
Abstract
Sarcoma is a rare malignancy with unfavorable prognoses. Accumulating evidence indicates that aberrant alternative splicing (AS) events are generally involved in cancer pathogenesis. The aim of this study was to identify the prognostic value of AS-related survival genes as potential biomarkers, and highlight the functional roles of AS events in sarcoma. RNA-sequencing and AS-event datasets were downloaded from The Cancer Genome Atlas (TCGA) sarcoma cohort and TCGA SpliceSeq, respectively. Survival-related AS events were further assessed using a univariate analysis. A multivariate Cox regression analysis was also performed to establish a survival-gene signature to predict patient survival, and the area-under-the-curve method was used to evaluate prognostic reliability. KOBAS 3.0 and Cytoscape were used to functionally annotate AS-related genes and to assess their network interactions. We detected 9674 AS events in 40,184 genes from 236 sarcoma samples, and the 15 most significant genes were then used to construct a survival regression model. We further validated the involvement of ten potential survival-related genes (TUBB3, TRIM69, ZNFX1, VAV1, KCNN2, VGLL3, AK7, ARMC4, LRRC1, and CRIP1) in the occurrence and development of sarcoma. Multivariate survival model analyses were also performed, and validated that a model using these ten genes provided good classifications for predicting patient outcomes. The present study has increased our understanding of AS events in sarcoma, and the gene-based model using AS-related events may serve as a potential predictor to determine the survival of sarcoma patients.
Similar content being viewed by others


Introduction
Sarcomas are rare and recalcitrant malignant mesenchymal tumors represented by more than 100 subtypes with different prognoses1. Sarcomas account for 1–3% of all malignancies and estimates suggest that approximately 15,610 new cases, and 6480 deaths, occur every year in the United States2. Sarcoma is often characterized by being occult, being difficult to diagnose early, having early and distant metastases, and having an overall 5-year survival rate of less than 15% for advanced-stage disease3. Despite the therapeutic advances being made, the heterogeneity of sarcomas makes them difficult to diagnose and their treatments complex and multidisciplinary. Therefore, a more in-depth understanding of sarcoma oncogenesis is essential to elucidate the underlying oncogenic mechanisms and to identify novel diagnostic and therapeutic biomarkers for sarcoma patients.
Accumulating evidence has demonstrated that aberrant regulation of gene expression is involved in sarcoma initiation and tumor progression4. Alternative splicing (AS) is a ubiquitous and important regulatory step in gene expression, enabling a fixed number of genes to generate a large variety of mature mRNAs. This increased transcript variety is also responsible for expanding the diversity of the transcriptome and the proteome. AS also has the potential to regulate gene expression, and therefore impact important molecular events for cellular differentiation and cell-type specific functions5. In the human genome, AS has been shown to play a vital role in the regulation of multiple molecular mechanisms and biological functions6. Aberrant AS events results in the inactivation of tumor-suppressing genes, promotion of angiogenesis, acceleration of proliferation, and inhibition of apoptosis7. Prominent in cancer, AS has now become a potential target for cancer therapeutics8. AS events are post-transcriptional processes that modulate gene expression, and are responsible for the increased proteomic diversity seen in a variety of cancers activities4,9. Aberrant AS events may also be relevant for explaining functional transformations in cancers7,10, often referred to as the "hallmarks of cancer" for prostate, ovarian, breast, colon, bladder, and lung cancer, among others11,12,13,14,15,16,17. AS events have also been reported to be involved in the regulation of apoptosis and autophagy genes18,19, tumor responses to chemotherapy, the orchestration of cancer stem-cell biology, and in epithelial-to-mesenchymal tumor transitions7,20,21. The regulators of RNA AS have the potential to become novel oncoproteins, contributing to dysregulation by modulating RNA isoforms in cancer-related pathways, and may be both biomarkers and targets for sarcoma diagnostics, prognostics, and treatments.
The Cancer Genome Atlas (TCGA) database contains a large number of gene profiles that can be used to investigate novel AS-related gene expression and prognosis data for cancer. In this study, we systematically analyzed AS events and constructed an AS-based gene model based on the sarcoma database in TCGA.
Results
The sarcoma landscape of AS events
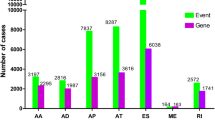
AS-event profiles were identified and analyzed in-depth from 236 sarcoma samples from the TCGA sarcoma cohort. We identified the seven AS patterns (ES, ME, RI, AP, AT, AD, and AA) listed in Fig. 1A. In total, we detected 9674 AS events in 40,184 genes, as illustrated in Fig. 1B. The ES pattern (15,311) was the most frequent AS-subtype, accounting for almost one-third of all events, followed by AT (8287), AP (7837), AA (3197), AD (2816), RI (2572), and ME events (164). Collectively, our findings demonstrate that these seven AS subtypes are frequently involved in sarcoma.

An overview of AS events in sarcoma. (A) The seven different subtypes of AS classification include ES, RI, AP, AT, AD, AA, and ME. (B) The relationship between AS events and their distribution among the seven subtypes. The ES events occurred in 15,311 genes, whereas the AT and AP events occurred in 8287 and 7837 genes, respectively. (C) Histogram showing the distribution among the seven types of splicing events that were significantly associated with overall survival prognoses. The ES, AT, and AP events accounted for the majority of the splicing events associated with overall survival prognoses. (D) Histogram showing the distribution among the seven types of splicing events that were associated with survival-related genes. The ES, AT and AP events presented a large amount of survival related genes. (E) Venn diagram showing the intersection between survival-related AS and survival-related genes. In total, we identified 267 genes related to survival.
Prognosis-associated AS events
To detect AS-event gene frequencies in sarcoma and survival, we integrated the clinical survival data from sarcoma patients. Using a univariate survival analysis, we obtained 2974 significant associations between prognoses and AS events (Fig. 1C), and 2311 significant associations between AS events and survival-related genes (Fig. 1D). We determined that 267 of these genes intersected between the AS-event cohort and the survival cohort (Fig. 1E). These results indicate that the majority of ES, AT, and AP events are significantly associated not only with overall prognoses for survival but also with survival genes.
AS-event subtypes indicate different prognoses
To determine the prognoses associated with the seven different AS-event subtypes, we calculated the relationships between AS subtypes and the landscape for overall survival genes. The seven AS-event subtypes were closely associated with overall survival prognoses (Fig. 2A), and an analysis of genes significantly related to AS events indicated that single genes could have a variety of AS events that are significantly associated with patient survival (Fig. 2B).

AS events are closely related to sarcoma prognoses. (A) The landscape of the seven subtypes of AS-associated genes that were significantly related to overall survival. (B) Distribution among the seven subtypes of AS-associated genes that were significantly related to survival genes. (C) AS-subtype area-under-the-curve (AUC) analyses for the classification of the top 15 AS-associated genes based on prognoses. (D) AS-subtype AUC analyses for prognosis classifications of the top 15 AS-associated genes based on multivariate modeling.
AS-related factors for prognosis predictions
To determine possible prognostic factors based on sarcoma AS events, we selected the 15 most significant genes to construct a survival regression model using area-under-the-curve (AUC) values. The results demonstrated that the seven AS-event subtypes all had high AUC values. For overall survival, the AUC values showed that AP (AUC = 0.902) was highly accurate and that AD (AUC = 0.879) and AT (AUC = 0.824) were well-suited for prognostic predictions. The other subtypes (AA: AUC = 0.777; RI: AUC = 0.761; ME: AUC = 0.796; and ES: AUC = 0.673) also had predictive AUC values (Fig. 2C), suggesting that AS-event subtypes associated with overall survival could serve as novel markers for prognostic predictions. The AP subtype also performed best in the variable-splicing associated with these 15 survival genes (Fig. 2D), indicating that analyses of AS events could be used as a new prognostic classification method for sarcoma.
AS event related gene interaction network construction
To further explore the interactions between survival-related AS events in sarcoma, we constructed a STRING database network using score values > 0.4. The 267 significantly associated overall survival genes, for the seven AS-event subtypes, were illustrated in Fig. 3. Using the protein–protein network construction for the corresponding genes involved in the regulatory network for each AS-event, the analysis showed that the AD and RI subtypes had the majority of interactions. The results also demonstrated that the prognosis-related genes associated with variable-splicing events had protein–protein interactions, with most of these genes involved in different biological functions.

The gene interaction network significantly related to overall survival. The different colors correspond to different AS-event subtypes, where yellow represents ES events and red represents ES events, according to the annotation on diagram. The ES event related genes, such as VEGFA, NME1, PTK2, and RFC5, showed significant interactions with the 267 genes associated with overall survival prognoses.
Gene functional analysis
To investigate the gene functions involved in the different types of variable-splicing events that were significantly related to patient prognoses, we performed a KEGG pathway-enrichment analysis22 (Fig. 4A). The ES and ME subtypes were involved in a variety of pathways. The ME subtype was correlated with platelet activation, focal adhesion, arginine and proline metabolism, and axon guidance, among others. The ES subtype was correlated with the regulation of the actin cytoskeleton, nucleotide excision repair, mismatch repair, and DNA replication, among others. The RI subtype was involved with ribosomes, the biosynthesis of amino acids, and Alzheimer’s disease, and the AP subtype was involved in metabolic pathways. The AA subtype was involved in amino-sugar and nucleotide-sugar metabolisms, and the AT subtype was involved in purine metabolism, cytosolic DNA-sensing pathways, and with RNA polymerase (Fig. 4B). These genes were also enriched in multiple disease-related pathways, suggesting their involvement in a multitude of biological functions through these seven subtypes of AS events that are significantly related to patient survival.

Functional annotations of AS events. (A) A KEGG enrichment analysis of the genes that were significantly related to overall survival. The AS events, such as ES, AP, RI, and ME, were the top 4 AS events that showed significant correlation with genes related to overall survival prognoses. (B) The KEGG enrichment results for the AS genes with the highest values for gene significance. AS events are closed linked to various biological functions. The results indicated that the ES and ME events are involved in complex pathway interactions, such as those related to nuclear, cancer, and immune cell pathways, among others.
Gene expression profiling and prognosis
To determine any correlations between gene expression and the prognoses for these variable-splicing events, that were significantly related to patient outcomes, we utilized a univariate survival analysis based on TCGA RNA-seq gene expression data. From this data, we used the 267 survival-related AS genes that were correlated with patient prognoses, and then Pearson correlations were performed to assess significance. We found that 149 (55.81%) of these genes were significantly related to variable-splicing (P < 0.05), indicating that the variable-splicing events of most genes were significantly related to their expression levels.
Hub gene selection
To determine the hub genes associated with AS events, we identified 21 genes with expression levels and AS-related Pearson correlation coefficients with absolute values > 0.5. A Cox multivariate analysis was then performed to identify independent prognostic factors. Using these methods, we identified ten genes (TUBB3, TRIM69, ZNFX1, VAV1, KCNN2, VGLL3, AK7, ARMC4, LRRC1, and CRIP1). To understand the relationship between potential hub genes and survival status, the risk scores for these 21 genes were calculated (Fig. 5A), and from these scores we established a ten-gene risk model. The resulting heat map revealed that genes ARMC4, LRRC1, and TUBB3 were overexpressed in the high-risk group, and that genes AK7 and CKNN2 were overexpressed in the low-risk group (Fig. 5B). Using the TCGA sarcoma dataset values, a survival status map was plotted to demonstrate the status for each sample (Fig. 5C). The risk scores, gene expression differences, and survival status for the ten hub genes, highlighted that AS events had the potential for prognosis predictions in sarcoma. We then constructed a multi-factor survival model and used it to analyze the variable-splicing events and expression-profile levels of these ten hub genes. The prognosis classifications are shown in Fig. 5D–G. This model demonstrated good prognostic classifications based on the RNA-seq and SpliceSeq data, and the AUC values were very high, suggesting that the ten genes of this model could be used as prognostic biomarkers for sarcoma.

AS gene-related risk scores and survival analyses. (A) Risk-score analysis of the sarcoma samples. (B) The gene expression heat map for the ten hub genes. Gene expression differences were significant between the high- and low-risk groups. (C) Relationship between survival time and survival status for each sarcoma sample. (D) Kaplan–Meier curves for overall survival and transcriptome gene expression levels. (E) Area-under-the-curve (AUC) multifactorial survival analysis related to overall patient survival based on gene expression at the transcriptome level. (F) Kaplan–Meier curves for overall survival and AS genes. (G) AUC multifactorial analysis of AS gene overall survival related to patient survival.
Hub gene expression and overall survival of patients with sarcoma
These ten hub genes also had predictive value for patient survival. As shown in the Fig. 6A–G, the elevated expression levels of KCNN2, CRIP1, AK7, ZNFX1, VAV1, TRIM69, and VGLL3 were significantly associated with a better overall patient survival. However, the Kaplan–Meier analyses showed that the increased expressions of LRRC1, ARMC4, and TUBB3 were significantly associated with unfavorable patient prognoses (Fig. 6H–J). Overall, the findings demonstrate that these hub genes may serve as novel biomarkers for use in prognostic predictions.

The association between the expression levels of the ten hub genes and overall survival of patients with sarcoma. (A–G) The upregulated expression levels of KCNN2, CRIP1, AK7, ZNFX1, VAV1, TRIM69, and VGLL3 were significantly associated with a better overall patient survival for sarcoma. (H–J) The upregulated expressions of LRRC1, ARMC4, and TUBB3 were significantly associated with poor prognoses in sarcoma patients.
Discussion
AS events frequently affect RNA binding, targeting specific RNA sequences or motifs which play important roles in gene expression regulation, cellular growth, development, tissue homeostasis, and RNA-species diversity8,14. AS events can be detected at the transcript level, using microarrays and RNA-seq data, and accumulating evidence indicates that aberrant RNA splicing patterns are associated with both the growth and progression of tumors23,24. Survival-related splicing factors are also important for AS events, as events that are favorable to overall survival were reported to be negatively associated with splicing factors in soft tissue sarcoma25. In Ewing sarcomas, hnRNPM-dependent AS promoted drug resistance and drove resistance to the inhibition of the PI3K/AKT/mTOR pathway26. It was reported that the majority of survival-associated AS events were also poor prognostic markers for sarcoma27. EWS-FLI1 was reported to play a crucial role in the AS-regulation process of Ewing sarcoma28. Consistent with previous studies, our analysis of the relationship between AS events and sarcoma-patient prognoses revealed that AS events were involved in sarcoma progression and may be potential predictors for sarcoma prognoses.
In this study we also identified ten prognosis-related genes (TUBB3, TRIM69, ZNFX1, VAV1, KCNN2, VGLL3, AK7, ARMC4, LRRC1, and CRIP1) closely associated with the occurrence and development of sarcoma. TUBB3, which codes for a microtubule protein, was overexpressed and linked to poor prognoses in a variety of cancers29,30. The function of TRIM69, a member of the tripartite motif (TRIM) family, has been reported to inhibit virus replication through a transcription-inhibition mechanism that prevented the synthesis of viral messenger RNAs31. ZNFX1, a novel lncRNA that has been shown to regulate cell proliferation, the cell cycle, cell migration, and cell invasion, may be a promising biomarker to predict poor prognoses in many cancers32,33,34. The ten hub genes were also involved with cancer initiation and progression, correlating with progression35. VAV1 has been reported to play a crucial role in the progression of human cancer, and AK7 expression has been positively correlated with malignant-cell proliferation in both acute lymphoblastic leukemia and Burkitt's B cell lymphoma36. ARMC4, an axonemal protein necessary for proper targeting, has also been identified among the novel genes associated with tumorigenesis in colorectal cancer37. LRRC1,a putative cell-polarity regulator, was significantly upregulated and considered to be a potential oncogene in hepatocellular carcinoma38. CRIP1 has been reported to be overexpressed in many cancer tissues and is considered to be an oncogene in tumor development and progression39.
The present ten gene-based model for prognostic predictions, focused on changes in AS events and changes in the expression levels of AS-related genes. The multivariate survival model assessed AS-related survival times for these ten hub genes, and successfully classified sarcoma prognoses. All of these hub genes are involved in a variety of oncogenesis and cancer progression functions, which indicates that this model may serve as a good predictor of survival prognoses for patients with sarcoma.
The present study verified that prognosis-associated AS events were ideal to construct a prognosis-prediction model. Using AS-related gene expression levels, we also identified the ten hub genes most relevant for this model. This novel model shows potential to contribute to both clinical and therapeutic approaches to better treat sarcoma.
Materials and methods
Data downloading and preprocessing
RNA-seq data from sarcoma AS events were downloaded from the SpliceSeq database (https://bioinformatics.mdanderson.org/TCGASpliceSeq/) compiled from TCGA. Data from a total of 261 samples were collected from the TCGA database (https://cancergenome.nih.gov/), including two normal tissue samples, and the RNA-seq gene expression profiles were from sarcomas and para-cancerous tissues. All represented patients had well-documented clinical information and follow-up data. The gene expression data in fragments per kilobase of exon model per million reads mapped (FPKM) were transformed into transcripts per million (TPM) values.
The corresponding gene profiles from the human genome, version GRCh38.p2 from GENECODE, were processed using gene ID conversion. In total, 236 protein-coding genes common to both the SpliceSeq and RNA-seq cohorts were identified. Lastly, we obtained the expression data for 19,754 genes for further analyses.
Analyses of RNA-seq data and AS events
SpliceSeq is a java application for the visualization and quantitation of splice junctions and exon proportions included in TCGA data. We defined seven AS subtypes for sarcomas: exon skip (ES), mutually exclusive exons (ME), retained intron (RI), alternate promoter (AP), alternate terminator (AT), alternate donor site (AD), and alternate acceptor site (AA).
Selection of survival-related AS events
Analyses of patient survival data were made using the survival package in R software (http://cran.r-project.org/package=survival), and the level of significance was set at P < 0.05.
Network analysis for AS-related genes
For the analysis of gene-network interactions, we used the STRING database (http://string-db.org/), and applied a threshold score > 0.4. Data visualization was performed using Cytoscape (https://cytoscape.org/).
A prognosis model based on ten hub genes
We performed a multivariate analysis of the gene expression data using the Cox proportional hazard-regression model. We identified the ten AS-related genes that demonstrated the most significant expression-profile changes, and then used these hub genes to create a prognostic model and for survival analyses. In addition, we determined the correlations between the expression profiles of these ten hub genes and overall patient survival based on Kaplan–Meier plots (https://kmplot.com/).
Correlations between AS subtypes and prognoses
We systematically explored patient prognoses associated with AS-subtype events and calculated AS-event distributions. We then determined the correlations between prognosis-related genes and these AS-event distributions.
Biological functions analysis
To determine the functions of the prognostic genes that were significantly related to AS events, we used KOBAS 3.0 (http://kobas.cbi.pku.edu.cn/), a web-based tool for gene/protein functional annotation and pathway-enrichment analysis.
Prognostic model construction
Characteristic genes were selected as being significantly prognostic with Pearson correlation coefficients of more than 0.5 or less than − 0.5. To construct our prognostic model, we used these prognostic genes and the frequencies of AS events, to develop a sarcoma prognostic index.
An analysis of prognosis-related genes
To determine correlations between levels of gene transcription and variable-splicing prognoses, we used a univariate analysis of AS-sequence profiles using TCGA RNA-seq data for associated genes to determine the impact of AS events on sarcoma prognosis.
Data availability
All of the data involved in this study are available in the public databases which are listed in the “Materials and methods” section.
References
Alavi, S., Florou, V., Tinoco, G., Trent, J. & Wilky, B. A precision medicine approach in sarcoma: Identification of patients who may benefit from early use of pazopanib. Discov. Med. 25, 131–144 (2018).
Karakousis, G. Update for ASCO 2015 sarcoma sessions. Surg. Oncol. 24, 369–370. https://doi.org/10.1016/j.suronc.2015.08.006 (2015).
Martin-Liberal, J. et al. Targeting protein kinase C in sarcoma. Biochim. Biophys. Acta 1846, 547–559. https://doi.org/10.1016/j.bbcan.2014.10.002 (2014).
Li, J., Shen, J., Wang, K., Hornicek, F. & Duan, Z. The roles of sox family genes in sarcoma. Curr. Drug Targets 17, 1761–1772. https://doi.org/10.2174/1389450117666160502145311 (2016).
Cieply, B. & Carstens, R. Functional roles of alternative splicing factors in human disease. Wiley Interdiscip. Rev. RNA 6, 311–326. https://doi.org/10.1002/wrna.1276 (2015).
Song, Q. et al. CRKL regulates alternative splicing of cancer-related genes in cervical cancer samples and HeLa cell. BMC Cancer 19, 499. https://doi.org/10.1186/s12885-019-5671-8 (2019).
Pradella, D., Naro, C., Sette, C. & Ghigna, C. EMT and stemness: Flexible processes tuned by alternative splicing in development and cancer progression. Mol. Cancer 16, 8. https://doi.org/10.1186/s12943-016-0579-2 (2017).
Urbanski, L., Leclair, N. & Anczuków, O. Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip. Rev. RNA 9, e1476. https://doi.org/10.1002/wrna.1476 (2018).
Hyung, D., Kim, J., Cho, S. & Park, C. ASpedia: A comprehensive encyclopedia of human alternative splicing. Nucleic Acids Res. 46, D58–D63. https://doi.org/10.1093/nar/gkx1014 (2018).
Montes, M., Sanford, B., Comiskey, D. & Chandler, D. RNA splicing and disease: Animal models to therapies. Trends Genet. TIG 35, 68–87. https://doi.org/10.1016/j.tig.2018.10.002 (2019).
Zhang, Y. et al. Pan-cancer analysis of clinical relevance of alternative splicing events in 31 human cancers. Oncogene 38, 6678–6695. https://doi.org/10.1038/s41388-019-0910-7 (2019).
Olender, J. & Lee, N. Role of alternative splicing in prostate cancer aggressiveness and drug resistance in African Americans. Adv. Exp. Med. Biol. 1164, 119–139. https://doi.org/10.1007/978-3-030-22254-3_10 (2019).
Zhu, J., Chen, Z. & Yong, L. Systematic profiling of alternative splicing signature reveals prognostic predictor for ovarian cancer. Gynecol. Oncol. 148, 368–374. https://doi.org/10.1016/j.ygyno.2017.11.028 (2018).
Yang, Q., Zhao, J., Zhang, W., Chen, D. & Wang, Y. Aberrant alternative splicing in breast cancer. J. Mol. Cell. Biol. 11, 920–929. https://doi.org/10.1093/jmcb/mjz033 (2019).
Xiong, W. et al. Genome-wide profiling of chemoradiation-induced changes in alternative splicing in colon cancer cells. Oncol. Rep. 36, 2142–2150. https://doi.org/10.3892/or.2016.5022 (2016).
Xie, R. et al. Polypyrimidine tract binding protein 1 promotes lymphatic metastasis and proliferation of bladder cancer via alternative splicing of MEIS2 and PKM. Cancer Lett. 449, 31–44. https://doi.org/10.1016/j.canlet.2019.01.041 (2019).
Chang, H. & Lin, J. SRSF1 and RBM4 differentially modulate the oncogenic effect of HIF-1α in lung cancer cells through alternative splicing mechanism. Biochim. Biophys. Acta Mol. Cell Res. 1866, 118550. https://doi.org/10.1016/j.bbamcr.2019.118550 (2019).
Paronetto, M., Passacantilli, I. & Sette, C. Alternative splicing and cell survival: From tissue homeostasis to disease. Cell Death Differ. 23, 1919–1929. https://doi.org/10.1038/cdd.2016.91 (2016).
Chen, Y. et al. Starvation-induced suppression of DAZAP1 by miR-10b integrates splicing control into TSC2-regulated oncogenic autophagy in esophageal squamous cell carcinoma. Theranostics 10, 4983–4996. https://doi.org/10.7150/thno.43046 (2020).
Pal, S., Gupta, R. & Davuluri, R. Alternative transcription and alternative splicing in cancer. Pharmacol. Ther. 136, 283–294. https://doi.org/10.1016/j.pharmthera.2012.08.005 (2012).
Bates, D., Morris, J., Oltean, S. & Donaldson, L. Pharmacology of modulators of alternative splicing. Pharmacol. Rev. 69, 63–79. https://doi.org/10.1124/pr.115.011239 (2017).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49, D545–D551. https://doi.org/10.1093/nar/gkaa970 (2021).
Climente-González, H., Porta-Pardo, E., Godzik, A. & Eyras, E. The functional impact of alternative splicing in cancer. Cell Rep. 20, 2215–2226. https://doi.org/10.1016/j.celrep.2017.08.012 (2017).
Martinez-Montiel, N., Rosas-Murrieta, N., Anaya Ruiz, M., Monjaraz-Guzman, E. & Martinez-Contreras, R. Alternative splicing as a target for cancer treatment. Int. J. Mol. Sci. https://doi.org/10.3390/ijms19020545 (2018).
Yang, X. et al. Determining the prognostic significance of alternative splicing events in soft tissue sarcoma using data from The Cancer Genome Atlas. J. Transl. Med. 17, 283. https://doi.org/10.1186/s12967-019-2029-6 (2019).
Passacantilli, I., Frisone, P., De Paola, E., Fidaleo, M. & Paronetto, M. hnRNPM guides an alternative splicing program in response to inhibition of the PI3K/AKT/mTOR pathway in Ewing sarcoma cells. Nucleic Acids Res. 45, 12270–12284. https://doi.org/10.1093/nar/gkx831 (2017).
Hong, W. et al. Genome-wide profiling of prognosis-related alternative splicing signatures in sarcoma. Ann. Transl. Med. 7, 557. https://doi.org/10.21037/atm.2019.09.65 (2019).
Selvanathan, S. et al. EWS-FLI1 modulated alternative splicing of ARID1A reveals novel oncogenic function through the BAF complex. Nucleic Acids Res. 47, 9619–9636. https://doi.org/10.1093/nar/gkz699 (2019).
Sekino, Y. et al. TUBB3 reverses resistance to docetaxel and cabazitaxel in prostate cancer. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20163936 (2019).
Sekino, Y. et al. TUBB3 is associated with high-grade histology, poor prognosis, p53 expression, and cancer stem cell markers in clear cell renal cell carcinoma. Oncology https://doi.org/10.1159/000506775 (2020).
Kueck, T. et al. Vesicular stomatitis virus transcription is inhibited by TRIM69 in the interferon-induced antiviral state. J. Virol. https://doi.org/10.1128/jvi.01372-19 (2019).
Xian, H., Zhuo, Z., Sun, Y., Liang, B. & Zhao, X. Circulating long non-coding RNAs HULC and ZNFX1-AS1 are potential biomarkers in patients with gastric cancer. Oncol. Lett. 16, 4689–4698. https://doi.org/10.3892/ol.2018.9199 (2018).
Wu, J., Zhang, G. & Sun, X. LncRNA ZNFX1-AS1 targeting miR-193a-3p/SDC1 regulates cell proliferation, migration and invasion of bladder cancer cells. Eur. Rev. Med. Pharmacol. Sci. 24, 4719–4728. https://doi.org/10.26355/eurrev_202005_21160 (2020).
Shi, L. et al. Long non-coding RNA ZNFX1-AS1 promotes the tumor progression and metastasis of colorectal cancer by acting as a competing endogenous RNA of miR-144 to regulate EZH2 expression. Cell Death Dis. 10, 150. https://doi.org/10.1038/s41419-019-1332-8 (2019).
Kang, L. et al. Elevated level of Vav1 was correlated with advanced biological behavior and poor prognosis in patients with gastric cancer. Int. J. Clin. Exp. Pathol. 11, 391–398 (2018).
Huang, P. et al. TGFβ promotes mesenchymal phenotype of pancreatic cancer cells, in part, through epigenetic activation of VAV1. Oncogene 36, 2202–2214. https://doi.org/10.1038/onc.2016.378 (2017).
Liang, Y. et al. Discovery of aberrant alteration of genome in colorectal cancer by exome sequencing. Am. J. Med. Sci. 358, 340–349. https://doi.org/10.1016/j.amjms.2019.07.012 (2019).
Li, Y., Zhou, B., Dai, J., Liu, R. & Han, Z. Aberrant upregulation of LRRC1 contributes to human hepatocellular carcinoma. Mol. Biol. Rep. 40, 4543–4551. https://doi.org/10.1007/s11033-013-2549-8 (2013).
He, G. et al. Cysteine-rich intestinal protein 1 silencing alleviates the migration and invasive capability enhancement induced by excessive zinc supplementation in colorectal cancer cells. Am. J. Transl. Res. 11, 3578–3588 (2019).
Acknowledgements
We would like to thank the investigators who participated in KEGG work.
Author information
Authors and Affiliations
Contributions
All authors had access to the data and reviewed the manuscript. H.L., J.Y. and N.W. designed the study. P.Q., J.R. and Y.M. collected data. H.L., J.Y. and G.Y. performed statistical analyses, H.L. and J.Y. drafted the manuscript. H.L. made the final decision of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, H., Yang, J., Yang, G. et al. Identification of prognostic alternative splicing events in sarcoma. Sci Rep 11, 14949 (2021). https://doi.org/10.1038/s41598-021-94485-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-94485-x
- Springer Nature Limited
This article is cited by
-
Outer dynein arm docking complex subunit 2 polymorphism rs7893462 modulates hepatocellular carcinoma susceptibility and can serve as an overall survival biomarker for hepatitis B virus-related hepatocellular carcinoma after hepatectomy: a cohort study with a long-term follow-up
World Journal of Surgical Oncology (2023)