
Abstract
Skeletal muscle fibers are primarily categorized into oxidative and glycolytic fibers, and the ratios of different myofiber types are important factors in determining livestock meat quality. However, the molecular mechanism for determining muscle fiber types in chickens was hardly understood. In this study, we used RNA sequencing to systematically compare mRNA and microRNA transcriptomes of the oxidative muscle sartorius (SART) and glycolytic muscle pectoralis major (PMM) of Chinese Qingyuan partridge chickens. Among the 44,705 identified mRNAs in the two types of muscles, 3,457 exhibited significantly different expression patterns, including 2,364 up-regulated and 1,093 down-regulated mRNAs in the SART. A total of 698 chicken miRNAs were identified, including 189 novel miRNAs, among which 67 differentially expressed miRNAs containing 42 up-regulated and 25 down-regulated miRNAs in the SART were identified. Furthermore, function enrichment showed that the differentially expressed mRNAs and miRNAs were involved in energy metabolism, muscle contraction, and calcium, peroxisome proliferator-activated receptor (PPAR), insulin and adipocytokine signaling. Using miRNA-mRNA integrated analysis, we identified several candidate miRNA-gene pairs that might affect muscle fiber performance, viz, gga-miR-499-5p/SOX6 and gga-miR-196-5p/CALM1, which were supported by target validation using the dual-luciferase reporter system. This study revealed a mass of candidate genes and miRNAs involved in muscle fiber type determination, which might help understand the molecular mechanism underlying meat quality traits in chickens.
Similar content being viewed by others


Introduction
Improving meat quality has long been a goal of broiler breeding programs, especially for Chinese native breeds1,2. However, meat quality is difficult to define because it is a complex trait influenced by numerous factors3. As the main tissue determining meat quality, skeletal muscle is a heterogeneous tissue composed of different types of muscle fibers, varying in their biochemical and structural characteristics. Previous studies have found that different types of muscle fibers can influence meat quality traits, including meat color, tenderness, water-holding capacity, juiciness, and flavor4,5. In chickens, myofiber can be divided into red and white fibers, which are referred to as oxidative (type I and IIA) and glycolytic fibers (type IIB), respectively. Oxidative fibers exhibit slow contractility and oxidative metabolism based on mitochondrial oxidative phosphorylation, whereas glycolytic fibers have fast contractility and glycolytic metabolism6,7. Although the differences between various muscle fiber types in physiology and functionality have been well studied, the molecular regulation of their specification and maintenance in chickens remains largely unknown8,9.
miRNAs are highly conserved non-coding small RNAs that regulate gene expression at the post-transcriptional level in most biological processes. Emerging evidence has demonstrated that miRNAs are involved in skeletal muscle differentiation and development10,11,12. Recent studies have shown that some muscle-specific miRNAs play an important role in the regulation of muscle fiber type specification and maintenance in certain vertebrate species13,14. For example, miR-499 has been reported to control muscle fiber composition by repressing transcriptional repressors of slow-switch contractile protein genes, such as SOX6 and FNIP115,16. microRNA-139-5p in mice has been identified to suppress the expression of myosin heavy chains I and IIa via inhibition of the calcineurin(CaN)/NFAT signaling pathway17. Until now, the knowledge of miRNAs in regulating the chicken muscle fiber phenotype remains limited. According to the only report from Ma et al., miR-1611 in chickens can mediate the expression of Six1, thereby affecting the proliferation and differentiation of myoblasts and transformation of muscle fiber types18.
Qingyuan partridge chicken, an important indigenous breed in China, is popular because of its good meat quality19. High oxidative metabolism of this breed leads to desirable muscle characteristics, such as a high red muscle ratio and appealing meat color and flavor. In the present study, we compared the miRNA and mRNA differences between pectoralis major muscle (PMM) of the glycolytic type and sartorius muscle (SART) of the oxidative type and identify the key miRNA-mediated mechanism for muscle fiber type regulation. This work would contribute to the understanding of chicken meat quality control and improvement.
Results
Overview of RNA sequencing
We used Illumina deep sequencing to analyze the protein-coding transcript abundance in eight libraries based on the PMM and SART extracted from four Qingyuan partridge hens. After lower-quality reads were filtered, a total of 676.3 million clean reads were generated, more than 79.3% of which were mapped to the reference genome (Table S1). The GC contents of the clean data ranged from 46.7 to 49.0%, and the clean read quality scores of Q20 and Q30 were above 98.6% and 95.2%, respectively, demonstrating that the reliability and quality of the sequencing data were adequate for further analysis. The PCA analysis showed all eight samples were found to form two distinct clusters which were consistent with their groups’ descriptions (Figure S1).
Using Galgal 6.0 as the reference genome, 35,873 known transcripts and 8,832 novel protein-coding transcripts were identified in the data (Table S2). After the transcript expression levels were quantified, the average expression of the novel mRNAs (5.26) approximated to half of the known mRNAs (10.24). A total of 36,004 mRNAs were co-expressed in both the PMM and SART, while 3,958 and 4,743 transcripts were expressed only in the PMM and SART, respectively. All protein-coding transcripts corresponded to 15,962 genes, with an average of 2.8 transcripts per gene locus. Novel transcripts were identified in 37.6% of protein-coding genes in the present study (Table S2).
Analysis of differentially expressed mRNAs
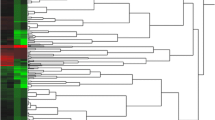
The analysis of differentially expressed genes in transcript level (DEGs) revealed a significant difference in multiple muscle tissues in chickens. There were 3,457 significant DEGs, including 2,364 up-regulated and 1,093 down-regulated mRNAs in the SART when compared to PMM (Table S3 & Fig. 1A). To further elucidate the functional roles of DEGs, we performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis for the DEGs. In biological processes, the enriched GO terms were associated with energy metabolism, blood circulation, muscle development, and contraction processes (Fig. 1B). The pathway enrichment revealed DEGs involved in several energy metabolism-related pathways, including oxidative phosphorylation, carbon metabolism and glycolysis/gluconeogenesis (Fig. 1B). Furthermore, the oxidative phosphorylation pathway was more often associated with up-regulated genes in SART, while glycolysis/gluconeogenesis was only enriched in PMM (Figure S2).

Gene and miRNA profiles between the pectoralis major muscle (PMM) and sartorius muscle (SART). (A) The hierarchical cluster analysis of differentially expressed mRNAs. (B) Top 20 significantly enriched GO terms of differentially expressed mRNAs in biological processes. (C) Top 20 significantly enriched KEGG pathways of differentially expressed mRNAs. The size and color of each bubble represent the amount of differentially expressed mRNAs enriched in the pathway and enrichment significance, respectively. (D) The hierarchical cluster analysis of differentially expressed miRNAs.
We also identified cardiac muscle contraction, adrenergic signaling in cardiomyocytes, focal adhesion, and the cGMP-PKG signaling pathway. Besides, most genes of CaN/NFAT signaling pathway, a core element of cGMP-PKG signaling pathway, were identified as DEGs in the present study.
Overview of small-RNA sequencing
A total of 102.5 Mb clean reads were obtained from eight libraries, representing a high ratio (> 90.8%) of clean reads (Table S4). After alignment with known small RNAs from Rfam, GenBank, and the reference genome, 76.1% of clean reads were identified as mature miRNAs, and the remaining small RNAs included rRNA, tRNA, scRNA, snRNA, snoRNA as well as exist-miRNA-edit (Fig. 2A). The size distribution of the reads was not significantly different in the PMM and SART libraries, and the majority of the reads had the lengths of 21–24 nt (Fig. 2B).

Overview of small RNA sequencing in chicken muscles. (A) The size distribution of all clean reads. (B) The distribution of raw reads mapped to the chicken genome. The percent of miRNA is approximately 76%, and the other 24% included rRNA, scRNA, snRNA, tRNA, snoRNA, repeat, exon sense/antisense, and intron sense/antisense.
Known chicken miRNAs that were identified were 509, including 405 co-expressed in the PMM and SART libraries and 104 expressed in only one library (Table S5). Ten mature miRNAs with the highest expression comprised approximately 75% of all known miRNAs’ reads, showing a relatively abundant distribution. Specially, three miRNAs, viz. gga-miR-1a-3p, gga-miR-26a-2-5p and gga-miR-26a-5p, had more than 10,000,000 reads. The number of novel miRNAs predicted in this study reached 189 (Table S5). The sequencing frequencies of the novel miRNAs were much lower as compared to those of the known miRNAs.
Analysis of differentially expressed miRNAs
A total of 67 differentially expressed miRNAs (DEMs) were identified, including 49 known and 18 novel miRNAs (Table S6 and Fig. 1D). From these, 42 miRNAs were up-regulated and 25 were down-regulated in SART. Among these DEMs, several related to muscle function were identified in this study, such as gga-miR-499, gga-miR-221, gga-miR-34a, and gga-miR-126. After target gene prediction, 5,862 target genes for the 67 DEMs were identified using a combination of three different tools. Among them, 1,537 mRNAs were found to be DEGs, which were assigned as intersection genes (Table S7). GO and KEGG pathway enrichment analysis of the intersection genes was performed to reveal the potential functions of the DEMs. The enriched GO terms mainly involved metabolic processes, cardiocyte differentiation and organ development biological processes (Figure S3A). The enriched pathways including the cGMP-PKG signaling pathway, adipocytokine signaling pathway, glycolysis/gluconeogenesis, insulin signaling pathway, AMPK signaling pathway, calcium signaling pathway and HIF-1 signaling pathway (Figure S3B).
Validation of differentially expressed mRNAs and miRNAs by qRT-PCR
To validate the differential expression results of mRNAs and miRNAs, the relative expression of eight mRNAs (MYH7B, NFATc3, PRKAG3, PPARGC1A, PPP3CA, CSRP3, SOX6, and CALM1) and four miRNAs (gga-miR-499-5p, gga-miR-196-5p, gga-miR-34a-5p and gga-miR-193a-3p) was quantified by qRT-PCR. In Fig. 3A, B, all selected DEGs and DEMs showed the concordant expression patterns between RNA-seq and qPCR results. Also, the computational and experimental fold changes in the present study showed a strong positive correlation with R2 = 0.9645 (Fig. 3C).

Validation of the differentially expressed mRNAs and miRNAs between the pectoralis major muscle (PMM) and sartorius muscle (SART). (A) Illustrating of qPCR confirmation results for eight selected differentially expressed mRNAs. (B) Illustrating of qPCR confirmation results for four selected differentially expressed miRNAs. (C) Regression analysis of log2(foldchange) values between RNA-seq and qPCR.
Construction of the miRNA-mRNA interaction network
By comparison with previous muscle-specific genes in pig, fish, and chicken8,20,21,22,23, more than 50 genes exhibiting similar expression patterns were screened in the current RNA-seq study (Table S8). These genes were involved in many functions related to muscle contraction and cytoskeleton, transcription factor, energy metabolism, Ca2+ homeostasis signaling and transport protein. Among these overlapped genes, most of the genes related to the myofiber characteristics in chickens, such as PPARGC1A, PRKAG3 and TGFB2, were also identified in the present study24,25. Besides, a list of important transcription factors including SOX6 and CSRP3 were found to be express in a fiber type-specific manner26,27.
To further understand and visualize the interactions of DEGs and DEMs related to muscle types, a miRNA-gene interaction network was constructed using the DEGs from Table S8 and all DEMs (Fig. 4). In the network, 92 interactions were identified. Two of the most significantly DEMs, gga-miR-196-5p and gga-miR-499-5p, were predicted to target CALM1 and SOX6, respectively. A dual-luciferase reporter system was used to verify the binding relationship between the identified miRNAs and mRNAs. Luciferase assay showed that gga-miR-196-5p and gga-miR-499-5p could reduce the luciferase activity by binding to the sites on CALM1 and SOX6 3′UTR, respectively (Fig. 5).

Interaction network of miRNAs and genes related to muscle fiber composition. The color of the node represents the expression regulation types of differentially expressed miRNAs and genes. Green means up-regulation and red means down-regulation in the SART.

Validation of the predicted miRNA-target interaction with the 3′UTR luciferase reporter system. (A) Luciferase assays were performed on DF-1 cells co-transfected with the SOX6 3′UTR-WT and gga-miR-499-5p mimics, or SOX6 3′UTR-MUT and gga-miR-499-5p mimics. (B) Luciferase assays were performed on DF-1 cells co-transfected with the CALM1 3′UTR-WT and gga-miR-196-5p mimics, or CALM1 3′UTR-MUT and gga-miR-196-5p mimics. Data were expressed as means ± SD. The P value was calculated using the t-test. **P < 0.01.
Discussion
Muscle fiber type composition is one of the key factors that affect meat quality. For example, the high content of oxidative fiber contributes more to juiciness and flavor28. Muscle fiber characteristics are affected by muscle types, locations, and functions within an animal. Compared to the research progress in mammals, the molecular mechanism underlying the muscle fiber characteristics in chickens is largely unknown, especially in miRNA regulation. In this work, we used the RNA-seq technology to compare mRNAs and miRNAs in order to identify transcriptomic differences between PMM and SART, as well as typical oxidative and glycolytic muscle tissues in chickens9,29. To our knowledge, our work was the first exploring the mechanism of chicken muscle fiber type regulation based on skeletal muscles using the RNA-seq approach.
In the present study, a total of 3,457 mRNAs were found differentially expressed between PMM and SART using high-throughput sequencing. By comparing with earlier transcriptome results related to different muscles, over 50 DEGs overlapped between multiple studies, indicating their roles in skeletal myofiber regulation8,20,21,22,23. As expected, the expression levels of genes related to energy metabolism, slow-type muscle protein-encoding, muscle contraction and cytoskeleton were significantly higher in oxidative muscles than in glycolytic muscles, implying their roles in meat quality regulation. For example, CSRP3 belongs to the cysteine and glycine-rich protein family, which play an important role in muscle fiber differentiation, is also highlighted as a fiber type-specific isoform in different species27,30. The mutation of another muscle-specific gene PRKAG3 has an influence on muscle metabolism and fiber type in pig, and have been shown to have a strong association with meat quality31,32. Similar to previous studies, the functional enrichment analysis demonstrated that these DEGs were involved in energy metabolism, muscle contraction, calcium homeostasis, peroxisome proliferator-activated receptor (PPAR), insulin and adipocytokine signaling. The newly identified DEGs might provide valuable information for the regulation of the chicken skeletal muscle phenotype.
Several groups have investigated miRNA expression between oxidative and glycolytic skeletal muscles in mammals and fish14,33,34,35,36. In this study, we have identified 899 chicken miRNAs in the two types of chicken muscles. Similar to other studies, most miRNA reads came from a few miRNAs37,38. Different from previous reports, the miRNAs with the highest expression were not differentially expressed between muscles. We detected 46 DEMs, many of which were related to muscle functions, including myofiber type switching. For example, as one of the most up-regulated miRNAs in the SART, miR-499 was shown to regulate the slow-twitch phenotype by targeting NFATc1/MEF2C and Fnip1/AMPK circuits15,39. Gga-miR-143 was another important miRNA. In swine, overexpression of miR-143 could induce the increase of slow fibers through targeting the HDAC4 expression40. Also, we found some DEMs identified here that involving muscle cell differentiation and energy metabolisms, such as gga-miR-221, gga-miR-34a and gga-miR-12641,42,43. Moreover, the biological functions of a few miRNAs in chicken muscles was revealed through the GO and pathway analysis of intersection genes. Our results provide novel information regarding the regulatory roles of these miRNAs.
To further understand the mechanism how miRNAs and their targets regulated muscle fiber types, an interaction network between miRNAs and mRNAs related to muscle fiber composition was constructed. One of the core genes in the network, SOX6 was targeted by seven miRNAs including gga-miR-499-5p and gga-miR-499-3p. As an important member of the SRY-related high mobility group (HMG) box (SOX) family of the transcription factors, SOX6 was highly expressed in skeletal muscle and acted as a repressor of fetal slow-twitch-specific gene expression in zebrafish and mice44,45. Further studies showed that SOX6 was a target of miR-499-5p16,46. As the top up-regulated miRNA in the SART, miR-196-5p was predicted to suppress CALM1 and MYLK4 expression. Based on KEGG enrichment analysis, CALM1 and MYLK4 were key elements of cGMP–PKG and calcium signaling pathways, which were vital in the regulation of muscle fiber type transformation47,48. In our previous work, PPARGC1A was reported being essential to slow muscle fiber formation in chicken myoblast cells, and its polymorphisms were associated with skeletal myofiber type traits8,24. We found that gga-miR-193-3p inhibited PPARGC1A in chicken muscles. Interestingly, our data on target validation provided strong evidence that SOX6 was a target of gga-miR-499-5p, whereas CALM1 was a target of gga-miR-196-5p. We therefore hypothesized that these miRNAs could target sequences in these genes to regulate the chicken muscle fiber phenotype.The CaN/NFAT signaling pathway provides an important link to calcium pattern dependent signaling and is implicated in the control of skeletal muscle fiber gene expression49,50. In the present study, several DEGs including CALM1, CALM2, PPP3CA, NFATc1 and NFATc3, were implicated in the CaN/NFAT signaling pathway, which was consistent with the previous studies8. Based on the miRNA-mRNA interaction network, various miRNAs were predicted to target these genes. Among these identified miRNAs, gga-miR-143-5p, gga-miR-499-5p and gga-miR-129-3p had multiple target genes, which were implicated in CaN/NFAT signaling, suggesting their roles in chicken muscle fiber regulation through the CaN/NFAT signaling pathway.
In summary, we characterized mRNA and miRNA transcript profiles of the PMM and SART muscles by RNA sequencing. We identified and characterized the DEGs and DEMs that were involved in chicken muscle fiber regulation. By analyzing these differentially expressed mRNAs and miRNAs, a miRNA-mRNA network associated with muscle fiber type composition was established. This study expanded our understanding of molecular mechanisms underlying meat quality traits in chickens.
Materials and methods
Ethics statement
All animal experiments were approved by the Animal Care and Use Committee at the Poultry Institute, Chinese Academy of Agricultural Science (Approval ID: S20180605). All these experiments followed relevant guidelines and regulations set by the Ministry of Agriculture and Rural Affairs of the People's Republic of China.
Animal, muscle sampling and RNA extraction
A total of 200 Qingyuan partridge chickens were obtained from Tinoo’s Foods Co., Ltd (Guangdong, China). All experimental birds were lived under the same environment and were raised using a standardized feeding method with free access to water. At the 140th day of age indicating sexual maturity, four female chickens with similar weights (1,428–1477 g, 1455 g averagely) were selected for transcriptome analysis. As previously described7, skeletal muscle samples were obtained from the intermediate section of the PMM and SART immediately after exsanguination, rapidly frozen in liquid nitrogen, and then stored at − 80 °C for further use.
Total RNAs were extracted from the PMM and SART muscle samples of the four hens using TRIzol reagent (Invitrogen, CA, USA) according to the manufacturer’s instructions. The RNA quality was checked using NanoDrop 2000 spectrophotometer (Thermo, USA) and Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA), respectively. RNA integrity number (RIN) ≥ 7 was set as the cutoff for RNA quality.
RNA sequencing and mRNA analysis
A total of eight cDNA libraries were constructed with four PMM and four SART muscle tissues, and 3 μg total RNA per sample was used as the input material for a cDNA library. After total RNAs were extracted, rRNAs were removed, and then the enriched RNAs were fragmented into short fragments and reverse transcribed into cDNAs. Double-stranded cDNAs were synthesized by replacing dTTPs with dUTPs in the reaction buffer used in second-strand cDNA synthesis. The resulting double-stranded cDNAs were ligated to adaptors after being end-repaired and A-tailed. Then, uracil-N-glycosylase (UNG) was used to digest the second-strand cDNAs. The digested products were size selected by agarose gel electrophoresis, PCR amplified and sequenced by Gene Denovo Biotechnology Co. (Guangzhou, China) using Illumina HiSeq 4000.
Raw data in FASTQ format were first processed with in-house scripts. The sequencing raw reads were obtained and used in further analysis after removing reads containing adapters, reads containing ploy-N, and low-quality reads from raw data. The clean reads of each sample were then mapped to the reference genome (Galgal 6.0) by Tophat2 (version 2.1.1)51. Known and novel transcripts from the TopHat alignment results were constructed and identified by the Reference Annotation Based Transcript (RABT) assembly of Cufflinks v2.1.152. Transcript abundance was quantified by the RSEM software53. The transcript expression levels were normalized by using the Fragments Per Kilobase of transcript per Million mapped reads (FPKM) method. mRNAs with a P value < 0.05 and fold change ≥ 2 were then identified as significant DEGs using the edgeR package54.
Small RNA sequencing and miRNA analysis
Eight small RNA libraries were also constructed using the same RNA samples as those used for RNA sequencing. After extracting total RNAs from muscle tissues, low molecular weight RNAs were separated by polyacrylamide gel electrophoresis (PAGE). Then, the 3′ adapters and 5′ adapters were ligated to the RNAs as well. The ligation products were reverse transcribed by PCR amplification, and the 140–160 bp PCR products were enriched to generate a cDNA library and sequenced using Illumina HiSeq 2500 by Gene Denovo Biotechnology Co., Ltd (Guangzhou, China).
After quality control processes, the clean reads were mapped to the chicken genome Galgal 6.0 using Bowtie255 and classified by aligning them against the GeneBank (Release 209.0), Rfam (11.0) and miRBase (21.0) databases as previously described37. Sequences matching Gallus gallus miRBase were considered known miRNAs. According to their genome positions and hairpin structures predicted by the software Mireap (v0.2), novel miRNA candidates were identified56. The miRNA expression level was calculated and normalized to transcripts per million (TPM). The criteria of fold change ≥ 1.5 and a P-value < 0.05 was used to screen the differentially expressed miRNAs (DEMs). The RNAhybrid (v2.1.2) + SVM_LIGHT (v6.01), Miranda (v3.3a) and TargetScan (version 7.0) software were used to predict targets of miRNAs57.
Function enrichment analysis
All DEGs and target genes of DEMs were annotated and classified by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis with the OmicShare tools (https://www.omicshare.com/tools/). The results with P value < 0.05 were considered significantly enriched.
Validation by real-time quantitative PCR
To validate the differential expression result from sequencing, eight mRNAs and four miRNAs were selected for qRT-PCR. The total RNAs for sequencing were reverse transcribed into cDNAs by the PrimeScript RT reagent kit (TaKaRa, Dalian, China). Then, qRT-PCR was conducted using KAPA SYBR Fast universal qPCR kit (Kapa Biosystems, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) genes were used as the internal reference. All primers are shown in Table S9. For miRNA expression, 3 µg of miRNA was subject to reverse transcription with the miRNA 1st-Strand cDNA Synthesis Kit (Vazyme, Nanjing, China). qRT-PCR was then performed by miRNA Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China). The U6 snRNA was used as the internal reference. All qRT-PCR reaction was carried out in the ABI 7,500 Real-Time PCR system (Applied Biosystem, CA, USA) with triplicate reactions for each sample. The relative expression levels of the genes and miRNAs were quantified using the 2(− ΔΔCt) method58. The independent sample T-test procedure of SPSS (Version 20.0) was used to assess expression differences between PMM and SART muscle samples.
Luciferase reporter assay
Luciferase reporter experiments were performed on DF-1 cells. The gga-miR-499-5p and gga-miR-196-5p mimics and the negative control mimic were purchased from Ribobio Co., Ltd (Guangzhou, China). Wild-type (WT) and mutant (MUT) luciferase reporter vectors were synthesized based on the PmiR-RB-Report vector by the same company (Table S9). Cells were transfected using either wild-type or mutant constructs, with the specific mimics or negative control mimic. And 48 h later, Dual-Glo Luciferase Assay System (E2920, Promega, MA, USA) was utilized to detect the luminescence.
Data availability
The data sets supporting the results presented here are available in the Sequence Read Archive (SRA) repository under accession number PRJNA578179.
References
Guan, R. F. et al. Meat quality traits of four Chinese indigenous chicken breeds and one commercial broiler stock. J. Zhejiang Univ. Sci. B 14, 896–902. https://doi.org/10.1631/jzus.B1300163 (2013).
Liu, J. et al. Protein profiles for muscle development and intramuscular fat accumulation at different post-hatching ages in chickens. PLoS ONE 11, e0159722. https://doi.org/10.1371/journal.pone.0159722 (2016).
Ismail, I. & Joo, S. T. Poultry meat quality in relation to muscle growth and muscle fiber Characteristics. Korean J. Food Sci. Anim. Resour. 37, 873–883. https://doi.org/10.5851/kosfa.2017.37.6.87 (2017).
Kim, G. D., Ryu, Y. C., Jeong, J. Y., Yang, H. S. & Joo, S. T. Relationship between pork quality and characteristics of muscle fibers classified by the distribution of myosin heavy chain isoforms. J. Anim. Sci. 91, 5525–5534. https://doi.org/10.2527/jas.2013-6614 (2013).
Hambrecht, E. et al. Preslaughter handling effects on pork quality and glycolytic potential in two muscles differing in fiber type composition. J. Anim. Sci. 83, 900–907. https://doi.org/10.2527/2005.834900x (2005).
Zierath, J. R. & Hawley, J. A. Skeletal muscle fiber type: influence on contractile and metabolic properties. PLoS Biol. 2, e348. https://doi.org/10.1371/journal.pbio.0020348 (2004).
Hakamata, Y., Watanabe, K., Amo, T., Toyomizu, M. & Kikusato, M. Characterization of mitochondrial content and respiratory capacities of broiler chicken skeletal muscles with different muscle fiber compositions. J. Poult. Sci. 55, 210–216. https://doi.org/10.2141/jpsa.0170141 (2018).
Shu, J. et al. Oxidative and glycolytic skeletal muscles show marked differences in gene expression profile in Chinese Qingyuan partridge chickens. PLoS ONE 12, e0183118 (2017).
Coudert, E. et al. Expression of glucose transporters SLC2A1, SLC2A8, and SLC2A12 in different chicken muscles during ontogenesis. J. Anim. Sci. 96, 498–509. https://doi.org/10.1093/jas/skx084 (2018).
Khatri, B. et al. MicroRNA profiling associated with muscle growth in modern broilers compared to an unselected chicken breed. BMC Genomics 19, 683. https://doi.org/10.1186/s12864-018-5061-7 (2018).
Ouyang, H. et al. Deep sequencing analysis of miRNA expression in breast muscle of fast-growing and slow-growing broilers. Int. J. Mol. Sci. 16, 16242–16262. https://doi.org/10.3390/ijms160716242 (2015).
Li, Z. et al. Systematic transcriptome-wide analysis of mRNA–miRNA interactions reveals the involvement of miR-142-5p and its target (FOXO3) in skeletal muscle growth in chickens. Mol. Genet. Genomics MGG 293, 69–80. https://doi.org/10.1007/s00438-017-1364-7 (2018).
Rooij, E. V. et al. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev. Cell 17, 662–673 (2009).
Ma, J. et al. The miRNA transcriptome directly reflects the physiological and biochemical differences between red, white, and intermediate muscle fiber types. Int. J. Mol. Sci. 16, 9635–9653. https://doi.org/10.3390/ijms16059635 (2015).
Liu, J. et al. Coupling of mitochondrial function and skeletal muscle fiber type by a miR-499/Fnip1/AMPK circuit. Embo Mol. Med. 8, 1212–1228 (2016).
Wang, X. Y. et al. MicroRNA-499-5p regulates porcine myofiber specification by controlling Sox6 expression. Anim. Int. J. Anim. Biosci. 11, 1–7 (2017).
Xu, M. et al. MicroRNA-139-5p suppresses myosin heavy chain I and IIa expression via inhibition of the calcineurin/NFAT signaling pathway. Biochem. Biophys. Res. Commun. 500, 930–936. https://doi.org/10.1016/j.bbrc.2018.04.202 (2018).
Ma, M. et al. lncRNA-Six1 is a target of miR-1611 that functions as a ceRNA to regulate Six1 protein expression and fiber type switching in chicken myogenesis. Cells 7, 243. https://doi.org/10.3390/cells7120243 (2018).
China National Commission of Animal Genetic Resources. Animal Genetic Resources in China: Poultry (China Agriculture Press, Beijing, 2011).
Zhu, J. et al. RNA-seq transcriptome analysis of extensor digitorum longus and soleus muscles in large white pigs. Mol. Genet. Genomics MGG 291, 687–701. https://doi.org/10.1007/s00438-015-1138-z (2016).
Li, Y., Xu, Z., Li, H., Xiong, Y. & Zuo, B. Differential transcriptional analysis between red and white skeletal muscle of Chinese Meishan pigs. Int. J. Biol. Sci. 6, 350–360. https://doi.org/10.7150/ijbs.6.350 (2010).
Campbell, W. G. et al. Differential global gene expression in red and white skeletal muscle. Am. J. Physiol. Cell Physiol. 280, C763-768. https://doi.org/10.1152/ajpcell.2001.280.4.C763 (2001).
Li, B. et al. Identification of candidate genes associated with porcine meat color traits by genome-wide transcriptome analysis. Sci. Rep. 6, 35224. https://doi.org/10.1038/srep35224 (2016).
Shu, J. T. et al. Transcriptional co-activator PGC-1α gene is associated with chicken skeletal muscle fiber types. Genet. Mol. Res. GMR 13, 895–905 (2014).
Chen, S. et al. Polymorphisms in AKT3, FIGF, PRKAG3, and TGF-β genes are associated with myofiber characteristics in chickens. Poult. Sci. 92, 325–330. https://doi.org/10.3382/ps.2012-02766 (2013).
Jackson, H. E. et al. The role of Sox6 in zebrafish muscle fiber type specification. Skelet. Muscle 5, 2. https://doi.org/10.1186/s13395-014-0026-2 (2015).
Schneider, A. G., Sultan, K. R. & Pette, D. Muscle LIM protein: expressed in slow muscle and induced in fast muscle by enhanced contractile activity. Am. J. Physiol. 276, C900-906. https://doi.org/10.1152/ajpcell.1999.276.4.C900 (1999).
Sibut, V. et al. Identification of differentially expressed genes in chickens differing in muscle glycogen content and meat quality. BMC Genomics 12, 112. https://doi.org/10.1186/1471-2164-12-112 (2011).
Barnard, E. A., Lyles, J. M. & Pizzey, J. A. Fibre types in chicken skeletal muscles and their changes in muscular dystrophy. J. Physiol. 331, 333–354. https://doi.org/10.1113/jphysiol.1982.sp014375 (1982).
Han, S. et al. Knockdown of CSRP3 inhibits differentiation of chicken satellite cells by promoting TGF-β/Smad3 signaling. Gene 707, 36–43. https://doi.org/10.1016/j.gene.2019.03.064 (2019).
Granlund, A., Jensen-Waern, M. & Essen-Gustavsson, B. The influence of the PRKAG3 mutation on glycogen, enzyme activities and fibre types in different skeletal muscles of exercise trained pigs. Acta Vet. Scand. 53, 20. https://doi.org/10.1186/1751-0147-53-20 (2011).
Uimari, P. & Sironen, A. A combination of two variants in PRKAG3 is needed for a positive effect on meat quality in pigs. BMC Genet. 15, 29. https://doi.org/10.1186/1471-2156-15-29 (2014).
Muroya, S. et al. Profiling of differentially expressed microRNA and the bioinformatic target gene analyses in bovine fast- and slow-type muscles by massively parallel sequencing. J. Anim. Sci. 91, 90–103 (2013).
Chu, W. et al. Proteomic and microRNA transcriptome analysis revealed the microRNA-SmyD1 network regulation in skeletal muscle fibers performance of Chinese perch. Sci. Rep. 7, 16498. https://doi.org/10.1038/s41598-017-16718-2 (2017).
Shen, L. et al. Genome-wide landscape of DNA methylomes and their relationship with mRNA and miRNA transcriptomes in oxidative and glycolytic skeletal muscles. Sci. Rep. 6, 32186. https://doi.org/10.1038/srep32186 (2016).
Liu, Y. et al. Identification of differences in microRNA transcriptomes between porcine oxidative and glycolytic skeletal muscles. BMC Mol. Biol. 14, 7–7 (2013).
Liu, Y. et al. Identification and differential expression of microRNAs in the testis of chicken with high and low sperm motility. Theriogenology 122, 94–101. https://doi.org/10.1016/j.theriogenology.2018.09.010 (2018).
Bekele, E. et al. Characterization of miRNA and their target gene during chicken embryo skeletal muscle development. Oncotarget 9, 17309–17324. https://doi.org/10.18632/oncotarget.22457 (2017).
Xu, M. et al. MicroRNA-499-5p regulates skeletal myofiber specification via NFATc1/MEF2C pathway and Thrap1/MEF2C axis. Life Sci. 215, 236–245. https://doi.org/10.1016/j.lfs.2018.11.020 (2018).
Zuo, J. et al. MicroRNA transcriptome profile analysis in porcine muscle and the effect of miR-143 on the MYH7 gene and protein. PLoS ONE 10, e0124873. https://doi.org/10.1371/journal.pone.0124873 (2015).
Liu, B. et al. miR-221 modulates skeletal muscle satellite cells proliferation and differentiation. In Vitro Cell. Dev. Biol. Anim. 54, 147–155. https://doi.org/10.1007/s11626-017-0210-x (2018).
Chen, Q. et al. miRNA-34a reduces neointima formation through inhibiting smooth muscle cell proliferation and migration. J. Mol. Cell. Cardiol. 89, 75–86. https://doi.org/10.1016/j.yjmcc.2015.10.017 (2015).
Wang, S. et al. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell 15, 261–271. https://doi.org/10.1016/j.devcel.2008.07.002 (2008).
Hagiwara, N., Yeh, M. & Liu, A. Sox6 is required for normal fiber type differentiation of fetal skeletal muscle in mice. Dev. Dyn. 236, 2062–2076. https://doi.org/10.1002/dvdy.21223 (2007).
von Hofsten, J. et al. Prdm1- and Sox6-mediated transcriptional repression specifies muscle fibre type in the zebrafish embryo. EMBO Rep. 9, 683–689. https://doi.org/10.1038/embor.2008.73 (2008).
Nachtigall, P. G., Dias, M. C., Carvalho, R. F., Martins, C. & Pinhal, D. MicroRNA-499 expression distinctively correlates to target genes sox6 and rod1 profiles to resolve the skeletal muscle phenotype in Nile tilapia. PLoS ONE 10, e0119804 (2015).
Young, M. E. & Leighton, B. Fuel oxidation in skeletal muscle is increased by nitric oxide/cGMP–evidence for involvement of cGMP-dependent protein kinase. FEBS Lett. 424, 79–83. https://doi.org/10.1016/s0014-5793(98)00143-4 (1998).
Mallinson, J., Meissner, J. & Chang, K. C. Chapter 2. Calcineurin signaling and the slow oxidative skeletal muscle fiber type. Int. Rev. Cell Mol. Biol. 277, 67–101. https://doi.org/10.1016/s1937-6448(09)77002-9 (2009).
Liu, Y., Shen, T., Randall, W. R. & Schneider, M. F. Signaling pathways in activity-dependent fiber type plasticity in adult skeletal muscle. J. Muscle Res. Cell Motil. 26, 13–21. https://doi.org/10.1007/s10974-005-9002-0 (2005).
Chin, E. R. et al. A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev. 12, 2499–2509 (1998).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. https://doi.org/10.1186/gb-2013-14-4-r36 (2013).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. https://doi.org/10.1038/nprot.2012.016 (2012).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 12, 323. https://doi.org/10.1186/1471-2105-12-323 (2011).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. https://doi.org/10.1093/bioinformatics/btp616 (2010).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. https://doi.org/10.1186/gb-2009-10-3-r25 (2009).
Hafner, M. et al. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods 44, 3–12. https://doi.org/10.1016/j.ymeth.2007.09.009 (2008).
Liu, L. et al. Whole-transcriptome analysis of atrophic ovaries in broody chickens reveals regulatory pathways associated with proliferation and apoptosis. Sci. Rep. 8, 7231. https://doi.org/10.1038/s41598-018-25103-6 (2018).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408. https://doi.org/10.1006/meth.2001.1262 (2001).
Acknowledgements
This research was funded by China Agriculture Research Systems (CARS-41), earmarked fund for Jiangsu Agricultural Industry Technology System (JATS[2018]249), Open Projects of Key Laboratory for Poultry Genetics and Breeding of Jiangsu Province (JQLAB-ZZ-201902) and Special Fund for Independent Innovation of Agricultural Science and Technology in Jiangsu Province of China (CX(15)1009).
Author information
Authors and Affiliations
Contributions
J.S., M.Z. and Y.L. conceived and designed the experiments. Y.L., M.Z., Y.T., Y.S.G.J. and J.X. performed the experiments and participated in data collection. Y.L., Z.S. and X.J. analyzed the data. Y.L. and M.Z. drafted the manuscript. J.Z. and J.S. revised the manuscript critically. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Y., Zhang, M., Shan, Y. et al. miRNA–mRNA network regulation in the skeletal muscle fiber phenotype of chickens revealed by integrated analysis of miRNAome and transcriptome. Sci Rep 10, 10619 (2020). https://doi.org/10.1038/s41598-020-67482-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-67482-9
- Springer Nature Limited
This article is cited by
-
MiRNA sequencing of Embryonic Myogenesis in Chengkou Mountain Chicken
BMC Genomics (2022)
-
Regulation of mRNA and miRNA in the response to Salmonella enterica serovar Enteritidis infection in chicken cecum
BMC Veterinary Research (2022)
-
Sox6, A Potential Target for MicroRNAs in Cardiometabolic Disease
Current Hypertension Reports (2022)
-
OIP5-AS1 Inhibits Oxidative Stress and Inflammation in Ischemic Stroke Through miR-155-5p/IRF2BP2 Axis
Neurochemical Research (2022)
-
Analysis of potential regulatory LncRNAs and CircRNAs in the oxidative myofiber and glycolytic myofiber of chickens
Scientific Reports (2021)