
Abstract
We investigated the genetic structure of three phenotypically distinct ecotypic groups of Norway spruce (Picea abies) belonging to three elevational classes; namely, low- (acuminata), medium- (europaea), and high-elevation (obovata) form, each represented by 150 trees. After rigorous filtering, we used 1916 Genotyping-by-Sequencing generated SNPs for analysis. Outputs from three multivariate analysis methods (Bayesian clustering algorithm implemented in STRUCTURE, Principal Component Analysis, and the Discriminant Analysis of Principal Components) indicated the presence of a distinct genetic cluster representing the high-elevation ecotypic group. Our findings bring a vital message to forestry practice affirming that artificial transfer of forest reproductive material, especially for stands under harsh climate conditions, should be considered with caution.
Similar content being viewed by others
Introduction
As a species of substantial economic and ecological importance, Norway spruce Picea abies (L.) Karst, has been the subject of numerous genetic diversity studies utilizing both biochemical and molecular markers1,2,3,4,5,6,7,8. Overall, most studies were conducted on the population level and aimed to identify the species extent of genetic differentiation9,10,11,12.
As genetic markers evolved, the answers on examined hypotheses improved in their precision, starting with allozyme1,2,3, through more informative microsatellite DNA markers13,14,15,16, to those relying on next-generation sequencing methods which made it possible to address more specific questions such as the impact of evolutionary factors on the genetic makeup of the species17,18,19,20,21,22. Historically, isoenzyme markers showed a low level of inter-population diversity2,23,24. Microsatellite markers are frequently used for population-level studies. However, due to their neutral nature, they are not affected by evolutionary forces such as selection and adaptation processes12,25,26.
At present, the availability of genomic-based genotyping platforms offers vast opportunities to address specific Norway spruce research questions, such as identifying candidate genes associated with drought response27, wood formation28, disease resistance29, morphological characteristics30 or local adaptations31.
Despite extensive gene flow and limited genetic differentiation among populations detected with neutral markers, Norway spruce populations possess strong local adaptation in phenological and climate-related traits along latitudinal gradients32 such as cold hardiness and bud set33, growth cessation34, or latitudinal gradient for red/far-red light ratio in connection to local adaptation for shade tolerance35. Also, a provenance effect on grown parameters and phenology was shown31,36. Only a few studies have paid attention to epigenetic factors in Norway spruce37,38 even that they may play an essential role in adaptative responses39,40.
The demographic history of Norway spruce has been widely studied, suggested current distribution originated from two main refugia7,8. A more recent study showed that the current distribution of Norway spruce genetic diversity is more admixtured due to repeated evolutionary events such as ancient splits, hybridization, and bottlenecks20. Norway spruce is a wind-pollinated species characterized by the high gene flow3,41. Along with the species outcrossing nature, other factors enhance gene flow level; namely, artificial spreading of spruce outside the natural grown area42 and man-influenced forest reproductive material transfer establishing non-autochthonous forests from non-local gene sources43.
Norway spruce possess noticeable variability in crown morphology, especially across substantial altitudinal gradients44. Despite a relatively continuous variance in crown parameters within stands, three prevalent morphotypes-ecotypic groups, are distinguished44,45,46 along an altitudinal gradient47. The low‐elevation ecotype, acuminata, is characterized by broad crowns, toothed cone scales, and comb‐like branches. In central Europe, this form occurs predominantly up to 500 m.a.s.l. In contrast, the high-elevation ecotype, obovata, typically has a narrow crown, round cone scales, and flat branches (500–1000 m.a.s.l) and the medium‐elevation ecotype, europaea, with rhombic toothed cone scales and a mostly conical crown with medium width and brush-type branches that are usually shorter, denser, and down hanging (1000 m.a.s.l up to tree line). It is essential to emphasize that these forms are not exclusively limited to one elevation or latitudinal zone, although any given Norway spruce ecotypic form is usually prevalent within particular stands44. Crown shape has been studied with association to altitude30; however, with the exception for our previous study utilizing microsatellite markers16, there is a paucity of information addressing the underlying genetic causes for the observed architectural differences among these various ecotypes.
Here, we aimed to elucidate the level of genetic differentiation among three phenotypically identified Norway spruce ecotypes determined by their crown morphology and the altitude of their stands using Genotyping-by-Sequencing (GBS) generated SNP data. GBS utilizes restriction enzyme digestion (RE) to focus on sequences in low-copy genomic regions while minimizing reads in the abundant repetitive sequences48 present in conifers49. By choosing appropriate REs, the repetitive regions of genomes can be avoided, and lower copy regions are targeted with higher efficiency50.
Using rigorously filtered data, we applied three widely used multivariate methods for genetic structure analysis. Results provided consistent outputs identifying a group of high-elevation individuals as a genetically distinct cluster. Additionally, we utilized the Redundancy Analysis (RDA51), a method that effectively detects even weak, multiloci molecular signatures to identify SNPs with genotype-environment association signals.
Material and methods
Study sites and plant samples
We focused on presumably autochthonous and naturally-regenerated stands composed from trees of morphologically predominant morphotypes (ecotypic forms) within the Czech Republic. Per each ecotype, 150 trees were sampled (N = 450). Stands representing the high- (HE), medium- (ME), and low-elevation (LE) ecotypic forms are located in the Giant Mountains National Park, the Jizera Mountains protected area, and the Bohemian Switzerland National Park, respectively (Table 1).
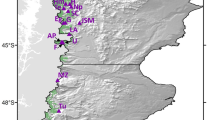
Generally, sampled trees were > 100-year-old with each tree's GPS coordinates recorded (supplementary Table S2). We targeted mature trees (DBH ≥ 25 cm) of the characteristic crown morphology to their respective ecotypic form (supplementary Figs. S1, S2, and S3). Samples were collected using a hole punch 15 mm in diameter, cutouts from the trunk were kept with silica gel in sealed plastic bags and stored at − 80 °C until further processing. Sampling design consists of transects or clusters with a 25 m as a minimum distance between adjacent trees (see Bínová16, for details). HE individuals were sampled across two subplots (Fig. 1).

DNA extraction
From each sample, approximately 50 mg of cambial and adjacent layers of wood tissue were scratched with a scalpel and frozen in liquid nitrogen, homogenized using a mixer mill MM400 (Retsch, Haan, Germany) for 3 min at 30 Hz. The genomic DNA was extracted by the DNeasy® Plant Mini Kit (Qiagen, Germany) according to the manufacturer's instructions. DNA concentration was measured using a NanoDropTM 2000 spectrophotometer (Thermo Fisher Scientific, Madison, USA). Both intact and HindIII (ThermoFisher, USA) digested DNA were run on 0.8% agarose gel to assure DNA quality.Undiluted aliquots of 40 µl DNA (mean concentration 127 ng/µl, 260/280 ratio between 1.47 and 1.91) were placed into 96-well PCR plates (95 samples + one negative control) and shipped under dry ice for analysis.
Genotyping
DNA samples were digested with the 6-base cutter restriction enzyme EcoT22I. Library and sequencing preparation were done in 96-plex, using 64 bp tag sequences. Sequencing ran on NextSeq 2000 (Illumina, San Diego, CA, USA) platform at the Cornell University sequencing facilities (Ithaca, NY, USA) according to GBS protocol56. Reads occurring at a minimal count of three were stored in a FASTQ file format.
Mapping and SNP calling
FASTQ file was aligned against the Picea abies reference genome v1.0., ftp://plantgenie.org/Data/ConGenIE/Picea_abies/v1.0/57,58 with Burrows-Wheeler Aligner (BWA, version 0.7.8-r45559). Aligned sequence tags were stored in SAM format file and SNP calling was performed using TASSEL-GBS Pipeline (version 3.0.174)60,61.
Low-quality SNPs were filtered out by minimum minor allele frequency (MAF) > 0.01 and missing data < 90% and the data were finally converted into Variant Call Format (VCF) file62.
Data filtration
VCF file (raw data) was subjected to a more stringent filtering process before analysis. All filtration steps were performed by VCFtools software (version 0.1.16)62 as follow: (1) only biallelic and non-indel SNPs were kept, (2) only loci with no-missing data were kept, (3) MAF was set > 0.05, (4) the Quality value threshold for loci was set ≥ 20, (5) only loci with mean depth values between 10 and 60 were retained, and (6) SNPs within 1000 bp distance were thinned.
Climatic data
Climatic data were downloaded from WorldClim database (http://www.worldclim.org/bioclim) in 0.5 arc-minute resolution (approx. 1 × 1 km). We utilized getData("worldclim", var = "bio", res = 0.5, lon = 15, lat = 5) function call from the raster package and assigned each tree stand with given climatic variables. After removing highly correlated variables, seven out of the original 19 climatic variables, were retained for further analysis (Table 2; PCA of selected climatic variables as a Supplement Fig. S4).
Data analysis
Primary data analysis was run with VCF tools. For Principal Component Analysis (PCA), as well as Discriminant Analysis of Principal Components (DAPC), we utilized functions implemented in R package adegenet63. For DAPC we used optim.a.score() function to control trade-off between the power of discrimination and over-fitting, to estimate the optimal number of PCs retained (n.pca = 37).
Bayesian clustering was done in STRUCTURE v2.3.4.64, the input file was generated with PGDSpider65 by VCF file conversion. To detect the most likely number of clusters, we generated K from 1 to 10 (a burn-in period of 10,000 iterations, followed by 100,000 iterations, admixture model scenario) using Evanno's approach66.
We used Nei's GST67, Jost's D68 and Hedrick's GST69 as measures of genetic differentiation among populations. We created genind objects for each pairwise comparison with the R package adegenet63 and performed 1000 bootstrap samples, with each subpopulation resampled according to its size, with the function chao_bootstrap() of the mmod R package70. Then, for each set of the permutated datasets, we obtained the observed genetic distance value and its normalized 95% confidence intervals (CI) (i.e., centered on the observed value and corrected with standard deviation across replicates) with the function summarise_bootstrap() in mmod package. We considered the genetic differentiation index to be statistically significant if their lower bound of the CI was greater than zero.
We employed the Mantel test from the ade4 package with 100,000 randomizations to estimate the correlation between the genetic distance matrix (provesti.dist() from poppr package71 and geographical and altitudinal distance matrices. The test uses Pearson's regression coefficient between geographic and genetic distance matrices. The statistics and the test's p value correspond to the proportion of times the randomized regression coefficient is equal or greater than the observed one72.
Redundancy analysis (RDA) implemented in vegan package73 was utilized to identify SNP loci associated with environmental variables for detecting probable adaptive variants51. The significance of the global RDA was evaluated by analysis of variance (ANOVA) with 1000 permutations. Then, we identified candidate loci based on locus scores (i.e., the loading of each locus in ordination space) that were ± 3 SD from the mean loading on the first three constrained ordination axes. We recognized the environmental variables exhibiting the strongest associations with each candidate adaptive locus using Pearson's correlation coefficient (r). We utilized a permutation test with 100,000 randomizations to estimate p value of such correlation and we adjusted α-level by Bonferroni correction (20 candidate SNPs and 7 climate variables) to value 0.00035.
Sample collection statements
The formal identification of the plant material used in our study was done by Jiri Korecky and confirmed by Jan Stejskal. We declare that the sample collection permissions were obtained from the responsible authorities. LE individuals were sampled in the Bohemian Switzerland National Park under the permission nr. SNPCS 0573/2015, HE individuals were sampled in the Giant Mountains National Park under the permission nr. OSML 38-11/2015, and ME individuals were collected in the Jizera Mountains protected area under the permission nr. LČR 8/2016. Norway spruce is not considered a species at risk of extinction or endangered species. Collection of plant material complied with institutional, national, and international guidelines and legislation. Trees in our study are categorized based on crown morphology (Supplementary Figs. S1, S2, and S3). GPS coordinates of every individual tree are provided in Supplementary Table S2; thus, every tree is traceable. Genomic data are available on-line (https://doi.org/10.6084/m9.figshare.16845445.v1).
Results
Genotyping
Four out of the 450 individuals failed in genotyping (one HE, one ME, and two LE), most likely due to the low DNA quality. Among the tested DNA restriction enzymes (EcoT22I, PstI, SbfI/MspI, SbfI/BfaI), EcoT22I, a rare cutter with the potential of good read depth for heterozygotes, was deemed the most appropriate enzyme based on fragment size optimization. There were 96 samples sequenced in one lane turning in approx. 5 M reads per sample. The total sequencing coverage (for 20 Gb Norway spruce genome) has been estimated approximately 0.025×.
Mapping and SNP calling
The FASTQ file was aligned against the Norway spruce reference genome and from 52,203,003 read tags present, 51.6 and 30.8% were uniquely and multiply aligned, whereas 17.6% tags remain unaligned. Variant calling performed on uniquely aligned tags generated 9,190,592 raw SNPs. These SNPs were filtered by minimum minor allele frequency (MAF) > 0.01 and missing data < 90%, kept 6,970,550 SNPs that were stored in VCF file (mean heterozygosity 0.074 ± 0.012).
Data filtration
The SNP data underwent a strict filtration in VCFtools software and was reduced from almost 7 million to 1,916 (0.03%) as follows:
-
(1)
only biallelic and non-indel SNPs were kept: 6,229,325 SNPs,
-
(2)
only loci with no missing data were kept: 36,313 SNPs,
-
(3)
MAF was set > 0.05: 16,982 SNPs,
-
(4)
the Quality value threshold for loci was set ≥ 20: 16,982 SNPs,
-
(5)
only loci with mean depth values between 10 and 60 were retained: 12,824 SNPs,
-
(6)
SNPs within 1000 bp distance were thinned: 1,916 SNPs.
The mean observed heterozygosity of the final SNP dataset was 0.271 ± 0.125 (expected heterozygosity 0.237 ± 0.114).
Data analysis
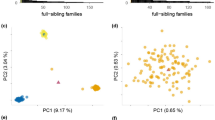
The PCA applied on the final dataset (1916 SNPs) separated the high-elevation from the medium and low-elevation ecotypes (Fig. 2). Although the percentage of explained variance is relatively low (PC1 explained 0.69% and PC2 explained 0.51%), the segregation between high-elevation and both ME and LE ecotypes (based on PC2) is visually apparent. It is interesting to note that ME and LE displayed no segregation pattern. Similarly, HE individuals from the two separate locations did not show clustering pattern.

Principal component analysis, PC1 and PC2 are plotted. Each dot represents genotyped individual; explained variance for each PC in the brackets.
For the DAPC analysis, the lowest BIC was not detected (the BIC curve constantly increased along with the increasing number of clusters), likely due to shallow population structure74. As a trade-off between the power of discrimination and over-fitting, we used optim.a.score() function and retained 37 PCs entering the analysis (n.pca = 37), n.da = 10. Similar to PCA analysis output, the DAPC differentiated the high-elevation ecotype from the low and medium elevation ecotypes. Again and similar to PCA analysis, individuals from both HE subplots overlapped and formed one cluster (Fig. 3).

Discriminant analysis of principal components (DAPC) scatterplot showing distinctiveness of the high-elevation (both HE1 and HE2 groups) ecotype in relation to the LE and ME.
Additionally, clustering using STRUCTURE (K = 5 detected as the most likely number of clusters, a graph for Evanno´s deltaK as Supplementary Fig. S5) showed shallow differentiation among populations; however, a HE Norway spruce ecotype cluster is noticeable (Fig. 4).

Summary barplot of estimated membership coefficients. Every single vertical line represents one Norway spruce individual in K-colored segments (K = 5) where 1, 2, and 3 represent the LE, ME, and HE ecotypic groups, respectively.
We estimated Nei's GST, Hedrick's GST and Jost's D genetic differentiation indices for each sampled population (Table 3). While the values of genetic differentiation among ecotypes was subtle, significant differences were observed (Table 3, Fig. 5) between the high-elevation and the other two ecotypes with values of one order of magnitude higher than LE–ME or HE1–HE2 pairs (Table 3).

Pairwise genetic differentiation indices among Norway spruce populations. The observed values displayed with normalized 95% confidence intervals.
The correlation of genetic distance with the geographical and altitudinal distances evaluated by the Mantel test were 0.062 and 0.068 (p < 10–5), respectively. While both correlations are low; they were significant, indicating that trees' genetic distance is correlated with altitudinal and geographical distances in an almost similar manner.
We used the Redundancy Analysis (RDA) method to select candidate SNPs to depict traces of local adaptations to climatic conditions. The first RDA component separates the high-elevation from medium- and low-elevation populations. In contrast, the second RDA component tends to emphasize differences in low- and medium-elevation populations and the third RDA depicted the difference between HE1 and HE2 populations (Fig. 6a,b). We subsequently detected 20 SNPs, that were ± 3 SD from the mean loading on the first three constrained ordination axes (Fig. 6c,d). Candidate SNPs were correlated with climate variables with 10 SNPs having the highest correlation to bio15 (precipitation seasonality, Pearson's correlation coefficient (r) ranging from − 0.05 to 0.13, yet no correlation was statistically significant), 7 SNPs with bio6 (min temperature of the coldest month, r ranging from 0.07 to 0.25, 2 correlations were statistically significant), 2 SNPs having the highest correlation with bio12 (annual precipitation, r being − 0.23 in both cases, both correlations were statistically significant), and 1 SNP with bio4 (temperature seasonality, r being 0.23, statistically significant). See Supplementary Table S1, for details.

(a, b) Individuals plotted by their score on the first, second, and third significant axes (a) RDA1 and RDA2; b: RDA1 and RDA3), population indicated by color; (c, d) 1916 SNPs plotted by their score on the first, second, and third significant axes (c) RDA1 and RDA2; (d) RDA1 and RDA3. Significant outlier SNPs (n = 20) are colored according to their most correlated climate variable (bio4 = temperature seasonality, bio6 = min temperature of coldest month; bio12 = annual precipitation, bio15 = precipitation seasonality).
Discussion
GBS data filtration
Genotyping-by-sequencing is a cost-effective genotyping technique that provides large datasets but is frequently of variable quality. Therefore, initial data filtration is needed, and one should set up the filtration criteria depending on the research question and overall genotyping quality. However, optimal settings are not straightforward and vary widely among studies75,76,77,78. The only exception is minor allele frequency (MAF) as in similar studies it is conventionally set to 0.0579,80,81. However, settings to different thresholds (0.01 and 0.02) are also reported. Our study inspected a variation attributable to most individuals within the respective group (ecotypic variance), so we set MAF to a 0.05 threshold.
We excluded multiallelic SNPs and indels during the first filtration step, which reduced the dataset to 89.4% of its original size. Further significant reduction occurred when excluded missing data (remaining approximately 0.5% of the original dataset size). Filtering for minor allele frequency additionally further reduced the data set by about one-half. To diminish the effect of linkage disequilibrium77, we thinned SNPs within a 1 kbp distance. Being aware of relatively low sequencing depth (mean depth for unfiltered 6.97 M SNPs data set is estimated 3.73), we set the minimum filtration depth for finally selected calling variants to 10, ensuring that only reliable SNPs are kept. We considered this to be the main reason for the low number of SNPs remained after filtering. Although we reduced the primary dataset significantly (preserving approximately 0.03% data out of the primary dataset), the remaining SNPs are robust and reliable.
Multivariate analyses
Although the interpopulation variation is usually blurred in unsupervised PCA due to summing the variability of both between and within groups82, in our data, separation of HE ecotype individuals from LE and ME groups on the first principal component is evident (Fig. 2). We were not able to obtain an unambiguous BIC number with find.clusters function;however, that is quite common for populations with shallow population structure83. It is noteworthy to emphasize that the direct geographical distance between the medium-elevation sampling area and subplot 1 of the high-elevation stand is approximately the same as between subplot 1 and subplot 2 of HE ecotype (Fig. 1). Based on both PCA and DAPC outputs, it seems that the genetic differentiation is more apparent along the altitudinal rather than the geographical distance. Mantel test is also showing a slightly higher genetic—altitude (0.068) than genetic—geographic (0.062) correlation. However, longitudinal and altitudinal clines are parallel and highly correlated in our setting (Fig. 1), making it difficult to disentangle their effects.
Also in the STRUCTURE barplot, fuzzy yet observable clustering of high-elevation ecotype based on estimated membership coefficients (Fig. 4) is attributable to generally low genetic differentiation indices among defined groups (Table 2, maximum value 0.02). For example, Latch et al.84 stated that the software STRUCTURE did not accurately distinguish among populations when the fixation index is lower than 0.03.
We looked for traces of local adaptations toward climatic conditions using RDA, which identified 20 candidate SNPs correlated to climate variables. However, a genome-wide association study conducted with climatic variables did not reveal any significantly associated SNP (not shown). This suggests that the inspected SNPs are not in a strong linkage with genes involved in local adaptation. Given that the Norway spruce genome size is 19.6 Gb57, and we have very low-density coverage, with one SNP per ca 800 kb, the likelihood of missing locally adapted regions is relatively high.
Genetic differentiation among stands
Overall, all genetic differentiation indices (Nei's GST, Jost's D, and Hendrick's GST) displayed significant differences for the LE–HE and ME–HE ecotypic groups (Fig. 5), whereas HE individuals from the two subplots showed significantly smaller genetic differences. Again, the HE genetic specificity is confirmed for both HE subplots indicating altitudinal rather than geographical differentiation effect. On the other hand, the eastmost HE2 population appears to be slightly more distant to both LE and ME than HE2, again indicating the impact of longitudinal differentiations.
Low genetic differentiation indices values in Norway spruce are common and frequently reported. For example, GST values among several German populations ranged from 0.002 to 0.03085, FST ranged from 0.0004 to 0.0035 among Austrian groups25, and from 0.00 to 0.04 among Italian Alpine populations26. These results are most likely a reflection of the species' intensive and long-distance pollen gene flow3,41,86. However, in contrast with these microsatellite-based studies, our SNP-based analysis might have detected an intrinsic effect of selection in high-elevation ecotype being manifested in our data. The fact that the high elevation samples were collected from two distinct locations provides additional credence to our observation.
Several studies investigated Norway spruce geographical variation along altitudinal gradient based on single nucleotide polymorphisms. Scalfi et al.87 with 384 SNPs did not detect any significant FST differences for six populations in a 200 m altitudinal gradient. With a set of 175 SNPs, Di Pierro et al.88 reported low but significant variation along elevational gradients (FST = 0.006). Romšáková et al.36 detected considerable differentiation among two altitudinally contrasting populations (700 versus 1450 m a.s.l). Subsequently, Hrivnák et al.89 replicated Romšáková's methodology for an altitudinal transect of several populations, reported genetic distinction only for individuals originating from the highest elevation stand (1500 m a.s.l).
It is essential to point out that several studies indicate that most variation in adaptive traits is complex and controlled by many loci of minor effects31,90. In our study, we did not detect individual SNPs contributing significantly to genetic grouping, hence we demonstrated the cumulative SNPs effect for ecotypic clustering by multivariate analysis methods.
Additionally, the effects of epigenetic mechanisms91 are also attributable to genetic variation, but this effect remained hidden in our study as the GBS approach could not detect them. Although epigenetic modifications are frequently overlooked50, it should be more reflected in genetic studies as not all of the variance in the phenotype can be accounted for those coded in DNA sequence92. In practice, that can be overcome only by using epigenetics mechanism-sensitive genotyping platforms.
Pollen transfer and natural barriers
Particularly in coniferous species, pollen gene flow is one of the most critical factors influencing genetic structure3. Medium-to-long distance pollen transport is commonly reported41,93, with the potential of pollen distribution hundreds of kilometers3. This fact reflects the shallow population differentiation revealed by genetic markers10,11,16. Conversely, the reproductive success of adjacent potential pollen donors is often limited3,94, which has negative consequences on seed orchard production95,96. According to Bucci and Vendramin97, the increase of the genetic divergence attributable to geographically formed variation is estimated in hundreds of kilometers (approximately 1800 km in Norway spruce). The distance between the sampled populations in our study is limited to circa one hundred kilometers without any natural geographical barriers (e.g., mountain ridges) in between. Therefore, we do not assume the effect of distance and geographical isolation to be factored but most likely selection mechanisms occurring in the HE group. Undoubtedly, also seed dispersal affects population structure, but its effect is more local98.
Although we have made every effort to target autochthonous Norway spruce populations, we could not entirely omit the historical effect of the human-mediated stand composition. Jansen et al.43 characterized Norway spruce as a highly translocated species for at least three centuries throughout Europe, including the Czech Republic. Sowing of Norway spruce seeds in the Jizera mountains (area of ME ecotype) has been recorded around 1820, but with local provenances. Although since the 1860s, the use of allochthonous forest reproductive material increased, cultivations were performed up to 1000 m a.s.l.99. In this regard, the HE ecotype might be considered more likely locally autochthonous. To minimize targeting non-natural regeneration in all three ecotypic groups, we intentionally placed sampling transects on less accessible slopes and rocky outcrops.
Phenological asynchrony
An additional aspect involved in the genetic differentiation of a given ecotypic group might be connected to reproductive phenology. In oak species, for example, phenological asynchrony driven by microclimatic variability determines the size of acorn crop100, in Eucalyptus flowering asynchrony build a natural reproductive barrier101. In spruce, local synchrony of pollen production and receptivity might promote short-distance pollination and preserve local-adapted genotypes102. Skrøppa and Steffenrem103 found clinal variation related to mean annual temperature. The intrapopulation variation was considerably larger in low altitude as compared to those from high-altitude populations. Thus, although the mean annual temperature difference of 2.3 °C between LE and ME ecotype is higher than the difference between ME and HE ecotype (1 °C), the narrower biological window of HE pollen receptivity might act as a barrier preventing non-local pollination.
Natural selection
During the twentieth century, numerous international Norway spruce provenance experiments were established across Europe, known as the Internation Union of Forest Research Organization (IUFRO) provenance trials104. Unfortunately, the information about the survival rate of lowland provenances planted in higher altitudes is scarce. Jurásek and Martincová105 observed a strong selection effect on Norway spruce seedlings in higher elevations with high mortality levels of lowland-originated seedlings compared to those from higher elevation (1000 m.a.s.l and above). Similar findings were reported by Hrdlička106 on a high-elevation testing plot (1070 m.a.s.l.) planted with two seedling sources. After a decade, seedlings originated from lower altitudes (560 m.a.s.l.) exhibit 95% mortality, whereas those from the higher altitude (970 m.a.s.l.) survived seamlessly.
Also in our study, we assume intensive natural selection among high-elevation individuals during the early growing stages as an intrinsic selection mechanism that eliminated unfavorable genotypes for adverse climatic conditions. As we focused on mature trees that persist on the stand despite the ontogenetic selection, we probably largely genotyped individuals that bear distinctive genetic pattern favorable to higher altitudes. Additionally, several ecophysiological studies indicated the resilient specific status of high-elevation ecotype such as enhanced drought tolerance107, higher nitrogenous content in needles108, and significantly different patterns in dehydrin gene expression109.
Conclusions
The current study aimed to dissect the genetic architecture of three Norway spruce ecotypic forms using SNP markers. We focused on presumably autochthonous stands composed from trees of morphologically predominant morphotypes, a.k.a ecotypes in a limited area of the Czech Republic. Due to such stringent selection criteria, we identified only one experimental area for LE, one area for ME, and two stands for high-elevation ecotypic form.
The data analysis revealed a conspicuous partition into two groups—low- and medium-elevation ecotypes being the one, and the high-elevation forming the second group. Being aware of the experimental limitations that do not allow to distinguish between local adaptation and IBD, we may only hypothesize reasons for such separation.
Although we did not detect any apparent biological (reproductive phenological asynchrony) or geographical (mountain ridges, long-distance separation) barriers, we assume the observed distinctive genetic structure of the high-elevation ecotype is a consequence of a natural selection. Target trees (> 100 years old) have already undergone not only phylogenetic but also ontogenetic selection pressure. Consistently to the long-term observations of forestry practitioners, we assume the intensive ontogenetic selection in a high-elevation ecotypic group promoting local paternal contributors mirrored in the distinctive overall genetic makeup of the extant population. Although our genome coverage was sparse to detect significant genotype-environment associations, we believe we obtained robust outputs in connection to ecotypic distinction. Our findings bring a vital message to forestry management practices. Under the current situation with bark beetle outbreaks across Europe causing increased demand on forest reproductive material for reforestation, some forestry practitioners appeal to relax forest reproductive material transfer rules. That should be considered with caution, especially in locations with extreme climates, as indigenous populations (seed donors) might have previously undergone an intensive selection and adaptation processes and be better adapted to these unfavorable conditions.
References
Lagercrantz, U. & Ryman, N. Genetic structure of Norway spruce (Picea abies): Concordance of morphological and allozymic variation. Evolution (N.Y.) 44, 38 (1990).
Gömöry, D. Effect of stand origin on the genetic diversity of Norway spruce (Picea abies Karst. populations. For. Ecol. Manage. 54, 215–223 (1992).
Burczyk, J., Lewandowski, A. & Chalupka, W. Local pollen dispersal and distant gene flow in Norway spruce (Picea abies [L.] Karst.). For. Ecol. Manage. 197, 39–48 (2004).
Vendramin, G. G., Anzidei, M., Madaghiele, A., Sperisen, C. & Bucci, G. Chloroplast microsatellite analysis reveals the presence of population subdivision in Norway spruce (Picea abies K.). Genome 43, 68–78 (2000).
Sperisen, C. et al. Tandem repeats in plant mitochondrial genomes: Application to the analysis of population differentiation in the conifer Norway spruce. Mol. Ecol. 10, 257–263 (2001).
Heuertz, M. et al. Multilocus patterns of nucleotide diversity, linkage disequilibrium and demographic history of Norway spruce [Picea abies (L.) Karst]. Genetics 174, 2095–2105 (2006).
Tollefsrud, M. M. et al. Genetic consequences of glacial survival and postglacial colonization in Norway spruce: Combined analysis of mitochondrial DNA and fossil pollen. Mol. Ecol. 17, 4134–4150 (2008).
Tollefsrud, M. M. et al. Combined analysis of nuclear and mitochondrial markers provide new insight into the genetic structure of North European Picea abies. Heredity (Edinb). 102, 549–562 (2009).
Maghuly, F., Pinsker, W., Praznik, W. & Fluch, S. Genetic diversity in managed subpopulations of Norway spruce [Picea abies (L.) Karst.]. For. Ecol. Manage. 222, 266–271 (2006).
Androsiuk, P. et al. Genetic status of Norway spruce (Picea abies) breeding populations for Northern Sweden. Silvae Genet. 62, 127–136 (2013).
Meloni, M., Perini, D. & Binelli, G. The distribution of genetic variation in Norway spruce (Picea abies Karst.) populations in the western Alps. J. Biogeogr. 34, 929–938 (2007).
Stojnić, S. et al. Assessment of genetic diversity and population genetic structure of Norway Spruce (Picea abies (L.) Karsten) at its Southern Lineage in Europe. Implications for conservation of forest genetic resources. Forests 10, 258 (2019).
Pfeiffer, A., Olivieri, A. M. & Morgante, M. Identification and characterization of microsatellites in Norway spruce (Picea abies K.). Genome 40, 411–419 (1997).
Schubert, R., Mueller-Starck, G. & Riegel, R. Development of EST-PCR markers and monitoring their intrapopulational genetic variation in Picea abies (L.) Karst. Theor. Appl. Genet. 103, 1223–1231 (2001).
Fluch, S. et al. Characterization of variable EST SSR markers for Norway spruce (Picea abies L.). BMC Res. Notes 4, 1–6 (2011).
Bínová, Z. et al. Genetic structure of Norway spruce ecotypes studied by SSR markers. Forests 11, 110 (2020).
Azaiez, A. et al. A catalog of annotated high-confidence SNPs from exome capture and sequencing reveals highly polymorphic genes in Norway spruce (Picea abies). BMC Genom. 19, 942 (2018).
Chen, Z. Q. et al. Accuracy of genomic selection for growth and wood quality traits in two control-pollinated progeny trials using exome capture as the genotyping platform in Norway spruce. BMC Genom. 19, 1–16 (2018).
Vidalis, A. et al. Design and evaluation of a large sequence-capture probe set and associated SNPs for diploid and haploid samples of Norway spruce (Picea abies). bioRxiv https://doi.org/10.1101/291716 (2018).
Chen, J. et al. Genomic data provide new insights on the demographic history and the extent of recent material transfers in Norway spruce. Evol. Appl. 12, 1539–1551 (2019).
Wang, X., Bernhardsson, C. & Ingvarsson, P. K. Demography and natural selection have shaped genetic variation in the widely distributed conifer Norway spruce (Picea abies). Genome Biol. Evol. 12, 3803–3817 (2020).
Bernhardsson, C., Zan, Y., Chen, Z., Ingvarsson, P. K. & Wu, H. X. Development of a highly efficient 50K single nucleotide polymorphism genotyping array for the large and complex genome of Norway spruce (Picea abies L. Karst) by whole genome resequencing and its transferability to other spruce species. Mol. Ecol. Resour. 21, 880–896 (2021).
Goncharenko, G. G., Zadeika, I. V. & Birgelis, J. J. Genetic structure, diversity and differentiation of Norway spruce (Picea abies (L.) Karst.) in natural populations of Latvia. For. Ecol. Manage. 72, 31–38 (1995).
Korshikov, I. I. & Privalikhin, S. N. Genetic structure of populations of Norway spruce (Picea abies (L.) Karst. from Ukrainian Carpathians. Russ. J. Genet. 43, 1364–1372 (2007).
Unger, G. M., Konrad, H. & Geburek, T. Does spatial genetic structure increase with altitude? An answer from Picea abies in Tyrol, Austria. Plant Syst. Evol. 292, 133–141 (2011).
Scotti, I., Paglia, G., Magni, F. & Morgante, M. Population genetics of Norway spruce (Picea abies Karst.) at regional scale: Sensitivity of different microsatellite motif classes in detecting differentiation. Ann. For. Sci. 63, 485–491 (2006).
Trujillo-Moya, C. et al. Drought sensitivity of Norway spruce at the species’ warmest fringe: Quantitative and molecular analysis reveals high genetic variation among and within provenances. G3 Genes Genomes Genet. 8, 1225–1245 (2018).
Baison, J. et al. Genome-wide association study identified novel candidate loci affecting wood formation in Norway spruce. Plant J. 100, 83–100 (2019).
Capador-Barreto, H. D. et al. Killing two enemies with one stone? Genomics of resistance to two sympatric pathogens in Norway spruce. Mol. Ecol. 30, 4433–4447 (2021).
Caré, O., Gailing, O., Müller, M., Krutovsky, K. V. & Leinemann, L. Crown morphology in Norway spruce (Picea abies [Karst.] L.) as adaptation to mountainous environments is associated with single nucleotide polymorphisms (SNPs) in genes regulating seasonal growth rhythm. Tree Genet. Genomes 16, 4 (2020).
Milesi, P. et al. Assessing the potential for assisted gene flow using past introduction of Norway spruce in southern Sweden: Local adaptation and genetic basis of quantitative traits in trees. Evol. Appl. 12, 1946–1959 (2019).
Chen, J. et al. Disentangling the roles of history and local selection in shaping clinal variation of allele frequencies and gene expression in Norway spruce (Picea abies). Genetics 191, 865–881 (2012).
Holliday, J. A., Ritland, K. & Aitken, S. N. Widespread, ecologically relevant genetic markers developed from association mapping of climate-related traits in Sitka spruce (Picea sitchensis). New Phytol. 188, 501–514 (2010).
Li, L., Chen, J. & Lascoux, M. Clinal variation in growth cessation and FTL2 expression in Siberian spruce. Tree Genet. Genomes 15, 82 (2019).
Ranade, S. S. & García-Gil, M. R. Molecular signatures of local adaptation to light in Norway spruce. Planta 253, 1–18 (2021).
Romšáková, I. et al. Nucleotide polymorphisms related to altitude and physiological traits in contrasting provenances of Norway spruce (Picea abies). Biologia (Bratisl). 67, 909–916 (2012).
Carneros, E., Yakovlev, I., Viejo, M., Olsen, J. E. & Fossdal, C. G. The epigenetic memory of temperature during embryogenesis modifies the expression of bud burst-related genes in Norway spruce epitypes. Planta 246, 553–566 (2017).
Mørtvedt Solvin, T. & Steffenrem, A. Modelling the epigenetic response of increased temperature during reproduction on Norway spruce phenology. Scand. J. For. Res. https://doi.org/10.1080/02827581.2018.1555278 (2019).
Amaral, J. et al. Advances and promises of epigenetics for forest trees. Forests 11, 1–21 (2020).
Yakovlev, I., Viejo, M. & Fossdal, C. G. in The Spruce Genome (eds. Porth, I. M. & la Torre, A. R.) 65–95 (Springer International Publishing, 2020). https://doi.org/10.1007/978-3-030-21001-4_5
Di-Giovanni, F., Kevan, P. G. & Arnold, J. Lower planetary boundary layer profiles of atmospheric conifer pollen above a seed orchard in northern Ontario, Canada. For. Ecol. Manage. 83, 87–97 (1996).
Spiecker, H. Silvicultural management in maintaining biodiversity and resistance of forests in Europe—Temperate zone. J. Environ. Manage. 67, 55–65 (2003).
Jansen, S., Konrad, H. & Geburek, T. The extent of historic translocation of Norway spruce forest reproductive material in Europe. Ann. For. Sci. 74, 1–17 (2017).
Morgenstern, E. K. Environmental Influences and Geographic Variation. Geographic Variation in Forest Trees (UBC Press, 1996).
Priehäusser, G. Die Fichten-Variationen und -Kombinationen des Bayr. Waldes nach phänotypischen Merkmalen mit Bestimmungsschlüssel. Forstwissenschaftliches Cent. 77, 151–171 (1958).
Samek, V. Metodika výzkumu morfologické proměnlivosti smrku z hlediska fytogeografického [Methodology of research of morphological variability of spruce from the phytogeographical point of view]. Zprávy Lesn. výzkumu [For. Rep.] 10, 18–25 (1964).
Geburek, T., Robitschek, K. & Milasowszky, N. A tree of many faces: Why are there different crown types in Norway spruce (Picea abies [L.] Karst.)? Flora Morphol. Distrib. Funct. Ecol. Plants 203, 126–133 (2008).
Beissinger, T. M. et al. Marker density and read depth for genotyping populations using genotyping-by-sequencing. Genetics 193, 1073–1081 (2013).
Bernhardsson, C., Wang, X., Eklöf, H. & Ingvarsson, P. K. in The Spruce Genome. Compendium of Plant Genomes (eds. Porth, I. & De la Torre, A.) 9–36 (Springer, 2020). https://doi.org/10.1007/978-3-030-21001-4_2
He, J. et al. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 5, 1–8 (2014).
Forester, B. R., Lasky, J. R., Wagner, H. H. & Urban, D. L. Comparing methods for detecting multilocus adaptation with multivariate genotype–environment associations. Mol. Ecol. 27, 2215–2233 (2018).
Cornes, R. C., van der Schrier, G., van den Besselaar, E. J. M. & Jones, P. D. An ensemble version of the E-OBS temperature and precipitation data sets. J. Geophys. Res. Atmos. 123, 9391–9409 (2018).
Hijmans, R. J. & van Etten, J. R Package raster—Geographic analysis and modeling with raster data. (2012). http://CRAN.R-project.org/package=raster. Accesed 24 Feb 2021.
Lacko, J. R package RCzechia. (2020). https://github.com/jlacko/RCzechia
GIMP Development Team. GIMP 2.10.28. (2019).
Elshire, R. J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6, 19379 (2011).
Nystedt, B. et al. The Norway spruce genome sequence and conifer genome evolution. Nature 497, 579–584 (2013).
Sundell, D. et al. The plant genome integrative explorer resource: PlantGenIE.org. New Phytol. 208, 1149–1156 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Bradbury, P. J. et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007).
Glaubitz, J. C. et al. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 9, e90346 (2014).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Jombart, T. & Ahmed, I. Adegenet 1.3–1: New tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071 (2011).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Lischer, H. E. L. & Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28, 298–299 (2012).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 70, 3321–3323 (1973).
Jost, L. GST and its relatives do not measure differentiation. Mol. Ecol. 17, 4015–4026 (2008).
Hedrick, P. W. A standardized genetic differentiation measure. Evolution (N. Y.) 59, 1633–1638 (2005).
Winter, D. J. MMOD: An R library for the calculation of population differentiation statistics. Mol. Ecol. Resour. 12, 1158–1160 (2012).
Kamvar, Z. N., Tabima, J. F. & Gr̈unwald, N. J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 1–14 (2014).
Séré, M., Thévenon, S., Belem, A. M. G. & De Meeûs, T. Comparison of different genetic distances to test isolation by distance between populations. Heredity (Edinb). 119, 55–63 (2017).
Oksanen, J. et al. vegan: Community Ecology Package. (2019). https://rdrr.io/cran/vegan/man/vegan-package.html. (Accessed 28 June 2021)
Miller, J. M., Cullingham, C. I. & Peery, R. M. The influence of a priori grouping on inference of genetic clusters: Simulation study and literature review of the DAPC method. Heredity 125, 269–280 (2020).
Pereira-Dias, L., Vilanova, S., Fita, A., Prohens, J. & Rodríguez-Burruezo, A. Genetic diversity, population structure, and relationships in a collection of pepper (Capsicum spp.) landraces from the Spanish Centre of diversity revealed by genotyping-by-sequencing (GBS). Hortic. Res. 6, 54 (2019).
Zhu, S., Niu, E., Shi, A. & Mou, B. Genetic diversity analysis of olive germplasm (Olea europaea L.) with genotyping-by-sequencing technology. Front. Genet. 10, 755 (2019).
Gardner, K. M. et al. Fast and cost-effective genetic mapping in apple using next-generation sequencing. G3 Genes Genomes Genet. 4, 1681–1687 (2014).
D’Agostino, N. et al. GBS-derived SNP catalogue unveiled wide genetic variability and geographical relationships of Italian olive cultivars. Sci. Rep. 8, 1–13 (2018).
Delfini, J. et al. Population structure, genetic diversity and genomic selection signatures among a Brazilian common bean germplasm. Sci. Rep. 11, 1–12 (2021).
Guajardo, V. et al. Genome-wide SNP identification in Prunus rootstocks germplasm collections using Genotyping-by-Sequencing: phylogenetic analysis, distribution of SNPs and prediction of their effect on gene function. Sci. Rep. 10, 1–14 (2020).
Zhu, S., Niu, E., Shi, A. & Mou, B. Genetic diversity analysis of olive germplasm (Olea europaea L.) with genotyping-by-sequencing technology. Front. Genet. 10, 1–11 (2019).
Jombart, T., Devillard, S. & Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 11, 1–15 (2010).
Jombart, T. & Collins, C. A Tutorial for Discriminant Analysis of Principal Components (DAPC) Using adegenet 1.3-6 (MRC Centre for Outbreak Analysis and Modelling, 2013).
Latch, E. K., Dharmarajan, G., Glaubitz, J. C. & Rhodes, O. E. Relative performance of Bayesian clustering software for inferring population substructure and individual assignment at low levels of population differentiation. Conserv. Genet. 7, 295–302 (2006).
Caré, O. et al. High morphological differentiation in crown architecture contrasts with low population genetic structure of German Norway spruce stands. Forests 9, 752 (2018).
Lindgren, D. et al. Can viable pollen carry scots pine genes over long distances? Grana 34, 64–69 (1995).
Scalfi, M. et al. Micro-and macro-geographic scale effect on the molecular imprint of selection and adaptation in Norway spruce. PLoS ONE 9, e115499 (2014).
Di Pierro, E. A. et al. Adaptive variation in natural Alpine populations of Norway spruce (Picea abies [L.] Karst) at regional scale: Landscape features and altitudinal gradient effects. For. Ecol. Manage. 405, 350–359 (2017).
Hrivnák, M., Krajmerová, D. & Gömöry, D. Lack of signals of selection at candidate loci at a small geographical scale along a steep altitudinal gradient in Norway spruce (Picea abies [L.] Karst.). Acta Biol. Cracoviensia Ser. Bot. 61, 43–51 (2019).
Savolainen, O., Pyhäjärvi, T. & Knürr, T. Gene flow and local adaptation in trees. Annu. Rev. Ecol. Evol. Syst. 38, 595–619 (2007).
Feil, R. & Fraga, M. F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 13, 97–109 (2012).
Banta, J. A. & Richards, C. L. Quantitative epigenetics and evolution. Heredity 121, 210–224 (2018).
Liu, Y. & El-Kassaby, Y. A. Ecological drivers of plant life-history traits: Assessment of seed mass and germination variation using climate cues and nitrogen resources in conifers. Ecol. Indic. 117, 106517 (2020).
Piotti, A., Leonardi, S., Piovani, P., Scalfi, M. & Menozzi, P. Spruce colonization at treeline: Where do those seeds come from. Heredity (Edinb). 103, 136–145 (2009).
Pakkanen, A., Nikkanen, T. & Pulkkinen, P. Annual variation in pollen contamination and outcrossing in a Picea abies seed orchard. Scand. J. For. Res. 15, 399–404 (2000).
Sønstebø, J. H. et al. Genetic diversity of Norway spruce (Picea abies (L.) Karst.) seed orchard crops: Effects of number of parents, seed year, and pollen contamination. For. Ecol. Manage. 411, 132–141 (2018).
Bucci, G., Vendramin, G. G. & Ellstrand, N. C. Delineation of genetic zones in the European Norway spruce natural range: Preliminary evidence. Mol. Ecol. 9, 923–934 (2000).
Di Pierro, E. et al. The effects of seed availability on habitat use by a specialist seed predator. Eur. J. Wildl. Res. 57, 585–595 (2011).
Matuszkiewicz, W. & Matuszkiewicz, A. Pflanzensoziologische Untersuchungen der Waldgesellschaften des Riesengebirges. Acta Soc. Bot. Pol. 29, 499–530 (1960).
Koenig, W. D., Knops, J. M. H., Carmen, W. J. & Pearse, I. S. What drives masting? The phenological synchrony hypothesis. Ecology 96, 184–192 (2015).
Barbour, R. C., Potts, B. M., Vaillancourt, R. E. & Tibbits, W. N. Gene flow between introduced and native Eucalyptus species: Flowering asynchrony as a barrier to F1 hybridisation between exotic E. nitens and native Tasmanian Symphyomyrtus species. For. Ecol. Manage. 226, 9–21 (2006).
Chmura, D. J. Phenology differs among Norway spruce populations in relation to local variation in altitude of maternal stands in the Beskidy Mountains. New For. 32, 21–31 (2006).
Skrøppa, T. & Steffenrem, A. Genetic variation in phenology and growth among and within norway spruce populations from two altitudinal transects in mid-Norway. Silva Fenn. 53, 10076 (2019).
Chmura, D. J. et al. Variation in growth of Norway spruce in the IUFRO 1972 provenance experimental series. Silvae Genet. 67, 26–33 (2018).
Jurasek, A. & Martincova, J. Specifika pěstování sadebního materiálu smrku ztepilého původem z horských poloh [Peculiarities of cultivating Norway spruce (Picea abies (L.) Karsten) planting originated from mountain localities]. Rep. For. Res. 50, 18-23 (2005).
Hrdlicka, O. Modravský smrk [Modrava Norway spruce]. Lesn. Pr. 84, 14–15 (2005).
Modrzyński, J. & Eriksson, G. Response of Picea abies populations from elevational transects in the Polish Sudety and Carpathian mountains to simulated drought stress. For. Ecol. Manage. 165, 105–116 (2002).
Oleksyn, J. et al. Growth and physiology of Picea abies populations from elevational transects: Common garden evidence for altitudinal ecotypes and cold adaptation. Funct. Ecol. 12, 573–590 (1998).
Čepl, J. et al. The dehydrins gene expression differs across ecotypes in Norway spruce and relates to weather fluctuations. Sci. Rep. 10, 20789 (2020).
Acknowledgements
We thank Dr. Charles Chen for his help and support during the GBS genotyping.
Funding
This work was supported by the National Agency of Agriculture research, Czech Republic (NAZV) [Grant number QK1910480]; and the EXTEMIT-K [Grant number CZ.02.1.01/0.0/0.0/15_003/0000433] financed by OP RDE.
Author information
Authors and Affiliations
Contributions
J.K.: conceptualization, investigation, writing—original draft, formal analysis; J.Č.: formal analysis, writing—original draft, visualization; J.S.: investigation, writing—review and editing; Z.F.: resources (lab activities), writing—review and editing; J.D.: resources (sampling, lab activities); M.L.: supervision, funding acquisition; Y.A.E.: writing—review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Korecký, J., Čepl, J., Stejskal, J. et al. Genetic diversity of Norway spruce ecotypes assessed by GBS-derived SNPs. Sci Rep 11, 23119 (2021). https://doi.org/10.1038/s41598-021-02545-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-02545-z
- Springer Nature Limited