
Abstract
Bacterial leaf steak (BLS) caused by Xanthomonas oryzae pv. oryzicola (Xoc) is a devastating disease in rice production. The resistance to BLS in rice is a quantitatively inherited trait, of which the molecular mechanism is still unclear. It has been proved that xa5, a recessive bacterial blast resistance gene, is the most possible candidate gene of the QTL qBlsr5a for BLS resistance. To study the molecular mechanism of xa5 function in BLS resistance, we created transgenic lines with RNAi of Xa5 (LOC_Os05g01710) and used RNA-seq to analyze the transcriptomes of a Xa5-RNAi line and the wild-type line at 9 h after inoculation with Xoc, with the mock inoculation as control. We found that Xa5-RNAi could (1) increase the resistance to BLS as expected from xa5; (2) alter (mainly up-regulate) the expression of hundreds of genes, most of which were related to disease resistance; and (3) greatly enhance the response of thousands of genes to Xoc infection, especially of the genes involved in cell death pathways. The results suggest that xa5 is the cause of BLS-resistance of QTL qBlsr5a and it displays BLS resistance effect probably mainly because of the enhanced response of the cell death-related genes to Xoc infection.
Similar content being viewed by others


Bacterial leaf streak (BLS) is a disease caused by the gram-negative bacterial pathogen Xanthomonas oryzae pv. oryzicola (Xoc) in rice. BLS is one of the most devastating quarantine diseases1 in the main rice-producing areas of the world and can cause significant yield loss. BLS resistance is quantitatively inherited in rice2. In contrast to qualitative disease resistance, which is controlled by major resistance (R) genes, race-specific, and easily defeated by co-evolving pathogens3, quantitative disease resistance is driven by multiple genes, generally non-race-specific and much more durable than qualitative resistance4,5,6,7. Therefore, breeding of disease-resistant cultivars is an effective strategy to control BLS. However, such effort has often been compromised due to lack of understanding of the underlying molecular mechanism for BLS resistance.
To date, at least 13 quantitative trait loci (QTLs) conferring BLS resistance have been mapped in rice2,8,9. In addition, a recessive gene bls1 showing race-specific resistance to BLS10 and a locus Xo1 conferring complete resistance to the African clade of Xoc strains of BLS11 are also reported. Interestingly, a non-host resistance gene Rxo1 from maize also displays qualitative resistance to BLS in rice12, which specifically activates multiple defense pathways related to hypersensitive response (HR) against Xoc, including some signaling pathways and basal defensive pathways such as the ethylene (ET) and salicylic acid (SA) pathways13.
Overexpression of Differentially Expressed Protein Gene 1 (DEPG1), a resistance gene that contains a nucleotide-binding site-leucine rich repeat (NBS-LRR) domain, results in increased susceptibility to Xoc strain RS105 and inhibition of some genes related to basal defensive pathways, implying its role of negative regulation for immunity in rice14. Similarly, suppression of some defense related (DR) genes, such as OsWRKY45-115, OsMPK616 and NRRB17, also presents negative regulation for immunity, increasing resistance to BLS. On the contrary, overexpression of genes OsPGIP4, GH3-2, and OsHSP18.0-CI significantly enhances resistance to BLS in rice, implying their positive regulation roles for resistance to Xoc18,19,20.
In our laboratory, a major QTL qBlsr5a conferring BLS resistance was previously mapped on rice chromosome 52 and further fine mapped to a 30-kb interval21. Three genes were annotated in the interval. Among them, LOC_Os05g01710 was considered to be the most possible candidate gene, which encodes a transcription initiation factor IIA gamma (TFIIAγ) protein. There were two nucleotides different between the LOC_Os05g01710 allele from the resistant parent and that from the susceptible parents, resulting in a substitution at the 39th amino acid between their encoding proteins21. Interestingly, the allele of LOC_Os05g01710 from the resistant parent was identical to xa5 in sequence, a recessive resistance gene against bacterial blight caused by Xanthomonas oryzae pv. oryzae (Xoo)22,23, implying that suppression of the dominant allele of LOC_Os05g01710 (Xa5) from the susceptible parent would increase the BLS resistance if it is really the gene responsible for the qBlsr5a effect. This implication has been verified by the result of RNA interference (RNAi) of Xa521,24, in which the Xa5-RNAi lines display enhanced resistance to BLS with little influence on agronomic traits (only slightly reducing plant height)24. Therefore, it is rational to infer that xa5 is the cause of the BLS-resistance effect of qBlsr5a, although the possibility of contribution from the other two genes in the interval cannot be completely excluded.
The present study aimed to investigate the molecular mechanism of xa5-mediated resistance to BLS in rice by analyzing the effects of Xa5-RNAi on gene expression regulation and transcriptional response to BLS pathogen Xoc. We revealed that Xa5-RNAi could greatly alter (mainly up-regulate) the expression of many genes related to disease resistance and enhance transcriptional response to Xoc, and Xa5 affects BLS resistance probably mainly by regulating the expression of genes involved in cell death.
Results
Xa5-RNAi plants and their BLS resistance
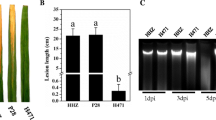
In total, 13 and 9 independent transgenic (T0) Xa5-RNAi (abbreviated as XR) plants were obtained from rice cultivars Nipponbare and Minghui 86 (MH86), respectively, and 8 T2 XR lines were subsequently derived, with 4 from Nipponbare (N-1/2/3/4) and MH86 (M-1/2/3/4) each. Inoculation test showed that the mean lesion length in the XR lines was significantly decreased in comparison with that in wild-type (WT) plants (Fig. 1A,B). qRT-PCR analysis indicated that Xa5 expression was significantly reduced in the XR lines (Fig. 1C). The lesion length and the Xa5 expression level were significantly correlated, with a correlation coefficient of 0.93 (P < 0.05) in the Nipponbare XR lines and 0.95 (P < 0.05) in the MH86 XR lines, respectively. These results reconfirmed that xa5 can enhance BLS resistance, and suggested again that xa5 is the cause of BLS-resistance of QTL qBlsr5a.

Performance of BLS resistance in Xa5-RNAi lines from Nipponbare and MH86. (A) Leaves of WT (NIP and MH86) and RNAi lines with BLS lesions at 15 days after Xoc inoculation, N and M represent RNAi lines derived from Nipponbare and MH86, respectively. (B) Lesion lengths of WT and T2 RNAi lines measured at 15 days after Xoc inoculation. (C) Relative expression levels of Xa5 in WT and T2 RNAi lines at 15 days after Xoc inoculation. RNAi lines of N-1/2/3/4 and M-1/2/3/4 were derived from Nipponbare and MH86, respectively. Letters indicate the statistical significance of difference at 0.01 level according to ANOVA-Tukey’s test.
Reads of RNA sequencing
Since the XR line M-4 from MH86 showed the most significant suppression of Xa5 expression and increase of BLS resistance (Fig. 1), it was used for RNA-seq together with MH86. In total, 12 RNA samples were sequenced, of which one sample was discarded due to poor sequencing quality. Therefore, a total of 11 RNA samples were analyzed (Table 1). More than 50 million reads were obtained in each sample, of which most (> 84%) were uniquely mapped to the Nipponbare reference genome (Table 1). These uniquely mapped reads were used for subsequent gene expression analysis. The RNA-seq data have been submitted to the database of the NCBI Sequence Read Archive (https://trace.ncbi.nlm.nih.gov/Traces/sra) under the accession number PRJNA558068.
To validate the RNA-seq data, qRT-PCR (Supplementary Table S1) was used to examine the expression of 6 genes encoding proteins related to disease resistance (including phytohormones, phyto-oxygenase, etc.) in the four treatments. The expression patterns of these six genes detected by qRT-PCR were consistent with those detected by RNA-seq (Fig. 2), indicating that the RNA-seq data were reliable.

Quantitative RT-PCR analysis of six genes to validate the RNA-seq data.
Differentially expressed genes
We analyzed differentially expressed genes (DEGs) in four pairs of comparison, namely, C1: XRM vs. WTM; C2: WTI vs. WTM; C3: XRI vs. XRM; and C4: XRI vs. WTI. C1 and C4 were used to examine the effect of XR on gene expression under normal condition and under the stress of Xoc infection, respectively, while C2 and C3 were used to examine the influence of XR on transcriptional response to Xoc infection. The four comparison combinations together would show the interaction between XR and Xoc infection on gene expression, which would help us understand the molecular mechanism underlying the BLS resistance mediated by Xa5.
A total of 317 DEGs were detected in C1 (Supplementary Table S2), among which ~ 3/4 (244) genes were up-regulated, suggesting that Xa5 functions mainly as a negative regulator for many genes. To examine whether, how many and what kinds of genes related to the resistance to biotic stress existed among the DEGs detected in C1, an overview of regulation and biotic stress showing the transcriptional changes basing on the software of MapMan was generated. MapMan analysis showed that many of the up-regulated genes were involved in the biological pathways related to biotic stress (Supplementary Fig. S1), including hormone signaling, cell wall, beta glucanase, proteolysis, defense genes (DR), redox state, signaling, Myb, secondary metabolites, etc. Most of these genes, such as those associated with cell wall function and phytohormones (salicylic acid, jasmonic acid and ethylene), have been found to play important roles in plant defenses against pathogens25, including Xoc in rice16,17,19.
There were 157 DEGs (125 up-regulated + 32 down-regulated) in C2 (Supplementary Table S3) and 3115 DEGs (1202 up-regulated + 1913 down-regulated) in C3 (Supplementary Table S4), respectively. The number of DEGs in C3 was ~ 19 times more than that in C2, suggesting that XR can dramatically enhance the transcriptional response to Xoc infection. However, the set of DEGs in C3 only covered ~ 1/3 (56/157) of that in C2, although most of the overlapped DEGs showed the same expression change directions in C2 and C3 (Fig. 3A). In addition, while there were more up-regulated genes than down-regulated genes in WT (C2), the trend was reversed in XR (C3; Fig. 3A). These results suggested that XR can also significantly alter the pattern of transcriptional response to Xoc infection.

Venn diagram of DEGs detected by RNA-seq in C2 (WTI vs. WTM) and C3 (XRI vs. XRM) (A) and that in C1 (XRM vs. WTM) and C4 (XRI vs. WTI) (B).
The number of DEGs identified in C4 was the highest, up to 5640 (2448 up-regulated + 3192 down-regulated; Supplementary Table S5), which was ~ 18 times as many as that in C1 (Fig. 3B). About 2/3 of the DEGs in C1 also existed in C4, displaying consistent expression change directions with only a few exceptions (Fig. 3B). These results indicated that Xoc infection greatly augmented the difference of gene expression profile between XR and WT.
Gene ontology enrichment
Gene ontology (GO) analysis showed that the DEGs in C1 were significantly enriched (P-value < 0.01) in 43 GO terms (Supplementary Table S6), including 29 terms on biological process (BP), 1 on cellular component (CC) and 13 on molecular function (MF). Among the BP terms, ~ 3/4 (22/29) were known to be related to plant disease resistance, which could be classified into several groups, including oxidation-reduction26, siderophore27, secondary metabolite28, cell death29 and so on. This was consistent with the result of MapMan analysis, suggesting that a main role of Xa5 is to regulate the expression of genes related to disease resistance.
In C2, 78 GO terms were significantly enriched (P-value < 0.01) with DEGs (Supplementary Table S7), including 41 on BP, 9 on CC and 28 on MF, respectively. Almost all of the BP terms were related to disease resistance, which could also be classified into several groups similar to those observed in C1 except for the group of cell death.
The DEGs in C3 were enriched (P-value < 0.01) in 66 GO terms (Supplementary Table S8), including 18 terms on BP, 21 on CC and 27 on MF, respectively. Among the BP terms, eight were related to plant disease resistance, including three about cell death, one about defense response, and four about thiamine metabolism. Thiamine is known to function as an activator of plant disease resistance30. The three terms about cell death were the most significant, with P-values (≤ 1.1 × 10−11) at least 105 times smaller than any other term, implying that this group of terms is particularly important for the function of Xa5 on BLS resistance.
Surprisingly, although there were much more DEGs in C3 than in C2, the number of enriched GO terms on BP in C3 was much smaller than that in C2. Comparison showed that there were no common BP terms between C2 and C3. However, they had 16 and 4 BP terms common with C1, respectively, among which the common terms between C1 and C2 were all about the basal defense processes, while those between C1 and C3 were about cell death only (Fig. 4).

Significant GO terms on biological process common in at least two of the four pairs of comparison.
C4 had 79 GO terms enriched (P-value < 0.01) with DEGs (Supplementary Table S9), including 33 on BP, 13 on CC and 33 on MF, respectively. Among the BP terms, nine were related to plant disease resistance, on the aspects of defense response (1 term), cell death (3 terms), immune (2 terms), and others. Similar to the case of C3, C4 had no BP terms common with C2, but four (the one about defense response and the three about cell death) common with C1, which were also common with C3 (Fig. 4). Besides, there was an additional BP term about photosynthesis common between C3 and C4. Noticeably, the three terms about cell death were also the most significant among all BP terms in C4, with P-values (≤ 5.8 × 10−15) at least 105 times smaller than any other term, implying again the special importance of the genes related to cell death in the Xa5-mediated BLS resistance.
Discussion
TFIIAγ is a component of the general transcription factor IIA and is involved in all polymerase II-dependent transcription in eukaryotes31,32. Two TFIIAγ-like genes, TFIIAγ1 and TFIIAγ5 (Xa5), have been found in rice. The recessive allele xa5, which encodes a V39E substitution variant of TFIIAγ5, is functionally confirmed to enhance the resistance to rice bacterial blight22,23. In addition, the results of fine mapping of QTL qBlsr5a21 and RNAi of Xa521,24 have suggested that xa5 is the most possible candidate gene of the QTL qBlsr5a conferring resistance to BLS. This study verified the Xa5-RNAi effect on BLS resistance (Fig. 1), further affirming the inference that Xa5 is responsible for the effect of qBlsr5a on BLS resistance.
It is generally considered that plants have evolved two different innate immune systems: the pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI) system and the effector-triggered immunity (ETI) system33. PTI belongs to a relatively weak immune response triggered by PAMPs, which depends on the basal defense to restrict the colonization of invading pathogens34. As PTI has no race specificity, it is predicted to confer durable and broad spectrum resistance35. ETI is mediated by polymorphic resistance proteins that can recognize the highly variable effectors from pathogens, with a fast and strong response usually accompanied with a hypersensitivity reaction (HR) and eventually programmed cell death (PCD) to restrict biotrophic cellular pathogens36. We have seen above that the DEGs in C1 were mainly enriched in four groups of GO terms on BP related to disease resistance, namely, oxidation–reduction, siderophore, secondary metabolite, and cell death (Supplementary Table S6). Among these GO terms, the former three groups belong to the mechanisms of basal defense, while the last group (cell death) belongs to the mechanism of HR. Therefore, they are responsible for PTI and ETI, respectively. This suggests that Xa5 regulates both PTI- and ETI-related genes.
It is interesting that some of the GO terms related to disease resistance in C1 were also detected in C2, C3 and C4 (Fig. 4). Among them, the PTI-related terms were only detected in C2, while the ETI-related terms were only detected in C3 and C4, and there was no overlap between C2 and C3 and between C3 and C4. In addition, the ETI-related terms were very highly significant and also the most significant in both C3 and C4 (Supplementary Tables S8, S9). These results indicated that the ETI-related terms became significant only under the Xa5-RNAi condition, no matter whether there was Xoc infection (in C3 and C4) or not (in C1), but Xoc infection could greatly increase the number of DEGs in these GO terms (Fig. 4). Hence, it is likely that the xa5-mediated BLS resistance is mainly due to the response of the genes involved in the cell death pathways to Xoc infection. It appears that the dominant allele Xa5 can inhibit the response of these genes to Xoc infection. Therefore, no GO terms about cell death could be detected in C2.
Certainly, PTI-related genes may also contribute to disease resistance. But why the above-mentioned PTI-related GO terms were not significant in C3 and C4? The possible reason could be that the response of the genes in these PTI-related GO terms to Xoc infection is similar to that to Xa5-RNAi (Fig. 4), and the effects of these two types of response are not additive but superimposed. Thus, since the response of the genes to Xa5-RNAi has already existed in an XR line, the response to Xoc infection is masked and therefore becomes undetectable.
Taken together, we have seen in this study that as a component of a general transcriptional factor, Xa5 plays an important role in the regulation of many genes related to disease resistance (including both PTI-related and ETI-related genes) in rice. Suppression of its expression can lead to defense-oriented reprogramming and thereby limit the multiplication or spread of the BLS pathogen Xoc through a stronger and more direct immune response like ETI to protect the plant. This defense strategy could accompany with a similar occurrence of a hypersensitivity reaction and eventually result in programmed cell death, cell death, or apoptosis to against the pathogen invasion, although the BLS resistance in rice is controlled by multiple genes and Xa5 functions only as a QTL.
It has been mentioned above that the recessive allele xa5 confers qualitative resistance to bacterial blight caused by Xoo22,23. Therefore, Xa5 has much stronger effect on bacterial blight resistance than that on BLS resistance. It is found that Xoc and Xoo induce almost completely different transcriptional changes in the host37, implying that xa5 might mediate different pathways to defend against Xoo and Xoc. However, considering that cell death is usually a result from hypersensitivity reaction occurring in qualitative resistance, we have reason to expect that the possible Xa5-mediated cell death mechanism for BLS resistance found in this study might probably also play a role in bacterial blight resistance.
Conclusions
The recessive gene xa5 is the cause of BLS resistance of QTL qBlsr5a. It displays BLS resistance effect probably mainly due to the enhanced response of the cell death-related genes to Xoc infection.
Material and methods
Plant materials
An indica rice cultivar Minhui 86 (MH86), which was created and released by Sanming Academy of Agricultural Sciences, Fujian, China, and a japonica rice cultivar Nipponbare, which is the cultivar used in the International Rice Genome Sequencing Project, were used in this experiment. Both cultivars were susceptible to BLS.
Vector construction and rice transformation
To construct Xa5-RNAi (abbreviated as XR) vector, a 515-bp fragment containing the whole coding sequence of LOC_Os05g01710 (Xa5) was amplified from the cDNA of Nipponbare by PCR. The amplified fragment was inserted into the pTCK303 vector downstream of the ubiquitin promoter, in both forward and reversed orientations (Fig. 5). The vector was introduced into Agrobacterium strain EHA105 using the freeze–thaw method, and further into the calli of Nipponbare and MH86 derived from mature embryos following the protocol of Ref.38. Positive transgenic lines were identified by PCR using the hygromycin specific primers. Ten positive T2 plants from each line were selected for quantitative real-time PCR (qRT-PCR) analysis and BLS-resistance assessment. All the primers used for XR vector construction and transgenic line identification are shown in Table 2.

Schematic diagram of Xa5-RNAi construct.
Pathogen infection and resistance assessment
The Xoc strain used for inoculation was kindly provided by Prof. Guoying Cheng of Huazhong Agricultural University. Rice plants were inoculated using the pricking inoculation method2 at the active tillering stage and lesion length was scored from three leaves of each plant 15 days after inoculation. The resistance ability of a line was indicated by the mean lesion length of 10 plants.
Quantitative real-time PCR analysis
Total RNA of leaves was extracted using TRIzol reagent (Invitrogen, https://www.invitrogen.com). First-strand cDNA synthesis was performed using PrimeScript RT Reagent Kit with gDNA Eraser (Takara, Japan) following the manufacturer’s instruction. The cDNA samples were then assayed by qRT-PCR using SYBR Premix Ex Taq (Takara). The gene-specific primers used for qRT-PCR analysis are listed in Supplementary information. Actin was used as an internal control. Two or more biological replicates and three technical replicates were tested. The relative expression level of a gene was calculated using the 2−ΔΔCt method39. The paired t-test method was used to examine the difference of gene expression level between different samples.
RNA sequencing and data analysis
Leaves of wild-type (WT) MH86 and one of its XR lines (M-4) at the active tillering stage were inoculated with Xoc or sterile water (mock inoculation, as control) and total RNA was extracted from the leaves 9 h after inoculation, according to the findings that the expression levels of genes related to BLS resistance drastically change in the time interval of 6–12 h after inoculation24,37. In total, there were four treatments: MH86 mock (denoted as WTM), MH86 inoculated (WTI), M-4 mock (XRM), and M-4 inoculated (XRI). Three biological replicates were set for each treatment. Therefore, there were 12 RNA samples in total. Each RNA sample was from a mixture of three 1-cm leaf segments next to the inoculation site. The RNA samples were sequenced on Illumina HiSeq 2000 performed by Biomarker Technologies (https://www.biomarker.com.cn/, BioMarker, Beijing, China). Reads (100 bp in length, paired-end) were mapped to the reference genome and genes available at the Rice Genome Annotation Project (https://rice.plantbiology.msu.edu). For gene expression analysis, the numbers of matched reads were normalized by the RPKM (reads per kb per million mapped reads) method40. DEGs were identified using the criteria of |log2(fold change)|≥ 2 and false discovery rate (FDR) ≤ 0.01. Gene ontology (GO) analysis was performed based on the Gene Ontology Database (https://www.geneontology.org/). The MapMan tool (https://MapMan.gabipd.org) was used for a graphical overview of pathways involving the DEGs.
References
Niño-Liu, D., Ronald, P. & Bogdanove, A. Xanthomonas oryzae pathovars: Model pathogens of a model crop. Mol. Plant Pathol. 7, 303–324 (2006).
Tang, D., Wu, W., Li, W., Lu, H. & Worland, A. J. Mapping of QTLs conferring resistance to bacterial leaf streak in rice. Theor. Appl. Genet. 101, 286–291 (2000).
Kou, Y. & Wang, S. Broad-spectrum and durability: Understanding of quantitative disease resistance. Curr. Opin. Plant Biol. 13, 181–185 (2010).
Wisser, R., Sun, Q., Hulbert, S., Kresovich, S. & Nelson, R. Identification and characterization of regions of the rice genome associated with broad-spectrum, quantitative disease resistance. Genetics 169, 2277–2293 (2005).
Pink, D. A. C. Strategies using genes for non-durable disease resistance. Euphytica 124, 227–236 (2002).
Poland, J., Balint-Kurti, P., Wisser, R., Pratt, R. & Nelson, R. Shades of gray: The world of quantitative disease resistance. Trends Plant Sci. 14, 21–29 (2009).
Kliebenstein, D. & Rowe, H. Plant science. Anti-rust antitrust. Science 323, 1301–1302 (2009).
Zheng, J. S., Li, Y. Z. & Fang, X. J. Detection of QTL conferring resistance to bacterial leaf streak in rice chromosome 2 (O. sativa L. spp. indica). Sci. Agric. Sin. 38, 1923–1925 (2005).
Chen, C. H., Wei, Z., Huang, X. M., Zhang, D. P. & Lin, X. H. Major QTL conferring resistance to rice bacterial leaf streak. Agric. Sci. China 5, 216–220 (2006).
Wen-Ai, H. E. et al. Identification of a resistance gene bls1 to bacterial leaf streak in wild rice Oryza rufipogon Griff. J. Integr. Agric. 11, 962–969 (2012).
Triplett, L. et al. A resistance locus in the American heirloom rice variety Carolina Gold Select is triggered by TAL effectors with diverse predicted targets and is effective against African strains of Xanthomonas oryzae pv. oryzicola. Plant J. 87, 472–483 (2016).
Zhao, B. et al. A maize resistance gene functions against bacterial streak disease in rice. Proc. Natl. Acad. Sci. U.S.A. 102, 15383–15388 (2005).
Zhou, Y. et al. Genome-wide gene responses in a transgenic rice line carrying the maize resistance gene Rxo1 to the rice bacterial streak pathogen, Xanthomonas oryzae pv. oryzicola. BMC Genom. 11, 78 (2010).
Guo, L. et al. Over-expression in the nucleotide-binding site-leucine rich repeat gene DEPG1 increases susceptibility to bacterial leaf streak disease in transgenic rice plants. Mol. Biol. Rep. 39, 3491–3504 (2012).
Tao, Z. et al. A pair of allelic WRKY genes play opposite roles in rice-bacteria interactions. Plant Physiol. 151, 936–948 (2009).
Shen, X. et al. Opposite functions of a rice mitogen-activated protein kinase during the process of resistance against Xanthomonas oryzae. Plant J. 64, 86–99 (2010).
Guo, L. et al. Suppression of expression of the putative receptor-like kinase gene NRRB enhances resistance to bacterial leaf streak in rice. Mol. Biol. Rep. 41, 2177–2187 (2014).
Fu, J. et al. Manipulating broad-spectrum disease resistance by suppressing pathogen-induced auxin accumulation in rice. Plant Physiol. 155, 589–602 (2011).
Feng, C. et al. The polygalacturonase-inhibiting protein 4 (OsPGIP4), a potential component of the qBlsr5a locus, confers resistance to bacterial leaf streak in rice. Planta 243, 1297–1308 (2016).
Ju, Y. et al. Overexpression of OsHSP18.0-CI enhances resistance to bacterial leaf streak in rice. Rice (N. Y.) 10, 12 (2017).
Xie, X. et al. Toward the positional cloning of qBlsr5a, a QTL underlying resistance to bacterial leaf streak, using overlapping sub-CSSLs in rice. PLoS ONE 9, e95751 (2014).
Iyer, A. & McCouch, S. The rice bacterial blight resistance gene xa5 encodes a novel form of disease resistance. Mol. Plant Microbe Interact. 17, 1348–1354 (2004).
Jiang, G. et al. Testifying the rice bacterial blight resistance gene xa5 by genetic complementation and further analyzing xa5 (Xa5) in comparison with its homolog TFIIAgamma1. Mol. Genet. Genom. 275, 354–366 (2006).
Yuan, M. et al. A host basal transcription factor is a key component for infection of rice by TALE-carrying bacteria. eLife 5, e19605 (2016).
Yang, Y., Ahammed, G., Wu, C., Fan, S. & Zhou, Y. Crosstalk among jasmonate, salicylate and ethylene signaling pathways in plant disease and immune responses. Curr. Protein Pept. Sci. 16, 450–461 (2015).
Matika, D. E., Frederickson, & Loake, G. J. Redox regulation in plant immune function. Antioxid. Redox Signal 21, 1373–1388 (2014).
Aznar, A. & Dellagi, A. New insights into the role of siderophores as triggers of plant immunity: What can we learn from animals?. J. Exp. Bot. 66, 3001–3010 (2015).
Piasecka, A., Jedrzejczak-Rey, N. & Bednarek, P. Secondary metabolites in plant innate immunity: Conserved function of divergent chemicals. New Phytol. 206, 948–964. https://doi.org/10.1111/nph.13325 (2015).
Coll, N. S., Epple, P. & Dangl, J. L. Programmed cell death in the plant immune system. Cell Death Differ. 18, 1247–1256 (2011).
Il-Pyung, A., Soonok, K. & Yong-Hwan, L. Vitamin B1 functions as an activator of plant disease resistance. Plant Physiol. 138, 1505–1515 (2005).
Høiby, T., Zhou, H., Mitsiou, D. & Stunnenberg, H. A facelift for the general transcription factor TFIIA. Biochim. Biophys. Acta 1769, 429–436 (2007).
Li, Y. F. et al. Characterization and functional analysis of Arabidopsis TFIIA reveal that the evolutionarily unconserved region of the large subunit has a transcription activation domain. Plant Mol. Biol. 39, 515–525 (1999).
Jones, J. D. G. & Dangl, J. L. The plant immune system. Nature 444, 323–329 (2006).
Zipfel, C. Plant pattern-recognition receptors. Trends Immunol. 35, 345–351 (2014).
Liu, W., Liu, J., Triplett, L., Leach, J. & Wang, G. Novel insights into rice innate immunity against bacterial and fungal pathogens. Annu. Rev. Phytopathol. 52, 213–241 (2014).
Dodds, P. & Rathjen, J. Plant immunity: Towards an integrated view of plant–pathogen interactions. Nat. Rev. Genet. 11, 539–548 (2010).
Cernadas, R. A. et al. Code-assisted discovery of TAL effector targets in bacterial leaf streak of rice reveals contrast with bacterial blight and a novel susceptibility gene. PLoS Pathog. 10, e1003972 (2014).
Nishimura, A., Aichi, I. & Matsuoka, M. A protocol for Agrobacterium-mediated transformation in rice. Nat. Protoc. 1, 2796–2802 (2006).
Livak, K. & Schmittgen, T. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Mortazavi, A., Williams, B., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008).
Acknowledgements
This study was supported in part by National Natural Science Foundation of China (No. 31501085), Fujian Agriculture and Forestry University innovation foundation (No. CXZX2019052G), Natural Science Foundation of Fujian Province (No. 2017J01438), National Key R&D Program of China (2017YFD0100103), and the International Sci-Tech Cooperation and Exchange Program of FAFU (Grant number: KXGH17014). The funding agencies had not involved in the experimental design, analysis, and interpretation of the data or writing of the manuscript.
Author information
Authors and Affiliations
Contributions
W.W. conceived and designed the experiments. X.X., Z.C., Z.B., H.G., Y.Z., T.L., J.Z. and M.Q. performed the experiments. X.X. analyzed the data. Z.C. contributed materials. X.X. and W.W. wrote the paper. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xie, X., Chen, Z., Zhang, B. et al. Transcriptome analysis of xa5-mediated resistance to bacterial leaf streak in rice (Oryza sativa L.). Sci Rep 10, 19439 (2020). https://doi.org/10.1038/s41598-020-74515-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-74515-w
- Springer Nature Limited
This article is cited by
-
Co-expression network analysis and identification of core genes in the interaction between wheat and Puccinia striiformis f. sp. tritici
Archives of Microbiology (2024)
-
The role of OsOFP8 gene in regulating rice leaf angle
Journal of Plant Biochemistry and Biotechnology (2023)
-
Exploiting the antibacterial mechanism of phenazine substances from Lysobacter antibioticus 13-6 against Xanthomonas oryzae pv. oryzicola
Journal of Microbiology (2022)