
Abstract
The complete mitochondrial genome (mitogenome) of animals can provide useful information for evolutionary and phylogenetic analyses. The mitogenome of the genus Exhippolysmata (i.e., Exhippolysmata ensirostris) was sequenced and annotated for the first time, its phylogenetic relationship with selected members from the infraorder Caridea was investigated. The 16,350 bp mitogenome contains the entire set of 37 common genes. The mitogenome composition was highly A + T biased at 64.43% with positive AT skew (0.009) and negative GC skew (− 0.199). All tRNA genes in the E. ensirostris mitogenome had a typical cloverleaf secondary structure, except for trnS1 (AGN), which appeared to lack the dihydrouridine arm. The gene order in the E. ensirostris mitogenome was rearranged compared with those of ancestral decapod taxa, the gene order of trnL2-cox2 changed to cox2-trnL2. The tandem duplication-random loss model is the most likely mechanism for the observed gene rearrangement of E. ensirostris. The ML and BI phylogenetic analyses place all Caridea species into one group with strong bootstrap support. The family Lysmatidae is most closely related to Alpheidae and Palaemonidae. These results will help to better understand the gene rearrangements and evolutionary position of E. ensirostris and lay a foundation for further phylogenetic studies of Caridea.
Similar content being viewed by others

Introduction
The Decapoda is an ecologically and economically important order of crustaceans comprising a wide variety of crabs, lobsters, prawns and shrimps totalling over 18,000 extant and fossil species1,2. It is also the most abundant and largest order of crustaceans. Shrimps of the infraorder Caridea are commonly found in marine and freshwater habitats and have attracted attention due to their high commercial value3,4,5. Currently, there are 15 superfamilies recognized in the Caridea6. The family Lysmatidae is shown to comprise five genera, viz. Lysmata Risso, 1816; Ligur Sarato, 1885; Mimocaris, Nobili, 1903; Lysmatella Borradaile, 1915 and Exhippolysmata Stebbing, 1915. In the past, genetic studies of caridean families indicated that Hippolytidae is not a monophyletic taxa7,8 but should be partitioned into at least two families. Thereafter, morphological and genetic studies have recovered the Hippolytidae as polyphyletic, and the family Lysmatidae was formally resurrected9. Lysmatid shrimps are unique among crustaceans because of their enigmatic sexual system. They are protandric simultaneous hermaphrodites: shrimps initially mature and reproduce solely as males and later in life become functional simultaneous hermaphrodites10. In addition, due to their wide diversity of lifestyles, shrimp from the genus Exhippolysmata are particularly special.
The species Exhippolysmata ensirostris (Kemp 1914), which is widely distributed in the Pacific region, extends from the coast of the East China Sea and South China Sea to the Indo-West Pacific. It is an important and commercially exploited species in the East China Sea and the South China Sea. However, research on the genus Exhippolysmata has been limited to its species investigation and morphological description. Most of the research in Lysmatidae has focused on the genus Lysmata, including their mitochondrial genes and evolutionary relationships11,12,13,14,15,16. Consequently, research on the mitochondrial genes of the genus Exhippolysmata has rarely been reported.
The complete mitochondrial genome (mitogenome) is typically extrachromosomal and characterized by maternal inheritance and with a high evolution rate17. A complete mitogenome is a powerful tool for analysing the evolutionary history and phylogeny of species18. The mitogenome can also provide direct molecular clues for gene rearrangement processes, which would reveal important information for phylogenetic analyses19. The mitogenome of most metazoans is a double-stranded closed circular molecule approximately 11–20 kb in length. It typically contains 37 genes, including 13 protein coding genes (PCGs), two ribosomal RNA genes (16S rRNA and 12S rRNA) and 22 transporter RNA genes20.
In this study, the first complete mitogenome of the genus Exhippolysmata was described for the first time. We first successfully determined the complete mitogenome sequence of E. ensirostris using Illumina sequencing technology. We also analysed the nucleotide composition, codon usage profiles of protein coding genes (PCGs), Ka/Ks ratios of 13 PCGs, tRNA secondary structures, gene order and investigate the evolutionary relationships within Caridea. The purpose of this study was to understand the characteristics of the E. ensirostris mitogenome and clarify the evolutionary relationships within the Caridea mitogenome.
Results and discussion
Genome organization and base composition
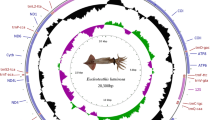
The complete mitogenome of E. ensirostris was found to be a typical circular molecule of 16,350 bp (Fig. 1), and the sequence was deposited in GenBank under accession number MK681888. The data that support the findings of this study are openly available in Microsoft OneDrive at (https://1drv.ms/w/s!Ag1aKdaw8CT3iHxX9f98FCkZvQ3n?e=BaRfdq). The newly sequenced mitogenome contains 13 PCGs, 22 tRNA genes, two rRNA genes and a large noncoding or control region (CR). Of the 37 genes, 23 were encoded on the heavy strand, and the other 14 were encoded on the light strand (Fig. 1, Table 1). The longest noncoding region was located between trnL2 and trnK, and the largest gene junction was located between trnL1 and 12S rRNA. The base compositions (Table 2) showed a high A + T content in the complete mitogenome (64.43%), PCGs (62.6%), tRNAs (66.04%), rRNAs (66.62%) and a CR (69.33%). The relative order of the nucleotide composition was A > T > C > G. The complete sequence had a positive AT skew (0.009) and a negative GC skew (− 0.199). As in other invertebrate mtDNAs, there were overlapping and noncoding bases between some genes.

Circular mitogenome map of Exhippolysmata ensirostris. Protein coding, ribosomal and tRNA genes are shown with standard abbreviations. Arrows indicate the orientation of gene transcription. The inner circles show the GC content and GC skew, which are plotted as the deviation from the average value of the entire sequence.
Protein coding genes and noncoding regions
The total length of the 13 PCGs was 10,852 bp and accounted for 66.3% of the whole E. ensirostris mitogenome. The 13 PCGs ranged from 165 bp (ATP8) to 1704 bp (nad5) (Tables 1, 2). Nine PCGs (cox1, cox2, cox3, nad2, nad3, nad6, atp8, atp6, and cob) were encoded on the heavy strand, and the other four PCGs (nad5, nad4, nad4 L and nad1) were encoded on the light strand (Table 1). Three genes (nad6, cox1 and cox3) were found to start with ATA, a further three (nad5, nad4 and nad4 L) with ATT, and the other seven with ATG. Eleven PCGs were found to end with the typical stop codon TAA, whereas cox1 and nad4 were found to end with TAG. Codon number and relative synonymous codon usage in the E. ensirostris mitochondrial genome are listed in Table 3. The patterns of codon usage of 13 PCGs are exhibited in Fig. 2A. The abundance of codon families and the relative synonymous codon usage (RSCU) in the PCGs were investigated for all available E. ensirostris mtDNAs, and the results are shown in Fig. 2B. The most frequently used codon was UUR (trnL2). There were 22 non-coding regions and eight overlaps of neighbouring genes in the mitochondrial genome of E. ensirostris. The largest non-coding region of E. ensirostris was identified as a putative control region. In addition, the position of the largest gene overlap (23 bp) was between trnL1 and 16S rRNA.

Codon usage patterns in the mitogenome of Exhippolysmata ensirostris CDspT, codons per thousand codons. Codon families are provided on the x axis (A); the relative synonymous codon usage (RSCU) (B).
To analyse the selection pressure on mitochondrial PCGs of the caridean shrimps, the ratio of the nonsynonymous and synonymous substitution rates (Ka/Ks) for the 13 PCGs from the six caridean species (E. ensirostris, Alpheus japonicas, Alvinocaris longirostris, Halocaridina rubra, Heterocarpus ensifer and Macrobrachium lanchesteri) was calculated. We found that the Ka/Ks values for all PCGs were lower than one (between 0.187 and 0.959), indicating that they are evolving under purifying selection (Fig. 3). Among all 13 caridean protein-coding genes, the average Ka/Ks of nad1 was the highest (0.959), and nad2 (0.941) and nad5 (0.927) also had very high average Ka/Ks values, indicating that these genes bear less selective pressure than other mitochondrial protein-coding genes.

The ratio of synonymous and nonsynonymous substitution rates (Ka/Ks) in all 13 mitochondrial PCGs of seven caridean shrimp. Ka: nonsynonymous substitution rate; Ks: synonymous substitution rate. The histogram represents the average Ka/Ks for each PCG.
Transfer and ribosomal RNA genes
The E. ensirostris mitochondrial genome encodes 22 tRNA genes, each of which was predicted to fold into a clover-leaf secondary structure that ranged in size from 64 bp (trnC) to 70 bp (trnV) of nucleotides (Table 1). The DHU arm of the trnS1 gene lacked any secondary structure (Fig. 4). The total length of the 22 tRNA genes in the E. ensirostris mitochondrial genome was 1446 bp. The overall A + T content of tRNA genes was 66.04%, which is similar to that of other carideans (Table 2)21. The mt tRNAs had a weakly positive AT skew (0.012) and positive GC skew (0.104). Fourteen tRNA genes (trnL2, trnK, trnD, trnG, trnA, trnR, trnN, trnS1, trnE, trnT, trnS2, trnI, trnM and trnW) were present on the heavy strand, and eight tRNA genes (trnF, trnH, trnP, trnL1, trnV, trnQ, trnC and trnY) were present on the light strand.

Putative secondary structures of tRNAs from the Exhippolysmata ensirostris mitogenome. The tRNAs are labelled with the abbreviations of their corresponding amino acids.
The 12S rRNA gene lay between trnL1 (CUN) and trnV, while the 16S rRNA gene lay between trnV and the putative control region, and both rRNA genes were encoded by the β-strand. As typically seen in other shrimp mitogenomes, the 16S rRNA and 12S rRNA genes of the E. ensirostris mitogenome were 1368 bp and 818 bp in length, respectively. The location and orientation of the rRNA genes were identical to the original arrangement of ancestral Caridea22. The A + T content of the two rRNA genes was 66.62%, and they had a negative AT skew (− 0.068, Table 2).
Gene rearrangement
Gene rearrangement in the Decapoda mitogenome commonly occurs and can be a tool to study phylogenetic relationships. Tan et al.19 gave an overview of mitochondrial gene orders (MGOs) of Decapoda, which revealed a large number of MGOs deviating from the ancestral arthropod ground pattern and unevenly distributed among infraorders. Here, we compared the MGOs of the Caridea mitogenomes with ancestral Decapoda and Caridea (Fig. 5). Among them, the MGOs in the mitogenomes of the families Pandalidae, Atyidae, and Alvinocarididae were identical to those of the ancestral Decapoda. However, fourteen carideans from the families of Lysmatidae, Alpheidae and Palaemonidae displayed gene rearrangements. This is in contrast with previous views that the gene order in Caridea is conserved23,24,25,26. Compared with the gene order of the ancestral Decapoda, E. ensirostris has a translocation, for which the gene order is trnL2-cox2 instead of cox2-trnL2 (Fig. 5C). Alpheus distinguendus, Alpheus hoplocheles, Alpheus inopinatus, Alpheus bellulus, Alpheus randalli and Alpheus japonicas in Alpheidae also undergo gene rearrangement, and trnE translocates and reverses with trnP27 (Fig. 5D). Alpheus lobidens has an extra duplication of trnQ located downstream of nad4l28 (Fig. 5E). In addition, the translocation of two tRNA genes was found in the mitochondrial genomes of Exopalaemon carinicauda, Palaemon annandalei, Palaemon capensis and Palaemon gravieri in Palaemonidae, wherein trnP or trnT were translocated, while the arrangement of other genes was identical29 (Fig. 5F). Palaemon sinensis in Palaemonidae has an extra translocation between trnG and trnE (Fig. 5G). The mitochondrial genome of Hymenocera picta in Palaemonidae bears a novel gene order, the gene block (nad1-trnL1-16S rRNA- trnV-12S rRNA-CR- trnI- trnQ) was rearranged from the downstream of trnS2 to the position downstream of nad4l (Fig. 5H). These data indicate that gene order is not conserved among caridean shrimp and could be useful for inferring phylogenetic relationships within Caridea when more mitochondrial data from Caridea become available in the future.

Linear representation of gene arrangements of an (A) ancestor of Decapoda; (B) ancestor of Caridea; (C) the Lysmatidae species Exhippolysmata ensirostris; (D) the Alpheidae species Alpheus distinguendus, Alpheus hoplocheles, Alpheus inopinatus, Alpheus bellulus, Alpheus randalli and Alpheus japonicas; (E) the Alpheidae species Alpheus lobidens; (F) the Palaemonidae species Exopalaemon carinicauda, Palaemon annandalei, Palaemon capensis and Palaemon gravieri; (G) the Palaemonidae species Palaemon sinensis; (H) the Palaemonidae species Hymenocera picta. All genes are transcribed from left to right. The green box indicated the duplicated gene. 16S rRNA and 12S rRNA are the large and small ribosomal RNA subunits, respectively.
Some mechanisms have been proposed to explain the rearrangement of genes in animal mitogenomes, including the tandem duplication/random loss model (TDRL)30, tandem duplication/non-random loss model (TDNL)31, and recombination32. Generally, TDRL is one of the most widely accepted mechanisms of mitochondrial gene rearrangement, which involves tandem duplication of gene regions caused by downstream chain mismatch during replication. TDNL attribute gene rearrangement to clustering by common polarity. The recombination within mitochondria mechanism involves the breaking and reconnecting of DNA double strands, leading to gene rearrangement and gene inversion33. Here, we propose that TDRL is more capable of explaining the cox2 and trnL2 translocations of the tRNA genes in the E. ensirostris mitochondrial genome.
Phylogenetic relationships
Many studies on the classification and evolutionary history of the Decapoda relied on morphological characteristics, which led to conflicting phylogenetic relationships. Under the best model, both ML and BI analyses of two data sets, based on the nucleotide sequences of the 13 PCGs and reconstruction of 53 species (including 51 Caridea species and two outgroup species) revealed the phylogenetic relationship between them. This study proposes a consistent phylogenetic relationship based on BI and ML methods; therefore, only one phylogenetic tree with both support values is presented (Fig. 6). Our results indicate that the mitochondrial genome sequence is robust for the inference of the relationships between shrimps. In addition, both ML and BI analyses of the two data sets show high branch support values. The phylogenetic tree based on the mitogenomes indicates that Palaemonidae and Alpheidae forme a monophyletic group and show a statistically significant relationship at the family level. Our complete mitogenome data suggest phylogenetic relationships among the major lineages of Caridea as ((((Alpheidae + Palaemonidae) + Lysmatidae) + Pandalidae) + Atyidae) + Alvinocarididae.

The phylogenetic tree was inferred from the nucleotide sequences of 13 mitogenome PCGs using BI and ML methods. Numbers on branches indicate posterior probability (BI) and bootstrap support (ML).
Although the main phylogenetic structures of our tree were consistent with those of previous result, some controversial findings were observed. Here, the families Alpheidae, Pandalidae, Lysmatidae and Palaemonidae clustered together as sister groups and were distantly related to Alvinocarididae, which supports the previous finding revealed by five nuclear genes (18S, Enolase, H3, NaK and PEPCK) in Li et al.8. However, Li et al.8 also revealed that Atyidae has been considered as basal lineages within the Caridea, which was conflict with our results. Based on both mitochondrial and nuclear genes (16S and 18S), Bracken et al. also revealed Atyidae represent basal lineages within the Caridea7. Meanwhile, in Sun et al.’s recent study, the phylogenetic relationship among Caridea was ((((Alpheidae + Palaemonidae) + Pandalidae) + Alvinocarididae) + Atyidae), which also considered Atyidae was distantly related to the four above families34. Furthermore, our result does not agree with Tan et al.35 and Wang et al.28, which state that Atyidae was the sister clade to Alvinocarididae. In our phylogenetic tree, most of the unstable and conflicting clades might have resulted from the limited taxon samples. The sequencing and assembly of the mitochondrial genome current result will promote the future work of further mitochondrial genome sequencing, and to increase in taxon sampling and genome sequencing which will help to resolve the classification of Caridea. Thus, more mitochondrial genome data will lead to a more comprehensive understanding of the phylogenetic relationships within Caridea and to resolve its classification.
Conclusions
Using next-generation sequencing methods, the mitogenome of E. ensirostris was determined to be a circular molecule of 16,350 bp. Compared with typical Decapoda mitogenomes, the gene order of this species had undergone a rearrangement, wherein cox2 and trnL2 were translocated to trnL2 and cox2. The gene rearrangement event occurring in E. ensirostris mitogenome can be explained by the TDRL model. The evolutionary patterns of PCGs were observed in the six caridean shrimp mitogenomes, which indicates that these genes were evolving under purifying selection. Phylogenetic analyses indicated the Caridea clades as monophyletic groups with strong bootstrap support. The family Lysmatidae is most closely related to Alpheidae and Palaemonidae. However, the lack of complete mitogenomes of other species of the Lysmatidae has limited the understanding of the evolution of this group at the genome level. Therefore, further studies are required to elucidate the phylogenetic status of species belonging to this group and their relationships.
Materials and methods
Sampling, identification and DNA extraction
An individual specimen of E. ensirostris was collected from Zhoushan, Zhejiang Province, China (30° 09′ 41″ N, 122° 35′ 10″ E) by bottom trawl fishery resource monitoring in November 2018. The specimen was identified morphologically and preserved in absolute ethanol. The total genomic DNA was extracted from muscle tissues of the specimen by the salt-extraction procedure with a slight modification36. Once extracted, the DNA was stored in 1 × TAE buffer at 4 °C. The extracted DNA was identified by 1.5% agarose gel electrophoresis and stored at − 20 °C.
Sequencing and assembly
The mitogenome of E. ensirostris was sequenced using next-generation sequencing by Origin Gene Co. Ltd., Shanghai, China. The mitogenome was sequenced from the total genomic DNA using an Illumina HiSeq X Ten platform to generate a library with an insert size of 400 bp. Then, the raw image data were converted into sequential data by base calling. A total of 5,515,049,137 bp of clean data and 37,141,698 clean reads were retrieved. Raw sequencing data were deposited into the Sequence Read Archive (SRA) database (SRR12199494) (http://www.ncbi.nlm.nih.gov/Traces/sra). De novo assembly of clean data without sequencing adapters was conducted using NOVOPlasty software (https://github.com/ndierckx/NOVOPlasty)37.
Mitochondrial genome annotation and analysis
Based on the sequence of the complete de novo assembled mitochondrial genome set, MITOS tools (http://mitos2.bioinf.uni-leipzig.de/index.py) was used for annotation with manual correction38. To ensure the accuracy of the assembled mitogenome, we first compared it to those of other Lysmatidae species and then further verified it using NCBI BLAST searches of the cox1 barcode sequence39. Base composition and relative synonymous codon usage (RSCU) values were calculated using MEGA v. 7.040. Identification of tRNA genes was verified using the MITOS WebServer. The rRNA genes were determined based on the locations of adjacent tRNA genes and by comparisons with other shrimp. Strand asymmetry was calculated using the formulae AT-skew = (A − T)/(A + T) and GC-skew = (G − C)/(G + C)41. The graphical map of the circular E. ensirostris mitogenome was drawn using the online mitochondrial visualization tool CGView Server42. In addition, we estimated the value of synonymous (Ks) and nonsynonymous substitutions (Ka) in the 13 mitochondrial PCGs using DnaSP 5.1.043. A Ka/Ks rate that is significantly less than one indicates negative (purifying) selective pressure, and a Ka/Ks rate that is significantly greater than 1 indicates positive selection44.
Phylogenetic analysis
A total of 51 caridean shrimp mitogenomes were downloaded from GenBank (https://www.ncbi.nlm.nih.gov/genbank/) for phylogenetic analysis (Table 4). The outgroup taxa were two Stenopodidea species: Stenopus hispidus and Spongiocaris panglao. We used the nucleotide sequences of the 13 protein coding genes (PCGs) to construct ML and BI phylogenetic trees. The 13 mitochondrial PCGs were aligned through MAFFT using default settings45, and then the resulting alignments were imported into Gblocks v. 0.91b (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) to select the conserved regions46. A substitution saturation analysis was performed in DAMBE v. 5.3.15 to test whether the dataset was suitable for constructing trees47. ML analysis was conducted using IQ-TREE v1.4.148 with the best-fit substitution model automatically selected by ModelFinder49 in the IQ-TREE package. GTR + I + G was selected as the best-fit model for nucleotide datasets under the Akaike Information Criterion (AIC) by MrModeltest 2.350, and then BI analysis was carried out using MrBayes 3.2.651 BI analysis was performed using default settings over four independent runs for 2 million generations sampled every 100 generations. The average standard deviation of split frequencies was < 0.01, the estimated sample size was > 200 and the potential scale reduction factor approached 1.0. The first 25% of samples were discarded as burn-in, and the remaining trees were used to calculate the Bayesian posterior probabilities for a 50% majority-rule consensus tree. All parameters were checked with Tracer v. 1.6 (http://tree.bio.ed.ac.uk/software/tracer/). The resulting phylogenetic trees were visualized in FigTree v. 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Data availability
The mitochondrial genome data has been submitted to NCBI GenBank under the following Accession Numbers MK681888.
References
De Grave, S. & Fransen, C. H. J. M. Carideorum Catalogus: The recent species of the dendrobranchiate, stenopodidean, procarididean and caridean shrimps (Crustacea: Decapoda). Zool. Med. Leiden 85(9), 195–588 (2011).
Shen, H., Braband, A. & Scholtz, G. Mitogenomic analysis of decapod crustacean phylogeny corroborates traditional views on their relationships. Mol. Phylogenet. Evol. 66(3), 776–789 (2013).
Chace, F. A. Jr. & Kensley, B. The cardiac notch in decapods. J. Crustacean Biol. 12(3), 442–447 (1992).
Holthuis, L.B., Fransen, C.H.J.M. & Van Achterberg, C. The recent genera of the Caridean and Stenopodidean shrimps (Crustacea, Decapoda): with an appendix on the order Amphionidacea. Nationaal Natuurhistorisch Museum, Leiden, pp. 6–328 (1993).
Martin, J. W. & Davis, G. E. An updated classification of the recent Crustacea. Los Angeles: Natural History Museum of County. Contrib. Sci. 39, 1–124 (2001).
De Grave, S. et al. A classification of living and fossil genera of decapod crustaceans. Raffles Bull. Zool. Suppl. 21, 1–109 (2009).
Bracken, H. D., De Grave, S. & Felder, D. L. Phylogeny of the infraorder Caridea based on mitochondrial and nuclear genes (Crustacea: Decapoda). Decapod Crustacean Phylogenet. 18, 274–298 (2009).
Li, C. P., De Grave, S., Lei, H. C., Chan, T. Y. & Chu, K. H. Molecular systematics of caridean shrimps based on five nuclear genes: Implications for superfamily classification. Zool. Anz. 250, 270–279 (2011).
De Grave, S., Li, C. P., Tsang, L. M., Chu, K. H. & Chan, T. Y. Unweaving hippolytoid systematics (Crustacea, Decapoda, Hippolytidae): Resurrection of several families. Zool. Scr. 43(5), 496–507 (2014).
Baeza, J. A. Protandric simultaneous hermaphroditism in the shrimps Lysmata bahia and Lysmata intermedia. Invertebr. Biol. 127(2), 181–188 (2008).
Baeza, J. A. & Bauer, R. T. Experimental test of social mediation of sex change in a protandric sequential hermaphrodite; the marine shrimp Lysmata wurdemanni (Crustacea: Caridea). Behav. Ecol. Sociobiol. 55, 544–550 (2004).
Xu, Y., Song, L. S. & Li, X. Z. The molecular phylogeny of Caridea based on 16S rDNA sequences. Mar. Sci. 29(9), 36–41 (2005).
Baeza, J. A. Testing three models on the adaptive significance of protandric simultaneous hermaphroditism in a marine shrimp. Evolution 60, 1840–1850 (2006).
Baeza, J. A. Molecular systematics of peppermint and cleaner shrimps: phylogeny and taxonomy of the genera Lysmata and Exhippolysmata (Crustacea: Caridea: Hippolytidae). Zool. J. Linn. Soc. Lond. 160, 254–265 (2010).
Baeza, J. A. Molecular phylogeny of broken-back shrimps (genus Lysmata and allies): A test of the ‘Tomlinson-Ghiselin’ hypothesis explaining the evolution of hermaphroditism. Mol. Phylogenet. Evol. 69, 46–62 (2013).
Baeza, J. A. & Prakash, S. An integrative taxonomic and phylogenetic approach reveals a complex of cryptic species in the ‘peppermint’ shrimp Lysmata wurdemanni sensu stricto. Zool. J. Linn. Soc. Lond. 185(4), 1018–1038 (2019).
Boore, J. L. Animal mitochondrial genomes. Nucleic Acids Res. 27(8), 1767–1780 (1999).
Liu, Q. N., Zhu, B. J., Dai, L. S., Wei, G. Q. & Liu, C. L. The complete mitochondrial genome of the wild silkworm moth, Actias selene. Gene 505(2), 291–299 (2012).
Tan, M. H. et al. Comparative mitogenomics of the Decapoda reveals evolutionary heterogeneity in architecture and composition. Sci. Rep. 9, 10756 (2019).
Gissi, C., Iannelli, F. & Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 101(4), 301–320 (2008).
Rich, A. & Rajbhandary, U. L. Transfer RNA: Molecular structure, sequence, and properties. Annu. Rev. Biochem. 45(1), 805–860 (1976).
Staton, J. L., Daehler, L. L. & Brown, W. M. Mitochondrial gene arrangement of the horseshoe crab Limulus polyphemus L.: Conservation of major features among arthropod classes. Mol. Biol. Evol. 14(8), 867–874 (1997).
Miller, A. D., Murphy, N. P., Burridge, C. P. & Austin, C. M. Complete mitochondrial DNA sequences of the decapod crustaceans Pseudocarcinus gigas (Menippidae) and Macrobrachium rosenbergii (Palaemonidae). Mar. Biotechnol. 7(4), 339–349 (2005).
Ivey, J. L. & Santos, S. R. The complete mitochondrial genome of the Hawaiian anchialine shrimp Halocaridina rubra Holthuis, 1963 (Crustacea: Decapoda: Atyidae). Gene 394(1–2), 35–44 (2007).
Wang, Z. et al. Complete mitochondrial genome of Parasesarma affine (Brachyura: Sesarmidae): Gene rearrangements in Sesarmidae and phylogenetic analysis of the Brachyura. Int. J. Biol. Macromol. 118, 31–40 (2018).
Lü, Z. M. et al. Complete mitochondrial genome of Ophichthus brevicaudatus reveals novel gene order and phylogenetic relationships of Anguilliformes. Int. J. Biol. Macromol. 135, 609–618 (2019).
Shen, X., Li, X., Sha, Z., Yan, B. & Xu, Q. Complete mitochondrial genome of the Japanese snapping shrimp Alpheus japonicus (Crustacea: Decapoda: Caridea): Gene rearrangement and phylogeny within Caridea. Sci. China Life Sci. 55(7), 591–598 (2012).
Wang, Q. et al. Characterization and comparison of the mitochondrial genomes from two Alpheidae species and insights into the phylogeny of Caridea. Genomics 112(1), 65–70 (2020).
Shen, X. et al. The complete mitochondrial genome of the ridgetail white prawn Exopalaemon carinicauda Holthuis, 1950 (Crustacean: Decapoda: Palaemonidae) revealed a novel rearrangement of tRNA genes. Gene 437(1–2), 1–8 (2009).
Moritz, C. & Brown, W. M. Tandem duplications in animal mitochondrial DNAs: Variation in incidence and gene content among lizards. Proc. Natl. Acad. Sci. 84(20), 7183–7187 (1987).
Lavrov, D. V., Boore, J. L. & Brown, W. M. Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: Duplication and nonrandom loss. Mol. Biol. Evol. 19(2), 163–169 (2002).
Lunt, D. H. & Hyman, B. C. Animal mitochondrial DNA recombination. Nature 387(6630), 247 (1997).
Dowton, M. & Campbell, N. J. H. Intramitochondrial recombination-is it why some mitochondrial genes sleep around?. Trends Ecol. Evol. 16(6), 269–271 (2001).
Sun, S., Cheng, J., Sun, S. & Sha, Z. Complete mitochondrial genomes of two deep-sea pandalid shrimps, Heterocarpus ensifer and Bitias brevis: Insights into the phylogenetic position of Pandalidae (Decapoda:Caridea). J. Oceanol. Limnol. 38(3), 816–825 (2020).
Tan, M. H., Gan, H. M., Lee, Y. P., Poore, G. C. & Austin, C. M. Digging deeper: new gene order rearrangements and distinct patterns of codons usage in mitochondrial genomes among shrimps from the Axiidea, Gebiidea and Caridea (Crustacea: Decapoda). Peer J. 5, e2982 (2017).
Aljanabi, S. M. & Martinez, I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 25, 4692–4693 (1997).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4), e18 (2017).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25(17), 3389–3402 (1997).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358 (1995).
Grant, J. R. & Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 36, 181–184 (2008).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Nielsen, R. Statistical tests of selective neutrality in the age of genomics. Heredity 86, 641–647 (2001).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14), 3059–3066 (2002).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17(4), 540–552 (2000).
Xia, X. & Xie, Z. DAMBE: Software package for data analysis in molecular biology and evolution. J. Hered. 92(4), 371–373 (2001).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32(1), 268–274 (2015).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Nylander, J. A., Ronquist, F., Huelsenbeck, J. P. & Nieves-Aldrey, J. Bayesian phylogenetic analysis of combined data. Syst. Biol. 53(1), 47–67 (2004).
Acknowledgements
This work was financially supported by National Key R&D Program of China (Grant No.: 2019YFD0901204), the Key R&D Program of Zhejiang Province (Grant No.: 2019C02056), the Open Foundation of Key Laboratory of Sustainable Utilization of Technology Research for Fishery Resource of Zhejiang Province (2020KF009) and the Project of Bureau of Science and Technology of Zhoushan Grant (No.: 2020C21026).
Author information
Authors and Affiliations
Contributions
Y.Y.Y. and K.D.X. designed the work, analyzed the data and wrote the paper, J.M and Y.H.G. analyzed the data, wrote the paper, and prepared the figures and tables. L.G. and L.H.J. analyzed the data, G.B.Y. and L.Z.M. reviewed drafts of the paper and supervised and directed the work. All authors gave final approval for the publication of the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ye, Yy., Miao, J., Guo, Yh. et al. The first mitochondrial genome of the genus Exhippolysmata (Decapoda: Caridea: Lysmatidae), with gene rearrangements and phylogenetic associations in Caridea. Sci Rep 11, 14446 (2021). https://doi.org/10.1038/s41598-021-93946-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-93946-7
- Springer Nature Limited
This article is cited by
-
Exploring the mitochondrial genome of Caridina pseudogracilirostris: a comparative analysis within the Atyidae Family
Molecular Biology Reports (2023)