shevade and Loboda49). Hence, based on a small population size and decline, we propose to list P. femoralis as Critically Endangered (C2a(i): <250 mature individuals, continuing population decline, and ≤50 mature individuals in each subpopulation).
Presbytis percura is only found in a number of isolated forests and faces extinction in the wild based on large-scale forest loss in Riau Province50. Riau experienced the highest rate of deforestation in Sumatra and 63% of the natural forest have been lost between 1985 and 200851. Additionally, forest fires linked to the ENSO events, and open burning of forest land for agricultural purposes destroy millions of hectares of land in Indonesia on an annual basis, and Riau is often one of the worst impacted areas, owing in part to its high concentration of peatland52. We thus infer that the area of occupancy, extent of occurrence and quality of habitat of P. percura have declined to such an extent that the population size has reduced by ≥80% over the last three generations since 1989 (30 years approximately; see Nijman and Manullang53 for the closely-related P. melalophos), thus fulfilling the IUCN criteria for Critically Endangered (A2cd A3cd A4cd).
Presbytis robinsoni ranges from northern Peninsular Malaysia through southern Thailand to southern Myanmar. There are no population estimates (neither recent nor in the past), but some of the species’ habitat continues to be converted for agriculture (primarily oil palm) and it is also targeted by the illegal pet trade. Overall, it cannot be evaluated based on population size and/or the restricted population criterion, but P. robinsoni is certainly a taxon of conservation concern and is here considered Near Threatened.
An urgent need for molecular data for additional Presbytis populations
Our results highlight the need for sampling multiple populations of Presbytis species and subspecies because even our limited fieldwork already provided strong evidence for the widespread presence of cryptic diversity or inappropriate synonymization within the genus. Additional data are also needed in order to be able to precisely assign samples and understand the distribution of Presbytis species. We collected one faecal sample that was suspected to come from an individual of P. siamensis cf. cana. However, its placement in the phylogeny reveals that it belongs to a genetically distinct lineage that is more closely related to P. melalophos + mitrata than P. s. siamensis (Supplementary Fig. S2). If the sample was indeed from P. s. cana, then the taxonomy of the pale-thighed langur P. siamensis, which currently comprises four subspecies, needs to be revisited unless the unexpected signal is due to introgression via the hybridization of two species. Regardless of the explanation, the taxon represented by the sample deserves species status, but the correct scientific name and range remain unclear because P. s. paenulata and P. s. rhionis lack molecular data. Even if the faecal sample originated from individual of P. melalophos/mitrata, its genetic distinctness suggests that these species require more attention. In addition, it would mean that the geographic ranges of these species need revision because the species are unknown from the place of sample collection. Overall, either explanation is reason for concern. Geographically, only P. s. siamensis (Fig. 5; left photo) has a wide distribution on the Malay Peninsula while the remaining three subspecies have narrow distributions (Fig. 6). Presbytis s. cana (Fig. 5; right photo) occurs in eastern Sumatra and on Kundur Island, P. s. paenulata is found mainly in a small-wedge of coastal forest in east-central Sumatra, and P. s. rhionis has only been found on the islands of Bintan and Batam (but may also be found on Galang Island) in the Riau Archipelago11. Given the extensive habitat loss to oil palm plantations in Sumatra and large-scale economic development in Bintan and Batam, these taxa are likely highly threatened. Study is urgently needed and we submit that faecal samples would be the best way to rapidly address the species limits and distributions. Particular attention should be given to sampling additional P. s. cana given the results of this study. Currently, we lack genetic data, even COI barcodes, for many subspecies of Presbytis and their distributions are poorly understood. Clearly, Southeast Asian langurs have received insufficient attention and broad surveys are needed that estimate population sizes while collecting faecal samples for a re-assessment of species boundaries54.

Presbytis siamensis siamensis (left image: Lee Zan Hui) and P. s. cana (right image: Andie Ang).

Distribution of four subspecies of Presbytis siamensis. Image: Lee Zan Hui.
Mitochondrial phylogeny of Presbytis
In the process of delimiting species, we re-examined the phylogenetic relationships within Presbytis by combining the data for mitochondrial genomes with the data generated by Meyer et al.12 for cyt-b and d-loop-HV1. This led to the resurrection of P. femoralis and P. percura which are here revealed to be sister species and more closely related to several other Presbytis species from Sumatra (and P. rubicunda in Borneo) than P. robinsoni from the Malay Peninsula. Note, that this placement of P. femoralis is in conflict with relationships proposed by Abdul-Latiff et al.28, who obtained a clade comprising of P. femoralis + P. robinsoni + P. siamensis which was sister species to the Presbytis species from Sumatra + P. rubicunda.
One limitation of our study is the lack of nuclear data for species delimitation and reconstructing relationships (e.g. Wang et al.39). Unfortunately, obtaining nuclear data from faecal samples remains challenging partially due to the low concentration of primate DNA in faecal samples. We assessed the primate nuclear DNA content in these metagenomes and found that it was only 0.09–3.13% of total DNA (Table 5). However, we would argue that the lack of nuclear data does not seriously challenge our conclusions. Firstly, we reveal deep mitochondrial splits of >2 million years between P. femoralis and P. percura. Secondly, our proposed species limits are consistent with morphological data that allow for assigning specimens unambiguously to one of the lineages that are here resurrected as species. Lastly, whatever limited information is available for Asian colobines does not point to widespread conflict between nuclear and mitochondrial signals. Wang et al.39 presents phylogenetic reconstructions based on mitochondrial and nuclear markers for Asian colobines. Four of the six congeneric nodes are congruent between nuclear and mitochondrial data although they came from different individuals (i.e., study used data from multiple sources). Nonetheless, it will be important to develop new approaches for obtaining nuclear data from faecal samples (see Chiou and Bergey55).
Non-invasive samples for species discovery
Seventeen (61%) of the 28 taxa of Presbytis are threatened (Vulnerable, Endangered, or Critically Endangered) while five (18%) taxa are Data Deficient5,11. Many taxa continue to be affected by habitat loss. This means that there is an urgent need to resolve species limits in order to better assess their conservation status and needs. Molecular data play a critical role in resolving species limits, and here faecal samples are particularly valuable because they allow for accelerating data collection. Yet, our literature survey suggests that faecal DNA remains underutilized for taxonomic research with only two other published studies explicitly using faecal DNA as evidence for justifying decision on species status: nine members from a brown lemur complex were given species rank34 and two subspecies of otters were elevated to species33. Faecal samples yield DNA that are valuable not only for taxonomic purposes but also population genetics, diet analyses, microbiome and parasite research, and should be routinely collected during field surveys. In groups such as Presbytis, faecal samples would be particularly useful as these animals are shy and many populations are highly threatened.
Conclusions
We here demonstrate the value of non-invasive faecal samples for addressing taxonomic questions that are of significant conservation importance. Based on mitochondrial DNA (mitogenomes, cyt-b and d-loop), we resurrect three species within the Presbytis femoralis group. The new species limits also led to a change in the conservation status of P. femoralis and P. percura which now have to be considered Critically Endangered. We further urge researchers to include the collection of non-invasive faecal samples into their field protocols8.
Materials and Methods
Literature survey
In order to assess how frequently faecal samples have been used for addressing taxonomic questions, we conducted a survey of mammalian literature for the last 41 years (between 1978–2018). We downloaded the records pertaining to Supertaxon Mammalia (ST = Mammalia) from Zoological Record (articles only). We then identified a subset containing valid mammalian species names by using the checklist by Burgin et al.56. This was done by searching the binomial name of 6,399 species in the list. We also searched for the genus and species names separately to include records that utilize genus abbreviations. We then retrieved the studies that involved DNA/molecular work on faecal samples by searching for the terms (feces/faeces/fecal/faecal/scat/scats) and (DNA/barcod/sequenc/molecul/genom/genetic/microsatellite). We lastly retrieved the records that were classified under Systematics/Taxonomy in the “Broad terms” field and examined the records manually.
Sample collection, DNA extraction, and sequencing
This study involved two types of datasets: (1) metagenomic data newly generated for faecal samples collected in Sumatra for Presbytis femoralis percura and one sample likely to belong to P. siamensis cana (herein referred as P. s. cf. cana) and (2) metagenomic data previously sequenced as part of Srivathsan et al.10 and reanalysed here because this study did not reconstruct multiple mitochondrial genomes.
Five faecal samples were collected for P. femoralis percura during an eight-day (25 April - 2 May 2018) survey in Riau Province50. We also collected one faecal sample believed to come from P. s. cana as the monkeys were seen on the same tree below which the fresh faecal sample was found. The samples were preserved following a two-step ethanol-silica method57 and subsequently stored at a −20 °C freezer at the Andalas University in Sumatra. Genomic DNA was extracted at Andalas University from 50 mg of faeces using QIAamp Fast DNA Stool Mini Kit (QIAGEN, Singapore). DNA was recovered in 30 μl of elution buffer (instead of 200 μl) in order to obtain a higher concentration of DNA. Each sample was also extracted 2–3 times and later pooled to recover more genomic DNA. Genomic DNA of these six samples were sent from Andalas University for Illumina HiSeq X (Illumina Inc., San Diego, CA) sequencing (150 PE) by a commercial provider (NovogeneAIT). A library was constructed for each faecal sample (fragment size 350 bp) using NEBNext Ultra II DNA Library Prep Kit. The five faecal samples for P. f. percura and one faecal sample of P. s. cf. cana were sequenced using HiSeq.
Bioinformatics for obtaining mitochondrial genomes
Raw reads generated for P. f. percura and P. s. cana in this study as well as those for six samples of P. f. femoralis from Srivathsan et al.10 were trimmed using Trimmomatic v.0.3335 under the following parameters: LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50, and the ILLUMINACLIP parameter was set at 2:30:1035. New mitochondrial reference genomes were assembled for P. f. percura and P. s. cana using MITObim v.1.958 using the available mitogenome of P. f. femoralis (KU899140: 16548 bp) from one sample each (ESBL1b and Pres2). Any redundancy due to the circular nature of mitochondrial genome was removed in the resulting assembly (details in readme file in Supplementary Material 3). We then obtained one mitochondrial genomes per faecal sample by mapping the quality-trimmed reads for the sample to the reference genome (ESBL samples to mitochondrial genome from ESBL1b, Pres2 to Pres2 and BLM1–6 to KU899140 or BLM5). Reads were mapped using Bowtie259 under paired end and --end-to-end mode. The resulting SAM files were converted to BAM files using SAMtools60. Variants were detected using LoFreq61 using parameters similar to Isokallio and Stewart62 with a few more stringent criteria: we included mapping quality in LoFreq’s model and also retained base-alignment quality. We furthermore applied a minimum allele frequency of 0.2 while filtering the vcf to accept heteroplasmy only if it was at a frequency of >0.2. Alternate mitochondrial genomes were reconstructed from the resulting vcf file using gatk63 FastaAlternateReferenceMaker and heteroplasmic sites were modified as ambiguous nucleotides using custom script. All mitochondrial genomes were annotated using MITOS64.
Lastly, we checked the BAM files for each mitochondrial genome for contamination with human reads. The reads were retrieved from BAM file corresponding to each mitochondrial genome and mapped back to reference human mitochondrial genome (NC_012920.1) using Bowtie2 under (--end-to-end, single end). No reads matched with 0 mismatches, a maximum of one read mapped with 1 bp mismatch across the datasets, while the remaining few reads (~3%) mapped at > = 2 bp. In order to assess if these reads were more similar to a Presbytis mitogenome than to a human mitogenome, we matched the sequences mapping to the human mitogenome against both human and P. f. robinsoni (DQ355299 or NC_008217.1) genomes using BLASTN and retained the best matching sequence. The P. f. robinsoni genome selected here is a curated RefSeq genome. Overall 0.06% of the mitochondrial reads preferentially matched to the human genome. The reference genomes for the three subspecies ESBL1b, Pres2 and BLM5 had 1/2874, 0/1054 and 13/22796 reads that were more likely human than Presbytis. In order to ensure that these reads did not affect the reconstruction of the reference mitochondrial genomes, we eliminated the reads from the BAM files using PICARD tools and recalled the consensus sequence as described before. The consensus sequence remained unchanged. The remaining samples also revealed negligible human contamination. ESBL 5, 6a, 8b (P. f. percura) and Pres2 (P. siamensis cf. cana) had no contaminating signals, while the number of reads for the shorter 76 bp datasets of P. f. femoralis revealed the following number of possible contaminating reads: BLM1: 2/4764, BLM2: 4/4307, BLM3: 1/3282, BLM4: 8/8220, BLM6: 3/2781. Overall, the mitochondrial genome reconstructions were not affected.
Phylogenetic reconstructions and species delimitations
The newly reconstructed mitochondrial genomes were combined with publicly available data from GenBank. For the latter, we downloaded all the mitochondrial sequences for colobine primates and then retained only the data for Asian colobines. We then curated the GenBank records by consulting the source publication and assessing the locality information in order to update the taxonomic names given that many subspecies are now considered species. We excluded those sequences for which the source information was incomplete. This curated set of sequences was used for downstream distance based (Automated Barcode Gap Discovery, ABGD37 and Objective Clustering38 and tree-based species delimitation analyses (Poisson Tree Processes or PTP)36. We also included data for d-loop HV1 obtained by Abdul-Latiff et al.28 for P. f. femoralis, P. f. robinsoni and P. s. siamensis from Malaysia. Given that these sequences were not submitted to GenBank, we used Nijman’s26 reconstruction of the sequences based on a table that lists all variable sites relative to a reference sequence.
For analyses with PTP, we used three datasets: (1) The Asian colobine mitogenome dataset based on genomes for Asian colobines (minimum length > 10,000 bp). The sequences for the 13 mitochondrial CDS, two ribosomal genes, and complete d-loop sequences were extracted, aligned, and concatenated. Here, Colobus guereza and Macaca sylvanus were selected as the outgroups. (2) A second dataset included the Presbytis mitogenome+cyt-b + HV1 dataset. This dataset covers more samples because Presbytis has been well sampled for cyt-b and the hypervariable region I (HV1) of d-loop (see Meyer et al.12). For the analyses of this dataset, we used Trachypithecus obscurus and T. barbei as outgroups. (3) The last dataset comprised HV1 sequences only (Presbytis HV1-only dataset). This included HV1 sequences obtained by Abdul-Latiff et al.28 for P. f. femoralis, P. f. robinsoni and P. s. siamensis.
All coding sequences were aligned in MEGA X65 based on amino acid translations (using Clustal). The ribosomal genes and d-loop sequences were aligned using MAFFT LINSI66. We ensured that only distinct haplotypes were retained and only used the longest sequence if identical sequences were found. The alignments were concatenated in SequenceMatrix 1.7.867. Maximum Likelihood reconstructions were carried out using RAxML v868. For the mitogenome and the Presbytis mitogenome+cyt-b + HV1 datasets, we determined best partitioning scheme by providing 42 different partitions to PartitionFinder69 corresponding to codon position for the 13 coding regions and 3 separate partitions for 12 S, 16 S and d-loop. RAxML was run using GTRGAMMA with the resulting partitioning scheme (no partitioning was done for the HV1 dataset) with 20 independent searches for the best tree. Multiparametric bootstrapping was conducted applying the automatic bootstopping criterion (autoMRE). The resulting trees were subjected to PTP-based species delimitation after excluding outgroups.
Distance-based species delimitation utilized ABGD based on uncorrected distances70. We assessed species delimitations under different parameters by varying the slope X = 0.1, X = 0.5 and X = 1 (X = 1.5 was not applicable to the dataset) and prior intraspecific divergences. Species delimitation was carried out based on the Asian colobine mitogenome dataset described above and the Presbytis cyt-b and HV1 alignments. The same datasets were also clustered using Objective Clustering as implemented in Species Identifier (Taxon DNA 1.6.2)38 at genetic distances of 2.0, 3.0 and 4.0%.
Divergence dating
Divergence dates between Asian colobine lineages were determined using BEAST v 2.6.071. We used Asian colobine mitogenome dataset but excluded d-loop for this analysis due its differing mutational patterns as done for previous studies12,72. For divergence estimates, we included the following genomes to the Asian colobine mitogenome dataset: Pongo pygmaeus (NC_001646), Pan troglodytes (NC_001643), Homo sapiens (NC_012920), Chlorocebus aethiops (NC_007009), Macaca sylvanus (NC_002764), Papio hamadryas (NC_001992), Theropithecus gelada (NC_019802). We also did a second analysis for Presbytis cyt-b as done by Meyer et al.12 and used a similar strategy for the various steps. Here, representative sequences from different Asian colobine genera were included: Trachypithecus obscurus (NC_006900), Nasalis larvatus (NC_008216), Rhinopithecus avunculus (NC_015485), Semnopithecus vetulus (NC_019582) in addition to the above-mentioned sequences for fossil-based calibration. The fossil calibration dates used in this study also followed the dates used by Meyer et al.12. For mitochondrial genomes, we analysed the data using the following partitioning schemes: by codon (5 partitions: 1,2,3 codon for coding genes, 12 S, 16 S) and by gene. We again tested partitioning by both gene and codon, but found the Effective Sample Size (ESS) to be low for multiple parameters. For cyt-b dataset, we used the 1 + 2 and 3 codon partitioning scheme12.
Divergence estimates were based on a relaxed log normal clock and a Yule prior. Site models were unlinked across partitions, and a model-averaging approach was used as implemented in bModelTest73. Two independent runs were conducted with 25 million generations with sampling at every 1000 generations. Tracer v 1.7.1 was used to assess convergence, LogCombiner 2.6.1 was used to combine the results and 10% burn-in removal was applied. TreeAnnotator v 2.6.0 was used to summarize the trees.
Estimation of host DNA content in faecal metagenomes
In order to estimate the amount of host nuclear DNA in the faecal metagenomes, we mapped the metagenomic reads to a colobine reference genome (Rhinopithecus roxellana: GCF_000769185.1) using bowtie-2 (--end-to-end and--very-sensitive mode). We next excluded reads that could represent contamination with human DNA. For this, the mapped reads were retrieved from the resulting bam files using samtools. These were mapped back to a combined reference dataset of R. roxellana and human genome (GCF_000001405.39, GRCh38) using Bowtie2. Resulting BAM file was filtered to exclude reads with any mismatches, and we then excluded all sequences that matched to human genome only.
Ethics statement
We followed the Code of Best Practices for Field Primatology (2014). All genetic material were obtained non-invasively through faecal samples; no animals were harmed in the process. We followed the rules and regulations of the Government of Indonesia and RISTEKDIKTI (research permit no. 3051/FRP/E5/Dit.KI/IX/2018).
References
IPBES. Summary for policymakers of the global assessment report on biodiversity and ecosystem services of the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services. In The IPBES Global Assessment on Biodiversity and Ecosystem Services (ed. Díaz, S. et al.) (IPBES secretariat, Bonn, Germany (2019).
Sodhi, N. S., Koh, L. P., Brook, B. W. & Ng, P. K. L. Southeast Asian biodiversity: an impending disaster. Trends Ecol. Evol. 19(12), 654–660 (2004).
Trimble, M. J. & van Aarde, R. J. Geographical and taxonomic biases in research on biodiversity in human-modified landscapes. Ecosphere 3(12), 1–16 (2012).
Titley, M. A., Snaddon, J. L. & Turner, E. C. Scientific research on animal biodiversity is systematically biased towards vertebrates and temperate regions. PLoS One 12(12), e0189577 (2017).
Mittermeier, R. A., Rylands, A. B. & Wilson, D. E. Handbook of the Mammals of the World, Vol. 3, Primates. Lynx Edicions, Barcelona, Spain (2013).
Mace, G. The role of taxonomy in species conservation. Phil. Trans. R. Soc. Lond. B Biol. Sci. 359(1444), 711–719 (2004).
Thomson, S. A. et al. Taxonomy based on science is necessary for global conservation. PLoS Biol. 16(3), e2005075 (2018).
Srivathsan, A., Nagarajan, N. & Meier, R. Boosting natural history research via metagenomic clean-up of crowdsourced feces. PLoS Biol. 17(11), e3000517 (2019).
Srivathsan, A., Sha, J., Vogler, A. P. & Meier, R. Comparing the effectiveness of metagenomics and metabarcoding for diet analysis of a leaf‐feeding monkey (Pygathrix nemaeus). Mol. Ecol. Res. 15(2), 250–261 (2015).
Srivathsan, A., Ang, A., Vogler, A. P. & Meier, R. Fecal metagenomics for the simultaneous assessment of diet, parasites, and population genetics of an understudied primate. Front. Zool. 13, 17 (2016).
Roos, C. et al. An updated taxonomy and conservation status review of Asian primates. Asian Primates J. 4(1), 2–38 (2014).
Meyer, D. et al. Mitochondrial phylogeny of leaf monkeys with implications for taxonomy and conservation. Mol. Phylogenet. Evol. 59(2), 311–319 (2011).
Oates, J. F., Davies, A. G. & Delson, E. The diversity of living colobines. In: Davies, A. G. & Oates, J. F. (eds). Colobine Monkey: Their Ecology, Behaviour and Evolution. Cambridge University Press (1994).
Nijman, V. Ecology of sympatric and allopatric Presbytis and Trachypithecus langurs in Sundaland. in The Colobines: Natural History, Behaviour and Ecological Diversity. (eds. Matsuda, I., Grueter, C. C. & Teichroeb, J. E). (Cambridge University Press, Cambridge, in press).
IUCN. The IUCN Red List of Threatened Species. https://www.iucnredlist.org (2008).
Groves, C. P. Primate Taxonomy. Smithsonian Institution Press, Washington (2001).
Martin, W. C. L. A monograph of the genus Semnopithecus (continued from page 326). Mag. Nat. Hist. 2, 434–441 (1838).
Raffles, S. T. Descriptive catalogue of a zoological collection made in the island of Sumatra and its vicinity. Trans. Linn. Soc. Lond. 13, 247 (1821).
Lyon, M. W. Jr Mammals collected in eastern Sumatra by Dr. W. L. Abbott during 1903, 1906, and 1907: with descriptions on new species and subspecies. Proc. US Natl. Mus. 34, 619–679 (1908).
Thomas, O. A new monkey from the Malay Peninsula. Proc. Zool. Soc. Lond. (Abstr. p. 26), 634-635 (1910).
Robinson, H. C. & Kloss, C. B. On six new mammals from the Malay Peninsula and adjacent islands. J. Fed. Malay States Mus. 4(2), 169–174 (1911).
Weitzel, V., Yang, C. M. & Groves, C. P. A catalogue of primates in the Singapore zoological reference collection. Raffles B. Zool. 36, 1–166 (1988).
Ang, A., Srivathsan, A., Md.-Zain, B. M., Ismail, M. R. B. & Meier, R. Low genetic variability in the recovering urban banded leaf monkey population of Singapore. Raffles B. Zool. 60, 589–594 (2012).
Ang, A. and Boonratana, R. Presbytis femoralis ssp. percura. In: The IUCN Red List of Threatened Species 2015 (in press).
Ang, A. Banded Leaf Monkeys in Singapore: Preliminary Data on Taxonomy, Feeding Ecology, Reproduction, and Population Size. MSc thesis, National University of Singapore (2010).
Nijman, V. Presbytis neglectus or P. femoralis, colobine molecular phylogenies, and GenBank submission of newly generated DNA sequences. Folia Primatol. 91(3), 228–239 (2019).
Vun, V. F., Mahani, M. C., Lakim, M., Ampeng, A. & Md.-Zain, B. M. Phylogenetic relationships of leaf monkeys (Presbytis; Colobinae) based on cytochrome b and 12S rRNA genes. Genet Mol Res 10(1), 368–381 (2011).
Abdul-Latiff, M. A. B., Baharuddin, H., Abdul-Patah, P. & Md.-Zain, B. M. Is Malaysia’s banded langur, Presbytis femoralis femoralis, actually Presbytis neglectus neglectus? Taxonomic revision with new insights on the radiation history of the Presbytis species group in Southeast Asia. Primates 60(1), 63–79 (2019).
Md.-Zain, B. M., Morales, J. C., Hassan, M. N., Jasmi, A. & Melnick, D. J. Is Presbytis a distinct monophyletic genus: inferences from mitochondrial DNA sequences. Asian Primates J. 1, 26–36 (2008).
Sterner, K. N., Raaum, R. L., Zhang, Y.-P., Stewart, C.-B. & Disotell, T. R. Mitochondrial data support an odd-nosed colobine clade. Mol. Phylogenet. Evol. 40(1), 1–7 (2006).
Schlegel, H. Monographie 40: simiae. Revue Methodique, Museum d’Histoire Naturelle des Pays-Bas 7, 1–356 (1876).
Low, M. E. Y. & Lim, K. K. P. The authorship and type locality of the banded leaf monkey. Presbytis femoralis. Nature Singapore 8, 69–71 (2015).
Koepfli, K.-P. et al. Establishing the foundation for an applied molecular taxonomy of otters in Southeast Asia. Conserv. Genet. 9, 1589 (2008).
Markolf, M. et al. True lemurs… true species - species delimitation using multiple data sources in the brown lemur complex. BMC Evol. Biol. 13, 233 (2013).
Bogler, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 30, 2114–2120 (2014).
Zhang, J., Kapli, P., Pavlidis, P. & Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29, 2869–2876 (2013).
Puillandre, N., Lambert, A., Brouillet, S. & Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 21, 1864–1877 (2012).
Meier, R., Shiyang, K., Vaidya, G. & Ng, P. K. L. DNA barcoding and taxonomy in Diptera: a tale of high intraspecific variability and low identification success. Syst. Biol. 55, 715–728 (2006).
Wang, X. P. et al. Phylogenetic relationships among the colobine monkeys revisited: new insights from analyses of complete mt genomes and 44 nuclear non-coding markers. PLoS One 7(4), e36274 (2012).
Napier, J. R. & Napier, P. H. A Handbook of Living Primates. Academic Press, London (1967).
Tilson, R. L. Infant coloration and taxonomic affinity of the Mentawai Islands leaf monkeys, Presbytis potenziani. J. Mammal. 57, 766–769 (1976).
Weitzel, V. A preliminary analysis of the dental and cranial morphology of Presbytis and Trachypithecus in relation to diet. MA thesis, Australian National University, Canberra (1983).
Roos, C., Nadler, T. & Walter, L. Mitochondrial phylogeny, taxonomy and biogeography of the silvered langur species group (Trachypithecus cristatus). Mol. Phylogenet. Evol. 47, 629–636 (2008).
Voris, H. K. Maps of Pleistocene sea levels in Southeast Asia: shorelines, river systems and time durations. J. Biogeogr. 27, 1153–1167 (2000).
Slik, J. W. et al. Soils on exposed Sunda Shelf shaped biogeographic patterns in the equatorial forests of Southeast Asia. PNAS 108, 12343–12347 (2011).
Tan, S. H. D., Ali, F., Kutty, S. N. & Meier, R. The need for specifying species concepts: how many species of silvered langurs (Trachypithecus cristatus group) should be recognized? Mol. Phylogenet. Evol. 49, 688–689 (2008).
Ang, A. Final Report (Phase 1: 2016-2018): Species Action Plan for the Conservation of Raffles’ Banded Langur (Presbytis femoralis femoralis) in Malaysia and Singapore. Unpublished Report, Wildlife Reserves Singapore (2018).
Shevade, V. S., Potapov, P. V., Harris, N. L. & Loboda, T. V. Expansion of industrial plantations continues to threaten Malayan tiger habitat. Remote Sens. 9(7), 747 (2017).
Shevade, V. S. & Loboda, T. V. Oil palm plantations in Peninsular Malaysia: Determinants and constraints on expansion. PLoS One 14(2), e0210628 (2019).
Rizaldi et al. Preliminary study on the distribution and conservation status of the East Sumatran banded langur (Presbytis femoralis percura) in Riau Province, Sumatra, Indonesia. Asian Primates J. 8(1), 25–36 (2019).
Uryu, Y. et al. Sumatra’s forests, their wildlife and the climate, windows in time: 1985, 1990, 2000, and 2009. WWF-Indonesia Technical Report Jakarta Indonesia. (2010).
World Bank. The cost of fire: An economic analysis of Indonesia’s 2015 fire crisis. Indonesia Sustainable Landscapes Knowledge Note. 1 (2016).
Nijman, V. & Manullang, B. Presbytis melalophos. In: The IUCN Red List of Threatened Species 2008: e.T18129A7666452. Accessed on 12 September 2019 (2008).
Ang, A., Ismail, M. R. B. & Meier, R. Reproduction and infant pelage colouration of the banded leaf monkey (Mammalia: Primates: Cercopithecidae) in Singapore. Raffles Bull. Zool. 58(2), 411–415 (2010).
Chiou, K. L. & Bergey, C. M. Methylation-based enrichment facilitates low-cost, noninvasive genomic scale sequencing of populations from feces. Sci. Rep. 8, 1975 (2018).
Burgin, C. J., Colella, J. P., Kahn, P. L. & Upham, N. S. How many species of mammals are there? J. Mammal. 99, 1–14 (2018).
Nsubuga, A. M. et al. Factors affecting amount of genomic DNA extracted from ape faeces and the identification of an improved sample storage method. Mol. Ecol. 13, 2089–2094 (2004).
Hahn, C., Bachman, L. & Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucleic Acids Res. 41, e129 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Wilm, A. et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 40, 11189–11201 (2012).
Isokallio, M. A. & Stewart, J. Low-frequency variant calling from high-quality mtDNA sequencing data. protocols.io https://doi.org/10.17504/protocols.io.nfkdbkw (2018).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Bernt, M. et al. MITOS: Improved de novo Metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Katoh, K., Kuma, K., Hiroyuki, T. & Miyata, T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518 (2005).
Vaidya, G., Lohman, D. J. & Meier, R. SequenceMatrix: concatenation software for the fast assembly of multi‐gene datasets with character set and codon information. Cladistics 27, 171–180 (2011).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T. & Calcott, B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773 (2016).
Srivathsan, A. & Meier, R. On the inappropriate use of Kimura‐2‐parameter (K2P) divergences in the DNA‐barcoding literature. Cladistics 28, 190–194 (2012).
Bouckaert, R. et al. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10(4), e1003537 (2014).
Stone, A. C. et al. More reliable estimates of divergence times in Pan using complete mtDNA sequences and accounting for population structure. Phil. Trans. R. Soc. Lond. B Biol. Sci. 365(1556), 3277–3288 (2010).
Bouckaert, R. R. & Drummond, A. J. bModelTest: Bayesian phylogenetic site model averaging and model comparison. BMC Evol. Biol. 17, 42 (2017).
Miller, G. S. Jr. The langurs of the Presbytis femoralis group. J. Mammal. 15, 124–137 (1934).
Elliot, D. G. S Pygathrix. In: A Review of the Primates, Vol. 3. American Museum of Natural History, New York (1913).
Miller, G. S. Jr Fifty-one new Malayan mammals. Smithsonian Miscellaneous Collections 61, 28 (1913).
Pocock, R. I. The monkeys of the genera Pithecus (or Presbytis) and Pygathrix found to the east of the Bay of Bengal. Proc. Zool. Soc. Lond. 104(4), 895–961 (1934).
Raven, H. C. Wallace’s Line and the Distribution of Indo-Australian Mammals. New York Press (1935).
Chasen, F. N. A handlist of Malayan mammals. B. Raffles Mus. 15, 1–209 (1940).
Hooijer, D. A. Quaternary langurs and macaques from the Malay Archipelago. Zoologische Verhandelingen Uitgegeven door het Rijksmuseum van Natuurlijke Historie te Leiden 55, 1–64 (1962).
Medway, L. The monkeys of Sundaland: ecology and systematics of the cercopithecids of a humid equatorial environment. In: Napier, J. R. & Napier, P. H. (eds). Old World Monkeys: Evolution. Academic Press, New York, 513–553 (1970).
Thorington, R. W. Jr. & Groves, C. P. An annotated classification of the Cercopithecoidea (1970). In: Napier, J. R. & Napier, P. H. (eds). Old World Monkeys: Evolution. Academic Press, New York (1970).
Wilson, C. C. & Wilson, W. L. Behavioral and morphological variations among primate population in Sumatra. Yearb. Phys. Anthropol. 20, 207–233 (1977).
Medway, L. The Wild Mammals of Malaya (Peninsular Malaysia) and Singapore. Oxford University Press, Oxford (1983).
Brandon-Jones, D. Colobus and leaf monkeys. In: MacDonald, I. D. (ed). Encyclopaedia of Mammals. George Allen and Unwin, London, 398-408 (1984).
Napier, P. H. Catalogue of Primates in the British Museum (Natural History) and elsewhere in the British Isles. Part III: Family Cercopithecidae, Subfamily Colobinae. British Museum (Natural History), London (1985).
Aimi, M. & Bakar, A. Taxonomy and distribution of Presbytis melalophos group in Sumatra, Indonesia. Primates 33, 191–206 (1992).
Brandon-Jones, D. et al. Asian primate classification. Int. J. Primatol. 25, 97–164 (2004).
Md.-Zain, B. M. Molecular systematic of the genus Presbytis. PhD thesis. Columbia University, New York (2001).
Acknowledgements
This research was partially funded by a grant of the Wildlife Reserves Singapore Conservation Fund to Dr. Andy Ang. We would like to thank the Government of Indonesia and RISTEKDIKTI for the research permit (3051/FRP/E5/Dit.KI/IX/2018) and the Government of Singapore and National Parks Board for the research permit (NP/RP16-092). We sincerely appreciate the assistance from Anugrah Viona Agesi, Wila Karlina, and Dyta Rabbani Aidil of the Genetic and Biomolecular Laboratory at Andalas University.
Author information
Authors and Affiliations
Contributions
A.A., A.S., R.M., V.N. wrote the main manuscript text; A.A., A.S., R., R.M., V.N. designed the study; A.A., R. conducted the field work; A.A., D.I.R. extracted DNA samples in D.I.R.’s laboratory; A.S., D.I.R. conducted bioinformatics analyses; and A.S. reconstructed the time-tree. All authors reviewed and edited the manuscript and gave final approval for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ang, A., Roesma, D.I., Nijman, V. et al. Faecal DNA to the rescue: Shotgun sequencing of non-invasive samples reveals two subspecies of Southeast Asian primates to be Critically Endangered species. Sci Rep 10, 9396 (2020). https://doi.org/10.1038/s41598-020-66007-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66007-8
- Springer Nature Limited
This article is cited by
-
Overcoming Challenges to Extracting and Sequencing Historical DNA to Support Primate Evolutionary Research and Conservation, with an Application to Galagos
International Journal of Primatology (2024)
-
Reconstruction of the personal information from human genome reads in gut metagenome sequencing data
Nature Microbiology (2023)