
Abstract
The objective of the present study was to discover the genetic variants, functional candidate genes, biological processes and molecular functions underlying the negative genetic correlation observed between body weight (BW) and egg number (EN) traits in female broilers. To this end, first a bivariate genome-wide association and second stepwise conditional-joint analyses were performed using 2586 female broilers and 240 k autosomal SNPs. The aforementioned analyses resulted in a total number of 49 independent cross-phenotype (CP) significant SNPs with 35 independent markers showing antagonistic action i.e., positive effects on one trait and negative effects on the other trait. A number of 33 independent CP SNPs were located within 26 and 14 protein coding and long non-coding RNA genes, respectively. Furthermore, 26 independent markers were situated within 44 reported QTLs, most of them related to growth traits. Investigation of the functional role of protein coding genes via pathway and gene ontology analyses highlighted four candidates (CPEB3, ACVR1, MAST2 and CACNA1H) as most plausible pleiotropic genes for the traits under study. Three candidates (CPEB3, MAST2 and CACNA1H) were associated with antagonistic pleiotropy, while ACVR1 with synergistic pleiotropic action. Current results provide a novel insight into the biological mechanism of the genetic trade-off between growth and reproduction, in broilers.
Similar content being viewed by others


Introduction
Reproductive traits in livestock species often show an antagonistic relationship with growth traits that is manifested as negative genetic correlation between single members of trait complexes. In chicken, a typical example is the negative genetic correlation (rg) estimated between body weight (BW) and egg number (EN) with rg estimates in the range from − 0.05 to − 0.551,2,3. In general, the most important source for genetic correlations is usually pleiotropy4, however, genetic correlations may also arise from linkage disequilibrium (LD) among distinct loci5.
One possible way of identifying plausible pleiotropic genetic loci is to perform multivariate or univariate GWAS of traits under interest. The resulting marker trait(s) associations are termed cross-phenotype (CP) associations6. While multivariate approaches7 allow for direct identification of CP associations, in the context of univariate analyses, detection of CP associations relies on aggregating results of single traits analyses via meta-analysis techniques8.
When searching for pleiotropic loci via GWAS, it is important to bear in mind that CP associations are based on statistical evidence regardless of the underlying cause9 and are not necessarily indicative of pleiotropic genetic variants.
On the contrary, pleiotropy occurs when a genetic locus truly affects more than one trait, simultaneously. When beneficial effects of a genetic factor on one trait are accompanied by negative effects on the other trait, antagonistic pleiotropy (AP) exists10,11. In contrast to AP, synergistic pleiotropy12,13 (SP) occurs when a genetic variant simultaneously either increases or decreases performance in two different traits.
Based on the mechanisms of action, pleiotropy can be distinguished in: biological (or horizontal), mediated (or vertical) and spurious pleiotropy6,14. Specifically, in biological pleiotropy, a genetic variant or a gene affects multiple phenotypes since causal variants for different phenotypes can be colocalized in the same gene. In mediated pleiotropy, there is a causal relationship between two phenotypes so as a variant exerts an effect on one phenotype through the another one while spurious pleiotropy refers to a falsely association between marker and phenotypes due to bias, misclassification or linkage disequilibrium (LD). To overcome the challenge of spurious pleiotropy in the latter case, approaches such as LD pruning and conditional and joint analysis (cojo) can be applied to alleviate the high SNP interdependency arising from LD and to select the LD independent SNPs.
In chicken, CP associations have already been reported by GWAS for various traits such as daily feed intake and efficiency15 and for egg weights at different ages16,17. Nevertheless, no GWAS has, so far, been reported with the aim to discover genetic variants associated with body weight (BW) and egg number (EN) in chickens.
Driven from the scarcity of relevant reports, we have elaborated the present study with the aim to identify genetic variants and genes simultaneously affecting BW and EN in chickens. To this end, first we conducted a bivariate GWAS to identify SNP signals associated with both traits. As LD could generate spurious pleiotropic associations (see reviews6,14) we then applied conditional and joint analysis (cojo) of the SNP signals obtained from bivariate analysis to identify LD-independent CP SNPs. Finally, we investigated the functional role of the candidate genes underlying the independent CP SNPs in attempts to propose the most relevant pleiotropic genes implicated in the genetic control of traits under study.
Results
Comparison of genome-wide significant SNPs found by bivariate analysis, BW univariate analysis and EN univariate analysis
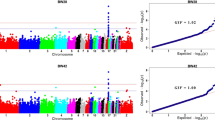
Estimations of the genomic inflation factors (univariate analyses: λBW = 0.86, λΕΝ = 0.95, bivariate analysis: λ = 0.85) were less than 1 indicating the absence of population structure or artifacts in the present data. Furthermore, the genomic genetic correlation (rg) between the two traits was estimated as high as − 0.183 ± 0.15 (results not shown). Figure 1 shows the profiles of the SNP p values (expressed as − log10 values) across the three GWAS. Specifically, a total number of 667 genome-wide significant SNPs (FDR p value < 0.10) were detected by BW univariate analysis and these SNPs were dispersed across the 28 autosomes (Figs. 2, 3). For EN univariate analysis, a total number of 10 SNPs across five autosomes (2, 3, 12, 26 and 28) were found to reach genome-wide significance (FDR p value < 0.10) (Figs. 2, 3). The bivariate analysis resulted in 630 genome-wide significant (FDR p value < 0.10) CP SNPs across the 28 autosomes (Figs. 2, 3).

Circular Manhattan plots showing the − log10(p values) of SNPs across the 28 autosomal chromosomes for body weight (BW) (a), egg number (EN) (b) and both traits (c), respectively. Red dots in Manhattan plots denote the genome-wide significant SNPs. Plots were constructed using the CMplot package (https://github.com/YinLiLin/R-CMplot) in R (http://www.r-project.org/).

Graphic depiction showing the positions of the genome-wide significant SNPs (denoted as points) and their p values from univariate and bivariate analyses aligned along each autosome. Black labels with number denote the chromosomes. Each SNP position is linked to at least one colored circle representing the associated trait (s) (i.e. body weight (BW): red circle, egg number (EN): blue circle and both traits: cyan circle). The common SNP between the three analyses (2 univariate and 1 bivariate analyses) is presented with purple color. Plot was constructed using the shinyChromosome (https://yimingyu.shinyapps.io/shinyChromosome59).

Venn diagrams showing the number of common significant SNPs between analyses. The left Venn diagram presents the number of common significant SNPs between bivariate association analysis (both traits: green color) and univariate association analyses (body weight (BW): blue color and egg number (EN): yellow color) while the right Venn diagram presents the number of common significant SNPs after conditional and joint (cojo) analyses. Some common SNPs (with their rsids) are also given with red color. Venn diagrams were constructed by VENNY 2.160.
Comparison of SNP signals across the three association analyses resulted in one common significant marker (rs313298834) on GGA12 (Figs. 2, 3). In addition, a total number of 475 SNPs were common between the BW univariate analysis and the bivariate analysis and all significant SNPs (n = 10) for EN were also detected by bivariate analysis (Fig. 3).
LD-independent significant SNPs
Cojo-GCTA analyses resulted in a total number of 49 independent CP SNPs while 21 and 5 independent significant SNPs were detected for BW and EN, respectively (Fig. 3, Supplementary Table S1). As observed in Fig. 3, no independent SNP was common between the three analyses. Nevertheless, markers were common between the BW univariate analysis and the bivariate analysis (Fig. 3).
Table 1 presents a detailed description of the 49 independent CP associations across the 25 autosomes (1–15, 17–21 and 23–28). The maximum number (n = 9) of independent markers were located on GGA1 while marker rs315329074 (GGA27) presented the lowest p value (6.13E-32) after ‘cojo’ analysis. Table 1 along with Supplementary Fig. S1 also shows the estimated marker effect sizes (β) on the two traits obtained from bivariate analysis. In accordance with antagonistic pleiotropic action i.e. positive effects on one trait and negative effects on the other trait, the estimated effect sizes for the majority (35 out of 49) of the CP associations displayed opposing signs (Supplementary Fig. S1).
Note that the estimated genomic rg (− 0.183) is a weighted average of effect sizes of markers exhibiting antagonistic, synergistic and non pleiotropic action and only in the extreme case of rg = − 1 all the implicated markers would exhibit opposing effects on both traits.
Effect prediction of the independent CP significant SNPs and identification of positional candidate genes and published QTLs
A total number of 40 positional candidate genes (of which 24 were annotated genes) were identified as lying within 33 independent SNPs while 16 SNPs were intergenic variants (Supplementary Table S2). Specifically, 33 SNPs were located within 26 protein coding genes and 14 long non-coding (lnc) RNA genes (Supplementary Table S2). Of these SNPs, one was a missense variant of gene ZC3H18, one a synonymous variant of gene EXTL1 and 23 were intron variants of annotated genes (CELF2, PTPRZ1, PTPRB, EIF1AX, NOL4, TMEM206, SLAIN2, SORCS2, FMN1, MARK3, CPEB3, EBF3, ACVR1, AMY2A, MAST2, TBL1XR1, CHSY1, CACNA1H, NPHP4, VPS11, PLXNA2 and CACNB1).
Figure 4 provides a view for the independent CP significant SNPs (n = 49) and the corresponding positional candidate gene(s) and published QTL(s). With regard to QTLs, 26 independent SNPs lied within 44 previously reported QTLs (Supplementary Table S2). As seen in Fig. 4, the majority of QTLs were related to body weight (hatch, at 21 or 36 days) or body parts weight (e.g. femur weight, proventriculus weight, breast muscle weight, ileum weight, wattles and comb weight), followed by QTLs related to Qualitative Traits (e.g. feathered feet, feather colour extended black and feather crested head) and finally QTLs related to feed, dry matter intake or feed conversion. Notably, one of the independent SNPs i.e. rs314529054 was located within a region of GGA6 where an ovary weight QTL and a body weight QTL are reported. Marker rs314529054 also lied within gene CPEB3 (Supplementary Table S2). Finally, rs317370260 lied within a region of GGA17 where a QTL related to egg production rate is reported. No positional gene was present in the area with only a lncRNA lying in proximity (distanced 5322 bp) to the marker.

Radial network showing the independent cross-phenotype significant SNPs (with rsids) and the corresponding positional candidate genes and/or published QTLs in the searched regions. Note that QTLs were grouped according to their trait relevance. Figure was constructed using data.tree and networkD3 R packages.

Radial network displaying the ACVR1, MAST2 and CACNA1H genes and their respective biological processes. Red color denotes the developmental process while reproduction is marked with green color. Figure was constructed using data.tree and networkD3 R packages.
Bioinformatics analyses
All annotated chicken genes were recognized by GeneCodis 4.0. MEA revealed 15 genes participating in significantly enriched concurrent GO BP and/or pathway annotations (Supplementary Table S3). Furthermore, the LAGO tool recognized all but two (NOL4 and ZC3H18) candidate genes and highlighted a total number of 120 enriched GO BPs (Supplementary Table S4).
Several candidate genes displayed growth functional relevance (e.g. GO:0032502: developmental process: participating genes ACVR1, CACNA1H, CHSY1, CPEB3, EBF3, EXTL1, FMN1, MAST2, NPHP4, PLXNA2, PTPRB, PTPRZ1, TBL1XR1ACVR1, PLXNA2, ACVR1, CACNA1H, CHSY1, CPEB3, EBF3, EXTL1, FMN1, NPHP4, PLXNA2, PTPRB, PTPRZ1, TBL1XR1). Nevertheless, only CPEB3, ACVR1, CACNA1H and MAST2 were found to participate in reproduction related processes. Specifically, CPEB3 was associated with three concurrent annotations related to reproduction (oocyte meiosis, regulation of translation and progesterone-mediated oocyte maturation, FDR p value = 0.01) (Supplementary Table S3). Jointly, the rest three genes (ACVR1, CACNA1H and MAST2) were associated with sexual reproduction (GO:0019953) while ACVR1 and MAST2 were further associated with cellular process involved in reproduction in multicellular organism (GO:0022412) (Supplementary Table S4; Fig. 5).
Of the four above genes, ACVR1 displayed the highest number of significant concurrent annotations (n = 55 GO BPs) and was also member of two pathways: positive regulation of BMP signaling (GO:0030513) and TGF-beta signaling pathway (gga04350). Based on functional relevance derived from ontological and functional analyses, the four above genes (ACVR1, CPEB3, MAST2 and CACNA1H) were nominated as most plausible pleiotropic candidate genes for BW and EN. The four above mentioned genes lied within respective numbers of SNPs i.e. CPEB3 (rs314529054, (GGA6), pcojo = 3.12E−07), ACVR1 (rs313879964 (GGA7), pcojo = 2.15E−22), MAST2 (rs317902708 (GGA8), pcojo = 3.88E−10) and CACNA1H (rs317631529, (GGA14), pcojo = 1.16E−11) (Table 1). Estimated effect sizes (β) obtained from bivariate analysis for the respective markers are indicative of antagonistic pleiotropic action for the three genes i.e. CPEB3 (βBW = 2.04, βEN = − 0.63), MAST2 (βBW = 4.93, βEN, = − 0.77) and CACNA1H (βBW = − 3.14, βEN = 0.74) and synergistic action for ACVR1 (βBW = 6.08, βEN = 0.26, Table 1). Of the three genes exhibiting antagonistic action, CPEB3 and MAST2 affected positively and negatively BW and EN, respectively, while a reverse trend was observed for CACNA1H.
With regard to molecular function, the four candidate genes showed GO slim terms of binding (GO:0005488) and protein binding (GO:0005515), ACVR1 and MAST2 of kinase activity (GO:0016301), catalytic activity (GO:0003824) and transferase activity (GO:0016740), CACNA1H of ion transmembrane transporter activity (GO:0015075), channel activity (GO:0015267) and transporter activity (GO:0005215) and finally CPEB3 of nucleic acid binding (GO:0003676) and translation regulator activity (GO:0045182) (results not shown).
Discussion
In the present study, marker trait(s) association analyses along with in silico exploration of the biological role(s) of the implicated genes were jointly applied to refine our understanding of the genetic trade-off between BW and EN, in broilers. The genetic antagonism between the two traits was confirmed at the genome-wide level, as the estimate of the genomic genetic correlation between BW and EN was as high as − 0.18 herein, in concordance with previous findings1,2,3. As this genome-wide estimate only describes the cumulative CP effects of all implicated causal loci, in this study we further attempted to identify genetic variants with strong statistical associations for both traits, quantify their patterns of pleiotropic effects and explore the involved biological processes and/or pathways.
Marker trait(s) association analysis verified previous results, by revealing one common SNP (rs313298834) on GGA12 that has been associated with EN in female broilers18 and five more markers (rs317668107 on GGA3, rs15608447 on GGA4, rs318098582 on GGA11, rs317414603 on GGA20 and rs315329074 on GGA27) associated with BW19 in broilers. We also noted a higher number of SNP signals passing the genome-wide FDR significance threshold (set up to 0.10 here) for BW when compared to EN. As the yield of GWAS critically depend on the underlying effect-size distribution of the implicated variants20, this is not a surprising finding and it may be attributed to the lower heritability estimate for EN (0.17) when contrasted to the respective estimate (0.30) for BW.
Bivariate analysis identified a vast number of genome-wide significant CP SNPs. To ensure that CP associations did not arise from LD between markers (spurious pleiotropy6), a critical step in the present study was to identify only LD-independent SNPs via stepwise cojo analysis, as in other GWAS21,22. After overcoming the LD challenge, our results disclosed several genetic variants simultaneously affecting BW and EN with some of them pointing to most promising pleiotropic genes, as we discuss below.
The first SNP that served as a proxy to a plausible pleiotropic gene was rs314529054 (GGA6). The marker is located within two reported QTLs related to ovary and body weight and lies within CPEB3 (cytoplasmic polyadenylation element binding protein 3) gene. Ontological and functional analysis suggested that the gene in question may be considered as a true pleiotropic gene, however, literature evidence on its functional relevance to the traits examined here is shortcoming. In mice, CPEB has been reported to control polyadenylation and translation during the dictyate stage of oocyte development and this regulation has also a profound influence on folliculogenesis23.
In contrast to the previous marker, rs313879964 (GGA7) pointed at a gene with a highly likely pleiotropic function. This specific marker lied within ACVR1 (serine/threonine-protein kinase receptor or activin receptor type I or activin a receptor, type 1, also known as ALK2) gene. This gene participates in several growth and reproduction related GO BPs and has well documented involvement in biological phenomena such as those examined here. As MEA showed, ACVR1 encodes for a bone morphogenetic protein (BMP) type I receptor of the transforming growth factor-beta (TGF-β) superfamily which plays a key role in cell growth while regulates several reproductive processes (such as follicular development and ovulation)24. In addition, ACVR1 regulates reproduction via the BMP and anti-Müllerian hormone (AMH) signaling25. In chickens, AMH is required for the urogenital development and germ cell migration26, is presented in early development of follicles and is expressed in small follicles27. So far, the chicken ACVR1 gene has been suggested as a positional candidate gene for body weight28, has a regulatory role during skeletal development in osteogenesis and chondrogenesis29 and is expressed within the chicken granulosa and thecal layers during ovarian follicle development30.
Two more markers, rs317902708 (GGA8) and rs317631529 (GGA14) pointed at two most promising pleiotropic genes i.e., MAST2 and CACNA1H, respectively. Specifically, MAST2 (microtubule associated serine/threonine kinase 2) has been previously detected by RNA-seq in visceral fat of broiler and layer females at the onset of sexual maturation31. Furthermore, human MAST2 gene has been reported to be involved in PI3K-AKT signaling pathway32 that regulates various cellular processes, such as proliferation, growth, apoptosis and cytoskeletal rearrangement33. On the other hand, CACNA1H (calcium voltage-gated channel subunit alpha1 H, also known as Cav3.2) encodes for Cav3.2 channel that is a member of the voltage-gated calcium channel family. This gene participates in the T-type Ca2+ channels which contribute to signal transduction pathways regulating protein synthesis, development, proliferation and cell differentiation34 that are mainly expressed during embryonic development34. Particularly, these channels are involved in the early stages of muscle differentiation in humans35 and mice36. Female Cav3.2-/- null mutant mice presented decreased body weight37 and reduced litter size38. Moreover, Cav3.2 may have a role in reproduction since it facilitates the influx of Ca2+ in mouse oocytes and eggs to maintain Ca2+ homeostasis during oocyte maturation and post fertilization38. The murine Cacna1h gene is also upregulated in the proestrus of the Gonadotropin-releasing hormone (GnRH) neurons39. GnRH determines the pattern of secretion of follicle stimulating hormone (FSH) and luteinising hormone (LH) that regulate the endocrine function and gamete maturation of gonads40. So far, the chicken CACNA1H gene has only been associated with egg quality41 and body weight42.
Based on the approach followed herein, three candidates (CPEB3, MAST2 and CACNA1H) were identified as ‘trade-off’ genes i.e. exhibiting antagonistic pleiotropy, while ACVR1 displayed synergistic pleiotropic action.
We hypothesize that the above four genes are indicative of horizontal pleiotropy although we acknowledge the scepticism of Jordan et al.43 who hypothesized that the pervasive horizontal pleiotropy observed in polygenic traits is, on some level, a logical consequence of widespread polygenicity. Present results seem to fairly support such a hypothesis as hundreds of markers with little individual effects on the traits could be detected in the present study, especially for BW.
Another interesting finding obtained herein was the presence of independent CP SNPs within long non-coding genes (lncRNAs: RNA transcripts greater than 200 bp in length). In nucleus, lncRNAs have been reported to function in-cis and in-trans whereby in-cis acting lncRNAs influence the expression of nearby genes44. Furthermore, lncRNAs can encode short peptides44,45 and function as molecular decoy for proteins or sponges for other transcripts (such as miRNAs)44. They can also regulate numerous functions such as epigenetic modification, transcription and post-transcription while playing a key role in tissue development, muscle contraction/relaxation44 and myogenesis45. In chickens, lncRNAs have been reported to regulate muscle development, lipid metabolism, egg production and disease resistance46.
To conclude, present results provide a novel insight in the genetic mechanism underlying antagonistic interplay between growth and reproduction in broilers. Further follow-up studies (e.g. fine mapping and gene expression studies) are warranted to experimentally verify present findings.
Methods
Data and quality control
Genotypic and phenotypic records were provided by Aviagen. The available data consisted of 2992 female broilers from a grand-grandparent (GGP) commercial line with phenotypic records on body weight (BW) at 35 days of age (average = 1822.7 g, SD = 143.6 g) and number of eggs (EN) per hen collected from 28 to 50 weeks of age (average = 132.4 eggs, SD = 29.8 eggs). Animals were genotyped using the 600 k Affymetrix HD SNP array47 resulting in a total number of 544,927 autosomal SNPs. Quality control (QC) was performed first at a sample and second at a marker level. At a sample level, 406 animals were excluded due to call rate < 0.99 and autosomal heterozygosity outside the 1.5 IQR (inter-quartile range: 0.013). At the marker level, 305,660 SNPs autosomal SNPs were excluded due to: call rate < 0.95, minor allele frequency (MAF) < 0.05 and LD pruning (r2 > 0.99 within windows of 1 Mb inter-marker distances). Finally, a total of 2586 samples and 239,267 autosomal SNPs were retained for further analyses. All QC criteria were applied using the SNP & Variation Suite software (http://www.goldenhelix.com).
Univariate and bivariate association analyses
First, we performed univariate analyses to detect significant SNP associations for individual traits. The following univariate linear mixed model was applied:
where y is a n × 1 vector of phenotypic values of BW or EN for n = 2586 animals, W is a n × 53 matrix of covariates of fixed effects including hatch (36 classes) and mating group (17 classes), α is a c × 1 vector of the corresponding coefficients, x is a n × 1 vector of marker genotypes (coded as 0, 1, and 2 according to the number of copies of the minor allele), β is the effect size of marker on BW or EN, u is a vector of random polygenic effects and e is a vector of random residuals. The random effects were assumed to be normally distributed with zero means and the following covariance structure:
where \(\sigma_{u}^{2}\) and \(\sigma_{e}^{2}\) are the polygenic and error variance components, I is the nxn identity matrix, and G is the n x n genomic relationship matrix. Univariate analyses were performed using the factored spectrally transformed linear mixed model (FaST-LMM48) software (C++ Version 2.07) that was available at github (https://fastlmm.github.io/). Apart from SNP p values, FaST-LMM automatically computed the SNP q values using the false-discovery rate (FDR49) correction method.
A bivariate linear mixed model was then applied to identify significant CP SNP associations with both traits. Specifically, the following bivariate linear mixed model was used:
with U ~ MNnx2(0, G, Vg) and
where Y is a n × 2 matrix of 2 phenotypes for n = 2586 animals, W is a n × 53 matrix of covariates (fixed effects) including hatch (36 classes) and mating group (17 classes); A is a c × 53 matrix of the corresponding coefficients including the intercept; x is a n-vector of marker genotypes (coded as 0, 1, and 2 according to the number of copies of the minor allele), β is 2-vector of marker effect sizes for the 2 phenotypes; U is a n × 2 matrix of random effects and E is a n × 2 matrix of random residuals. Furthermore, G is the n x n genomic relationship matrix (estimated as centered genomic matrix7,50), Vg is a 2 × 2 symmetric matrix of genetic (co)variance, I is a n × n identity matrix, Ve is a 2 × 2 symmetric positive definite matrix of residual variance component and MNn×2(0, V1, V2) denotes the n × 2 matrix normal distribution with mean 0, row covariance matrix V1 (n × n) and column covariance matrix V2 (2 × 2).
Association of each SNP with both traits was assessed by testing the null hypothesis that the marker effect sizes for both phenotypes are zero i.e. H0: β = 0, where β is a vector of the two marker effects, against the general alternative hypothesis H1: β ≠ 0. The Wald test statistic was used to infer the significant CP SNP associations. The genetic correlation (rg) was also estimated between the two traits. Bivariate analysis was performed using the GEMMA51 software (version 0.98.1).
For each association analysis, the estimation of the genomic inflation factor (λ) was used to assess potential systematic bias due to population structure or the analytical approach52. If the λ value was greater than 1, it provided evidence for some systematic bias52. If the λ value was less than or equal to 1, no adjustment was needed53. λ was estimated using the SNP & Variation Suite software (http://www.goldenhelix.com).
Multiple-testing correction
For univariate association analyses, FaST-LMM corrected SNP p values for multiple comparisons, so there was no need for an additional correction, however, for bivariate analysis the FDR49 correction method was applied using R (http://www.r-project.org/). For all analyses, SNPs with FDR p values lower than 0.10 were considered as genome-wide significant.
Selection of LD-independent SNPs
Results obtained from univariate and bivariate analyses were further subject to stepwise conditional and joint (cojo) analysis using the ‘cojo-slct’ option and the GCTA54 tool to select the independent SNPs. The cojo-GCTA analysis corrects β and p values of neighboring SNPs (in a sliding window of 10 Mb) based on the LD between the SNPs. This ensures that the SNP with the lowest p value is selected first for conditioning the effect on neighboring loci based on the LD between the neighboring SNPs and the selected SNP. Following LD-based correction of effect, all SNPs that remained significant under a p value threshold (4.2e−06) are run through the same process in a stepwise manner. A p value threshold as high as 4.2e−06 (i.e. 1/total number of analyzed SNPs) was used here to declare the independent significant SNPs. In short, cojo analysis identifies: (1) the number of independent SNP signals in a region and (2) association signals due to the joint effect of several SNPs. To identify the independent CP associations, we used as input in the cojo-GCTA analysis the summary-level statistics obtained by the bivariate analysis. Specifically, the b estimates along with their standard errors were used to estimate t values for the SNPs and t values were finally converted to p values using R code (http://www.r-project.org/).
Effect prediction of the independent CP significant SNPs and detection of positional candidate genes and published QTLs
To predict the consequences of the independent CP significant SNPs on genes, transcripts, protein sequence and regulatory regions, the Variant Effect Predictor (VEP, https://www.ensembl.org/Tools/VEP55) tool was employed with the latest release (Ensembl release 102, accessed: 18 December 2020).
Physical positions of SNPs were also obtained by the VEP tool using the GRCg6a assembly (https://www.ensembl.org/Gallus_gallus/Info/Annotation, GenBank Assembly ID: GCA_000002315.5, accessed: 18 December 2020). The VEP tool was also used to search for positional candidate genes and for published QTLs including the independent CP significant SNPs. Note that both Ensembl and NCBI RefSeq transcript databases were used. With regard to published QTLs, VEP retrieves information via connections with Animal QTL database (Animal QTLdb) and Online Mendelian Inheritance in Animals (OMIA) database for Gallus gallus.
Bioinformatics analyses
We conducted ontological and functional analysis of the positional candidate genes in efforts to elucidate their functional role and relevance to the traits under study. First, modular enrichment analysis (MEA) using GeneCodis 4.0 (https://genecodis.genyo.es/56,57) was carried out. MEA removes the redundant terms and produces genes and annotations grouped in modules (or metagroups) which are functionally coherent and are ranked by their significance and relevance57. For MEA, we selected the species of Gallus gallus for the input genes and searched for Gene Ontology (GO) biological processes (BPs) as well as KEGG pathway significantly enriched concurrent annotations. Here, concurrent annotations with FDR p value lower than 0.05 were considered as significantly enriched.
Second, the LAGO tool (https://go.princeton.edu/cgi-bin/LAGO58) was used to infer the GO BP terms of the candidate genes. Since there were unknown genes during exploration of chicken GO annotations, the human GO annotations were used here. Computation of p values was based on the hypergeometric distribution and a p value cut-off equal to 0.10 was set as denoting significantly enriched terms. Candidate genes associated with enriched GO BP and pathways relevant to growth and reproduction processes were considered as functionally relevant to the traits under study and were thus nominated as candidate pleiotropic genes. Finally, the GO term Mapper (https://go.princeton.edu/cgi-bin/GOTermMapper) was employed to infer the GO slim molecular function terms of the pleiotropic candidate genes using human genes as input.
Ethical approval
All animals included in this study were not subjected to any invasive procedures.
Data availability
The data that support the findings of this study are available from Aviagen but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of Aviagen.
References
Sang, B. D. et al. Estimation of genetic parameters for economic traits in Korean native chickens. Asian-Australasian J. Anim. Sci. 19, 319–323 (2006).
Lwelamira, J., Kifaro, G. C. & Gwakisa, P. S. Genetic parameters for body weights, egg traits and antibody response against Newcastle Disease Virus (NDV) vaccine among two Tanzania chicken ecotypes. Trop. Anim. Health Prod. 41, 51–59 (2009).
Tongsiri, S. et al. Genetic parameters and inbreeding effects for production traits of Thai native chickens. Asian-Australasian J. Anim. Sci. 32, 930–938 (2019).
Cheverud, J. M. The Genetic architecture of pleiotropic relations and differential epistasis. In The Character Concept in Evolutionary Biology 411–433 (Elsevier, 2001). https://doi.org/10.1016/b978-012730055-9/50030-6.
Falconer, D. S. & Mackay, T. F. C. Introduction to Quantitative Genetics. (1996).
Solovieff, N., Cotsapas, C., Lee, P. H., Purcell, S. M. & Smoller, J. W. Pleiotropy in complex traits: Challenges and strategies. Nat. Rev. Genet. 14, 483–495 (2013).
Zhou, X. & Stephens, M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat. Methods 11, 407–409 (2014).
Li, X. & Zhu, X. Cross-phenotype association analysis using summary statistics from GWAS. In Methods in Molecular Biology, Vol 1666 455–467 (Humana Press Inc, 2017).
Tyler, A. L., Crawford, D. C. & Pendergrass, S. A. The detection and characterization of pleiotropy: Discovery, progress, and promise. Brief. Bioinform. 17, 13–22 (2016).
Medawar, P. B. An Unsolved Problem of Biology (H.K. Lewis, 1952).
Williams, G. C. Pleiotropy, natural selection, and the evolution of senescence. Evolution (N. Y.) 11, 398 (1957).
Ostrowski, E. A., Rozen, D. E. & Lenski, R. E. Pleiotropic effects of beneficial mutations in Escherichia coli. Evolution 59, 2343–2352 (2005).
Leiby, N. & Marx, C. J. Metabolic erosion primarily through mutation accumulation, and not tradeoffs, drives limited evolution of substrate specificity in Escherichia coli. PLoS Biol. 12, 20 (2014).
van Rheenen, W., Peyrot, W. J., Schork, A. J., Lee, S. H. & Wray, N. R. Genetic correlations of polygenic disease traits: From theory to practice. Nat. Rev. Genet. https://doi.org/10.1038/s41576-019-0137-z (2019).
Yuan, J. et al. Genome-wide association studies for feed intake and efficiency in two laying periods of chickens. Genet. Sel. Evol. 47, 82 (2015).
Liu, Z. et al. Genome-wide association analysis of age-dependent egg weights in chickens. Front. Genet. 9, 128 (2018).
Yi, G. et al. Genome-wide association study dissects genetic architecture underlying longitudinal egg weights in chickens. BMC Genom. 16, 746 (2015).
Tarsani, E. et al. Deciphering the mode of action and position of genetic variants impacting on egg number in broiler breeders. BMC Genom. 21, 512 (2020).
Tarsani, E., Theodorou, G., Palamidi, I. & Kominakis, A. Identification of candidate genes for body weight in broilers using extreme-phenotype genome-wide association study. Int. J. Genet. Genom. 8, 29 (2020).
Park, J. H. et al. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat. Genet. 42, 570–575 (2010).
Karlsson Linnér, R. et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat. Genet. 51, 245–257 (2019).
Revez, J. A. et al. Genome-wide association study identifies 143 loci associated with 25 hydroxyvitamin D concentration. Nat. Commun. 11, 1647 (2020).
Racki, W. J. & Richter, J. D. CPEB controls oocyte growth and follicle development in the mouse. Development 133, 4527–4537 (2006).
Li, Q. Transforming growth factor β signaling in uterine development and function. J. Anim. Sci. Biotechnol. 5, 210 (2014).
Valer, Sánchez-de-Diego, Pimenta-Lopes, Rosa, & Ventura,. ACVR1 function in health and disease. Cells 8, 1366 (2019).
Lambeth, L. S. et al. Anti-Müllerian hormone is required for chicken embryonic urogenital system growth but not sexual differentiation1. Biol. Reprod. 93, 20 (2015).
Johnson, P. A., Kent, T. R., Urick, M. E. & Giles, J. R. Expression and regulation of anti-Mullerian hormone in an oviparous species, the Hen1. Biol. Reprod. 78, 13–19 (2008).
Moreira, G. C. M. et al. Unraveling genomic associations with feed efficiency and body weight traits in chickens through an integrative approach. BMC Genet. 20, 20 (2019).
Lin, S., Svoboda, K. K. H., Feng, J. Q. & Jiang, X. The biological function of type i receptors of bone morphogenetic protein in bone. Bone Res. 4, 20 (2016).
Lovell, T. M., Knight, P. G. & Gladwell, R. T. Differential expression of mRNAs encoding the putative inhibin co-receptor (betaglycan) and activin type-I and type-II receptors in preovulatory and prehierarchical follicles of the laying hen ovary. J. Endocrinol. 188, 241–249 (2006).
Bornelöv, S. et al. Comparative omics and feeding manipulations in chicken indicate a shift of the endocrine role of visceral fat towards reproduction. BMC Genom. 19, 295 (2018).
Wang, L. et al. Cross-cancer pleiotropic analysis reveals novel susceptibility loci for lung cancer. Front. Oncol. 9, 1492 (2020).
Vivanco, I. & Sawyers, C. L. The phosphatidylinositol 3-kinase-AKT pathway in humancancer. Nat. Rev. Cancer 2, 489–501 (2002).
Ertel, S. I., Ertel, E. A. & Clozel, J. P. T-type Ca2+ channels and pharmacological blockade: Potential pathophysiological relevance. Cardiovasc. Drugs Ther. 11, 723–739 (1997).
Carter, M. T., McMillan, H. J., Tomin, A. & Weiss, N. Compound heterozygous CACNA1H mutations associated with severe congenital amyotrophy. Channels (Austin). 13, 153–161 (2019).
Berthier, C., Monteil, A., Lory, P. & Strube, C. α1H mRNA in single skeletal muscle fibres accounts for T-type calcium current transient expression during fetal development in mice. J. Physiol. 539, 681–691 (2002).
Lundt, A. et al. Cav3.2 T-type calcium channels are physiologically mandatory for the auditory system. Neuroscience 409, 81–100 (2019).
Bernhardt, M. L. et al. CaV3.2 T-type channels mediate Ca2+ entry during oocyte maturation and following fertilization. J. Cell Sci. 128, 4442–4452 (2015).
Vastagh, C., Solymosi, N., Farkas, I. & Liposits, Z. Proestrus differentially regulates expression of ion channel and calcium homeostasis genes in GnRH neurons of mice. Front. Mol. Neurosci. 12, 137 (2019).
Marques, P., Skorupskaite, K., George, J. T. & Anderson, R. A. Physiology of GNRH and gonadotropin secretion. Endotext 20, 20 (2018).
Zhang, Q. et al. Integrating transcriptome and genome re-sequencing data to identify key genes and mutations affecting chicken eggshell qualities. PLoS One 10, e0125890 (2015).
Mebratie, W., Reyer, H., Wimmers, K., Bovenhuis, H. & Jensen, J. Genome wide association study of body weight and feed efficiency traits in a commercial broiler chicken population, a re-visitation. Sci. Rep. 9, 20 (2019).
Jordan, D. M., Verbanck, M. & Do, R. HOPS: A quantitative score reveals pervasive horizontal pleiotropy in human genetic variation is driven by extreme polygenicity of human traits and diseases. Genome Biol. 20, 14 (2019).
Pasut, A., Matsumoto, A., Clohessy, J. G. & Pandolfi, P. P. The pleiotropic role of non-coding genes in development and cancer. Curr. Opin. Cell Biol. 43, 104–113 (2016).
Militello, G. et al. A novel long non-coding RNA myolinc regulates myogenesis through TDP-43 and Filip1. J. Mol. Cell Biol. 10, 102–117 (2018).
Li, Z., Ren, T., Li, W. & Han, R. Regulatory mechanism and application of lncRNAs in poultry. In Poultry (ed. Sath, D.) (IntechOpen, 2019). https://doi.org/10.5772/intechopen.83800.
Kranis, A. et al. Development of a high density 600K SNP genotyping array for chicken. BMC Genom. 14, 59 (2013).
Lippert, C. et al. FaST linear mixed models for genome-wide association studies. Nat. Methods 8, 833–835 (2011).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300 (1995).
Zhou, X., Carbonetto, P. & Stephens, M. Polygenic modeling with Bayesian sparse linear mixed models. PLoS Genet. 9, 20 (2013).
Zhou, X. & Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 44, 821–824 (2012).
Yang, J. et al. Genomic inflation factors under polygenic inheritance. Eur. J. Hum. Genet. 19, 807–812 (2011).
Hinrichs, A. L., Larkin, E. K. & Suarez, B. K. Population stratification and patterns of linkage disequilibrium. Genet. Epidemiol. 33, 20 (2009).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Carmona-Saez, P., Chagoyen, M., Tirado, F., Carazo, J. M. & Pascual-Montano, A. GENECODIS: A web-based tool for finding significant concurrent annotations in gene lists. Genome Biol. 8, R3 (2007).
Tabas-Madrid, D., Nogales-Cadenas, R. & Pascual-Montano, A. GeneCodis3: A non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. 40, W478–W483 (2012).
Boyle, E. I. et al. GO::TermFinder—Open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20, 3710–3715 (2004).
Yu, Y., Yao, W., Wang, Y. & Huang, F. shinyChromosome: An R/shiny application for interactive creation of non-circular plots of whole genomes. Genom. Proteom. Bioinform. 17, 535–539 (2019).
Oliveros, J. C. VENNY. An interactive tool for comparing lists with Venn Diagrams. http://bioinfogp.cnnb.csic.es/tools/venny/index.html (2015).
Acknowledgements
Aviagen is acknowledged for data provision and financial support.
Author information
Authors and Affiliations
Contributions
E.T. performed all the analyses and drafted the main manuscript text. A.K.R. and G.M. conducted data collection and preparation and contributed to writing the manuscript. A.L.H.T. assisted in identification of positional candidate genes and published QTLs. A.K. conceived and supervised the study and contributed to writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Tarsani, E., Kranis, A., Maniatis, G. et al. Detection of loci exhibiting pleiotropic effects on body weight and egg number in female broilers. Sci Rep 11, 7441 (2021). https://doi.org/10.1038/s41598-021-86817-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-86817-8
- Springer Nature Limited
This article is cited by
-
Simulation of dual-purpose chicken breeding programs implementing gene editing
Genetics Selection Evolution (2024)
-
Genetic and phenotypic parameter estimates for selection within Ugandan indigenous chickens
Tropical Animal Health and Production (2023)