
Abstract
Despite effective lipid reduction and corresponding benefits for cardiovascular disease prevention and treatment, statins have pleiotropic effects potentially increasing the risk of ischemic heart disease (IHD), particularly by increasing body mass index (BMI). We assessed whether the effects of genetically mimicked statins on IHD were strengthened by adjusting for BMI in men and women. We also assessed if increasing BMI was specific to statins in comparison to other major lipid-lowering treatments in current use, i.e., proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors and ezetimibe. Using univariable and multivariable Mendelian randomization (MR) we found genetically mimicked effects of statins increased BMI (0.33, 95% confidence interval (CI) 0.28 to 0.38), but genetically mimicked PCSK9 inhibitors and ezetimibe did not. Genetically mimicked effects of statins on IHD reduction in both sexes (odds ratio (OR) 0.55 per unit decrease in effect size of low-density lipoprotein cholesterol (LDL-c), 95% confidence interval (CI) 0.40 to 0.76), was largely similar after adjusting for BMI, in both men (OR 0.48, 95% CI 0.38 to 0.61) and women (OR 0.66, 95% CI 0.53 to 0.82). Compared with variations in PCSK9 and NPC1L1, only variation in HMGCR was associated with higher BMI. The effects on IHD of mimicking statins were similar after adjusting for BMI in both men and women. The BMI increase due to statins does not seem to be a concern as regards the protective effects of statins on IHD, however other factors driving BMI and the protective effects of statins could be.
Similar content being viewed by others


Introduction
Statins reduce low-density lipoprotein cholesterol (LDL-c) by inhibiting 3-hydroxy-3-methylglutaryl–coenzyme A reductase (HMGCR), giving a corresponding reduction in morbidity and mortality from cardiovascular disease (CVD)1,2. Despite effective lipid modification, pleiotropic effects, possibly increasing the risk of CVD3,4 have also been noticed during long-term use of statins. Specifically, statin treatment may increase risk of type 2 diabetes5 and is associated with higher body mass index (BMI)3,4, both of which increase CVD risk6,7. Effects on adiposity and glucose metabolism may influence the choice of lipid-lowering treatment3 and statin adherence8.
Genetically predicted proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, ezetimibe and statins similarly reduce the risk of cardiovascular events per unit reduction in LDL-c5, but only statins are thought to increase BMI. Given statins increase BMI, this suggests greater effects of statins than other lipid modifiers per unit change in LDL-c. More importantly, whether these associations manifest differently by sex remains unknown, although differences by sex are evident for some effects of statins9. Sex-specific information may contribute to understanding the pleiotropic effects of statins and inform their use, particularly for prevention where the risks and benefits can be finely balanced, and differ by sex10.
We used Mendelian randomization (MR) to obtain unconfounded sex-specific estimates of the effects of genetically mimicked statins on IHD allowing for their effects on BMI using the largest available sex-specific genome-wide association studies (GWAS). Specifically, for IHD and LDL-c we used GWAS of the UK Biobank cohort study provided by Neale lab (http://www.nealelab.is/uk-biobank/) and for BMI a GWAS of the UK Biobank and the Genetic Investigation of ANthropometric Traits (GIANT) consortium11. To identify if the BMI increase was unique to statins, we also sex-specifically assessed whether this effect was evident for the other two common lipid modifiers, PCSK9 inhibitors and ezetimibe. Finally, we replicated the analysis using genetic predictors of LDL-c and genetic associations with IHD from other GWAS.
Results
We used a single nucleotide polymorphisms (SNP) rs12916 to mimic statins. In sensitivity analysis, the six correlated SNPs from HMGCR (Supplementary Table S1-2, r2 > 0.13) were used taking account of their correlations. Of the 7 SNPs mimicking effects of PCSK9 inhibitors from PCSK9 (Supplementary Table S1 and Supplementary Table S3), the three independent (r2 < 0.05) SNPs (rs11206510, rs2149041 and rs7552841) were used in the main analysis (Fig. 1). Of the 5 correlated SNPs mimicking effects of ezetimibe from NPC1L1 (Supplementary Table S1 and Supplementary Table S4, r2 > 0.05), rs10260606 (proxy of rs2073547, r2 = 0.99) was used in the main analysis given it had the strongest association with LDL-c (Fig. 1). These associations with LDL-c for the SNPs mimicking statins, PCSK9 inhibitors and ezetimibe are given overall (Supplementary Table S5), in men (Supplementary Table S6) and in women (Supplementary Table S7).

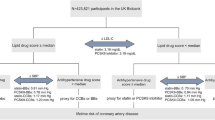
Selection of genetic instruments for lipid modifiers and BMI.
Instrument strength
The F statistic for most SNPs used to genetically mimic the effects of statins, PCSK9 inhibitors and ezetimibe were > 10 for men, women and overall (Supplementary Table S5-7). The F statistics for the 264 SNPs (men), 314 SNPs (women) and 530 SNPs (overall) predicting BMI were all greater than 10, with median 49.8, 42.2 and 41.8 respectively.
Associations of genetically mimicked lipid modifiers with BMI
Genetically mimicked effects of statins increased BMI in men and women whereas genetically mimicked effects of PCSK9 inhibitors and of ezetimibe were not associated with BMI (Fig. 2). Further analysis for PCSK9 inhibitors and ezetimibe was not conducted, given their lack of association with BMI.

Flowchart of the analyses and populations used in the study.
Associations of genetically mimicked effects of statins with BMI and IHD
Genetically mimicked effects of statins reduced the risk of IHD overall (odds ratio (OR) 0.55, 95% CI 0.40 to 0.76), and in men and women (Table 1). Genetically predicted BMI was positively associated with IHD for both sexes, with a slightly greater effect in women (OR 1.65, 95% CI 1.45 to 1.89) than in men (OR 1.40, 95% CI 1.26 to 1.54). However, MR-Egger intercepts for BMI suggested pleiotropic effects for women (P intercept = 0.005), where the MR-Egger estimate was much smaller than the IVW estimate (Table 1). However, the IGX2 was low (less than 90%; 89.8% overall, 85.0%, 84.9% for men and women, respectively), suggesting the violation of the NOME assumption for the MR-Egger estimates which were biased towards the null12, and that the MR-Egger estimates may not be reliable for one-sample MR13. The weighted median, consistent between one-sample and two-sample MR13, showed positive associations of BMI with IHD in both sexes (Table 1).
In multivariable MR the conditional F-statistics for effects of genetically mimicked statins were 10.8 overall, 5.7 for men and 11.9 for women and for BMI were 61.8 overall, 31.6 for men and 56.6 for women. The multivariable MR-Egger intercept was significant overall and for men (P = 0.002, 2.33*10–8, respectively) and the corresponding modified Q statistic was also significant (877.5 and 435.3, respectively; P values < 0.001), so we focused on the MR-Egger estimates overall (OR 0.60, 95% CI 0.52 to 0.69) and for men (OR 0.48, 95% CI 0.38 to 0.61). The Q statistic (399.6, P < 0.001) also suggested pleiotropic effects for women, althought the MR-Egger intercept was not significant, where the MR-Egger estimate was also used (OR 0.66, 95% CI 0.53 to 0.82).
Compared with the univariable estimates for genetically mimicked effects of statins on IHD, the multivariable estimates (OR) adjusted for BMI were slightly smaller for men using MR-Egger, and were slightly larger for women and overall using MR-Egger (Table 2 and Fig. 4). Sensitivity analysis including correlated SNPs showed similar estimates as above for both univariable and multivariable MR (Figs. 3, 4 and Tables 1, 2).

Forest plot for univariable MR estimates comparing the effects of genetically mimicked statins, PCSK9 inhibitors and ezetimibe on BMI. The red colour represents the main analysis using independent SNPs and the blue colour represents the sensitivity analysis using correlated SNPs. Assuming the UK Biobank standard deviations for LDL-c (0.871 mmol/L), BMI (4.77 kg/m2) and height (1.70 m), for each one mmol/L lower LDL-c from statin use for overall (Beta statin-BMI = 0.33), BMI increases by 1.81 kg/m2, and weight increases by 5.2 kg.

Forest plot for main analysis comparing the univariable MR estimates with the multivariable MR estimates for the effects of genetically mimicked statins and BMI on IHD. In the main analysis, the lead SNP, rs12916 was used for mimicking statins. The MR-Egger method was used for presenting the univariable MR estimates, given that the multivariable MR-Egger estimates were suggested for overall, men and women.
Replication
We replicated the primary results using a different GWAS, i.e., GLGC for LDL-c, UK Biobank and GIANT14 seperately for BMI and CARDIoGRAMplusC4D for IHD which had fewer SNPs for BMI. Similarly, genetically mimicked effects of statins increased BMI, whereas these effects were not evident for PCSK9 inhibitors or ezetimibe (Table 3). Genetically mimicked effects of statins were negatively associated with IHD, while BMI were positively associated with IHD (Table 4). Compared with the univariable estimates, the multivariable estimates (OR) for effects of statins on IHD adjusting for BMI were largely similar (Table 5).
Discussion
Consistent with previous findings our study provides genetic evidence that statins raise BMI3,4, while PCSK9 inhibitors or ezetimibe do not affect BMI3,15. BMI increases the risk of IHD in both sexes16,17. Our study adds by showing that the BMI increase caused by statins may not detract from the protective effects of mimicking statins on IHD in both men and women.
Although statins are known to increase BMI, the underlying mechanism remains unknown. Previous studies have shown dietary changes, particularly higher caloric intake for statin users18. Compared with statins, the other two lipid modifiers, PCSK9 inhibitors and ezetimibe however, did not affect BMI, which implies that higher BMI is not a consequence of lipid lowering, particularly of LDL-c lowering. Differences in effects on BMI for the three lipid modifiers may be related to their mechanisms of action. PCSK9 inhibitors enhance the clearance of LDL-c by increasing LDL receptors19, while ezetimibe reduces the cholesterol absorption20. In contrast, statins inhibit cholesterol synthesis21, an upstream bioprocess involving de novo synthesis in Leydig cells, which affects steroidogenesis22,23. Genetic scores for HMGCR, PCSK9 and NPC1L1 have been similarly associated with a slightly increased risk of diabetes24, and LDL-c lowering genetic variants located around genes targeted by lipid-lowering therapy have been positively associated with risk of type 2 diabetes25. Taken together these studies indicate detrimental pleiotropic effects of these variants on diabetes as a possible consequence of lipid lowering, which is different from our findings, suggesting statins specifically increase BMI, possibly indicating a different underlying pathway.
Although the estimates for BMI on IHD were not entirely consistent, it is likely that BMI increases the risk of IHD, with possibly more convincing estimates for men than women, which may also be partially explained by the hormone changes resulting from higher BMI. BMI raises plasma insulin in both sexes26, resulting in testosterone stimulation in vivo27, possibly via gonadotropin releasing hormone28. Testosterone stimulation is more relevant in men than in women given that men have higher testosterone than women29. Meanwhile, testosterone increases the risk of IHD more in men than women9. An MR study has also shown insulin raises the risk of CVD in men but not in women30. Conversely, BMI reduces testosterone in men31. As such, the overall effects of statins mediated by BMI in men may be relatively neutral due to these pleiotropic effects.
The effects of genetically mimicking statins on IHD were largely similar after adjusting for BMI in both men and women, suggesting that BMI may not necessarily mediate or undermine the effects of statins on IHD, despite the positive association of BMI with IHD. BMI is not a well-defined intervention reflecting measurable factors, such as diet and exercise, where different ways of changing BMI may have different effects32. Thus, a BMI increase due to statin therapy, but may not necessarily lead to IHD. However, although the association of statins with IHD does not seem to be mediated by BMI, the increase of BMI per 1 mmol/L LDL-c corresponds to an increased risk of IHD of OR 1.13 95% CI 1.07 to 1.19. Statins also increase the risk of type 2 diabetes, however, we did not adjust for type 2 diabetes because type 2 diabetes may be a downstream effect of BMI, whereby higher BMI increases the risk of type 2 diabetes33, likely resulting in higher risk of IHD6. Similarly, we did not adjust for HbA1c which may also partially mediate the effects of BMI on IHD34.
Despite providing sex-specific estimates and showing sex differences for the direct effects of statins on IHD allowing for BMI, this study has some limitations. First, instruments should satisfy three key assumptions, i.e., the instrument is strongly associated with the exposure, not related to factors confounding the exposure-outcome relation, and influences the outcome only via effects on the exposure. The genetic variants used are well established mimics of statins, PCSK9 inhibitors and ezetimibe24. However, despite the high conditional F-statistics for BMI in the multivariable MR, the conditional F-statistics for genetically mimicked statins were low, and the Q statistic was high for overall estimate and men in the multivariable MR, suggesting pleiotropic effects for these two analyses, which were addressed by using multivariable MR-Egger. Pleiotropic effects for multivariable MR were not evident for women in this study. We cannot exclude the possibility that the SNPs mimicking effects of statins influence IHD through other pathways given the potential pleiotropic effects of these SNPs35.
Our findings, mainly based on populations of European descent, may not be transportable to other populations. Nevertheless, causes are consistent across populations, although relevance of the causal mechanism may vary by population36. Third, we replicated the results based on other large GWAS consortia for both exposure and outcome. However, due to lack of availability of sex-specific summary statistics for IHD in relevant populations, this study lacks sex-specific replication. Fourth, we assumed a linear relation for the association of lipid modifiers and BMI with IHD37. Fifth, linear regression was used for a binary outcome in the GWAS of IHD from Neale lab, which may generate false positives particularly when case numbers are small or minor allele frequency (MAF) is low (http://www.nealelab.is/blog/2017/9/11/details-and-considerations-of-the-uk-biobank-gwas). However, the number of IHD cases was large and all SNP MAFs were over 1%. Sixth, our exposure statin use is similar to a dichotomized binary exposure, which does not have a clear interpretation38. However, statin use was instrumented by a number of SNPs and our estimates may correspond to different levels of statin use. Seventh, the estimates here do not necessarily reflect the exact risk reduction from pharmacologic treatments39, but the lifelong effects of endogenous exposures per unit decrease in LDL-c estimated by Mendelian Randomization9, which are usually greater than the short-term effects of pharmacologic intervention from an RCT40.
Methods
Genetic predictors mimicking effects of lipid modifiers
We used well-established genetic variants mimicking effects of statins (rs12916, rs10066707, rs17238484, rs2006760, rs2303152 and rs5909), PCSK9 inhibitors (rs11206510, rs2149041, rs7552841, rs2479409, rs2479394, rs10888897 and rs562556) and ezetimibe (rs10260606 (proxy of rs2073547, r2 = 0.99), rs217386, rs7791240, rs10234070 and rs2300414)24, from the corresponding genes encoding the target protein for each lipid modifier (HMGCR, PCSK9 and NPC1L1, respectively). As the few SNPs mimicking each lipid modifier are correlated, independent (r2 < 0.05) genetic predictors of the lipid modifiers strongly associated with LDL-c (p < 5*10–8) were used in the primary analysis, i.e., rs12916 for statins, rs11206510, rs2149041, rs7552841 for PCSK9 inhibitors and rs10260606 for ezetimbe. For sensitivity analysis all correlated SNPs given above for each lipid modifier were used24 with a correlation matrix between SNPs for Europeans obtained from the 1000 Genomes catalogue using LDlink (https://ldlink.nci.nih.gov). MR estimates are presented in effect sizes of LDL-c reduction to mimic the effects of statins, PCSK9 inhibitors and ezetimibe, extracted from LDL-c genetic summary statistics from the UK Biobank where a sex-specific GWAS is available (http://www.nealelab.is/uk-biobank). The summary statistics are based on white British people and were adjusted for age, age2, sex, age*sex, age2*sex and the first 20 principal components for both sexes, and were adjusted for the first 20 principal components, age and age2 for sex-specific GWAS. For replication, the associations of the relevant SNPs with LDL-c were also taken from the Global Lipids Genetics Consortium (GLGC), a meta-analysis of 188 577 participants mainly of European descent adjusted for age and sex, and excluding people known to be on lipid lowering medication41 (Fig. 2).
Genetic predictors of BMI
SNPs that were strongly (p < 5*10–8) and independently (r2 < 0.001) associated with BMI were also obtained from the largest sex-specific GWAS of the meta-analysis11 of the UK Biobank GWAS and the GIANT consortium (Fig. 1), which included 806,834 participants of European ancestry, including 374,756 men and 434,794 women adjusted for age, sex, recruitment centre, genotyping batches and 10 principal components. GIANT14 with 339,224 participants of European ancestry was adjusted for age, age2, and any study specific covariates, such as principal components. In GIANT estimates for unrelated individuals were stratified by sex and case/control status, while for family based studies sex was included as a covariate.
Genetic associations with IHD
Sex-specific associations of selected SNPs with IHD were also extracted from a UK Biobank GWAS provided by Neale lab, which concerned up to 361,194 participants with 20,857 cases of IHD, including 167,020 men with 15,056 cases of IHD and 194,174 women with 5,801 cases of IHD. Sex-combined associations with IHD were from the UK Biobank were taken from SAIGE genetic summary statistics42. Replication was based on CARDIoGRAMplusC4D (Coronary ARtery DIsease Genome wide Replication and Meta-analysis (CARDIoGRAM) plus The Coronary Artery Disease (C4D) Genetics) consortium, a meta-analysis of GWAS of IHD mainly based on people of European descent including 60,801 cases and 123,504 controls adjusted for study specific covariates and over-dispersion43 (Fig. 2).
Statistical analysis
Instrument strength was assessed by the F statistic using an established approximation, i.e., the square of beta of the SNP-exposure association divided by the square of its standard error12.
Genetic associations for each SNP were aligned on the same effect allele for exposure and outcome. Inverse-variance weighting (IVW) with multiplicative random effects44 combining SNP-specific Wald estimates (SNP-outcome divided by SNP-exposure) was used to obtain overall and sex-specific estimates of the associations of genetically predicted effects of lipid modifiers (i.e., statins, PCSK9 inhibitors and ezetimibe) with BMI and IHD, and estimates of the associations of genetically predicted BMI with IHD.
Multivariable MR was used to assess overall and sex-specific associations of genetically predicted effects of lipid modifiers with IHD adjusted for BMI. We combined the genetic variants for lipid modifiers and BMI in the multivariable MR, removed any BMI variants correlated with statins SNPs (r2 < 0.001) and extracted their associations with LDL-c, BMI and IHD using corresponding GWAS. We used the conditional F-statistic to give an estimate of instrument strength conditional on the other exposure in multivariable MR45. We also used the modified Q statistic to assess horizontal pleiotropic effects using the WSpiller/MVMR package45, and used the MR-Egger estimate when a significant Q statistic46,47 suggested violation of exclusion restriction assumption.
Sensitivity analysis
In sensitivity analysis, we used all SNPs mimicking effects of statins, PCSK9 inhibitors and ezetimibe using correlation matrices obtained from LDlink (https://ldlink.nci.nih.gov/). We used MR-Egger as sensitivity analysis for the univariable analysis given its less stringent assumptions assuming the instrument strength is independent of the direct effect48, while IVW assumes balanced pleiotropy. However, we did not use MR-Egger for correlated instruments due to concerns about interpretability47. We used the IGX2, an adaptation of the I2 statistic, to assess the violation of the no measurement error (NOME) assumption, where a low IGX2 (< 90%) suggests a violation of the assumption for MR-Egger12. We also used the weighted median as it provides valid estimates when more than 50% of the information comes from valid instruments49.
All statistical analyses were conducted using R (version 4.0.3; R Foundation for Statistical Computing, Vienna, Austria). The univariable and multivariable MR estimates were obtained using the MendelianRandomization R package and the WSpiller/MVMR package to obtain the conditional F-statistic and the modified Q statistic.
Ethics approval and consent for publication
Ethics approval was not required as only publicly available data was used in this study.
Conclusion
Compared with variations in PCSK9 and NPC1L1, HMGCR was uniquely associated with higher BMI for both sexes. Similar positive associations of BMI with IHD were found for men and women. The effects of mimicking statins on IHD per unit decrease in LDL-c were similar after adjusting for the harmful effects of BMI in both men and women.
Data availablity
The datasets generated and analysed during the current study are available in the UK Biobank repository, (http://www.nealelab.is/uk-biobank).
Abbreviations
- BMI:
-
Body mass index
- CARDIoGRAMplusC4D:
-
Coronary artery disease genome wide replication and meta-analysis plus the coronary artery disease genetics
- CVD:
-
Cardiovascular disease
- GIANT:
-
Genetic investigation of anthropometric traits
- GLGC:
-
Global lipids genetics consortium
- GWAS:
-
Genome-wide association studies
- HbA1c:
-
Glycated haemoglobin
- IHD:
-
Ischaemic heart disease
- IVW:
-
Inverse-variance weighting
- LD:
-
Linkage disequilibrium
- LDL:
-
Low-density lipoprotein
- MR:
-
Mendelian randomization
- OR:
-
Odds ratio
- PCSK9:
-
Proprotein convertase subtilisin/kexin type 9
- SD:
-
Standard deviation
- SNP:
-
Single nucleotide polymorphisms
References
Michos, E. D., McEvoy, J. W. & Blumenthal, R. S. Lipid management for the prevention of atherosclerotic cardiovascular disease. N. Engl. J. Med. 381, 1557–1567. https://doi.org/10.1056/NEJMra1806939 (2019).
Ray, K. K., Corral, P., Morales, E. & Nicholls, S. J. Pharmacological lipid-modification therapies for prevention of ischaemic heart disease: current and future options. Lancet (London, England) 394, 697–708. https://doi.org/10.1016/s0140-6736(19)31950-6 (2019).
Ferrières, J. et al. Body mass index impacts the choice of lipid-lowering treatment with no correlation to blood cholesterol - findings from 52 916 patients in the Dyslipidemia International Study (DYSIS). Diabet. Obes Metab 20, 2670–2674. https://doi.org/10.1111/dom.13415 (2018).
Swerdlow, D. I. et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet (London, England) 385, 351–361. https://doi.org/10.1016/s0140-6736(14)61183-1 (2015).
Ference, B. A. et al. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N. Engl. J. Med. 375, 2144–2153. https://doi.org/10.1056/NEJMoa1604304 (2016).
Juutilainen, A. et al. Gender difference in the impact of type 2 diabetes on coronary heart disease risk. Diabetes Care 27, 2898–2904. https://doi.org/10.2337/diacare.27.12.2898 (2004).
Eeg-Olofsson, K. et al. New aspects of HbA1c as a risk factor for cardiovascular diseases in type 2 diabetes: an observational study from the Swedish National Diabetes Register (NDR). J. Intern. Med. 268, 471–482. https://doi.org/10.1111/j.1365-2796.2010.02265.x (2010).
Hope, H. F. et al. Systematic review of the predictors of statin adherence for the primary prevention of cardiovascular disease. PLoS ONE 14, e0201196–e0201196. https://doi.org/10.1371/journal.pone.0201196 (2019).
Schooling, C. M., Zhao, J. V., Au Yeung, S. L. & Leung, G. M. Investigating pleiotropic effects of statins on ischemic heart disease in the UK Biobank using Mendelian randomisation. Elife 9, e58567. https://doi.org/10.7554/eLife.58567 (2020).
Yebyo, H. G., Aschmann, H. E., Kaufmann, M. & Puhan, M. A. Comparative effectiveness and safety of statins as a class and of specific statins for primary prevention of cardiovascular disease: A systematic review, meta-analysis, and network meta-analysis of randomized trials with 94,283 participants. Am. Heart J. 210, 18–28. https://doi.org/10.1016/j.ahj.2018.12.007 (2019).
Yengo, L. et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum. Mol. Genet. 27, 3641–3649. https://doi.org/10.1093/hmg/ddy271 (2018).
Bowden, J. et al. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int. J. Epidemiol. 45, 1961–1974. https://doi.org/10.1093/ije/dyw220 (2016).
Minelli, C. et al. The use of two-sample methods for Mendelian randomization analyses on single large datasets. Int. J. Epidemiol. https://doi.org/10.1093/ije/dyab084 (2021).
Locke, A. E. et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206. https://doi.org/10.1038/nature14177 (2015).
Schmidt, A. F. et al. PCSK9 genetic variants and risk of type 2 diabetes: a mendelian randomisation study. Lancet Diabetes Endocrinol. 5, 97–105. https://doi.org/10.1016/s2213-8587(16)30396-5 (2017).
Nordestgaard, B. G. et al. The effect of elevated body mass index on ischemic heart disease risk: causal estimates from a Mendelian randomisation approach. PLoS Med. 9, e1001212. https://doi.org/10.1371/journal.pmed.1001212 (2012).
Mongraw-Chaffin, M. L., Peters, S. A. E., Huxley, R. R. & Woodward, M. The sex-specific association between BMI and coronary heart disease: a systematic review and meta-analysis of 95 cohorts with 1·2 million participants. Lancet Diabetes Endocrinol. 3, 437–449. https://doi.org/10.1016/s2213-8587(15)00086-8 (2015).
Sugiyama, T., Tsugawa, Y., Tseng, C. H., Kobayashi, Y. & Shapiro, M. F. Different time trends of caloric and fat intake between statin users and nonusers among US adults: gluttony in the time of statins?. JAMA Intern. Med. 174, 1038–1045. https://doi.org/10.1001/jamainternmed.2014.1927 (2014).
Page, M. M. & Watts, G. F. PCSK9 inhibitors - mechanisms of action. Aust. Prescr. 39, 164–167. https://doi.org/10.18773/austprescr.2016.060 (2016).
Phan, B. A. P., Dayspring, T. D. & Toth, P. P. Ezetimibe therapy: mechanism of action and clinical update. Vasc Health Risk Manag. 8, 415–427. https://doi.org/10.2147/VHRM.S33664 (2012).
Stancu, C. & Sima, A. Statins: mechanism of action and effects. J. Cell Mol. Med. 5, 378–387. https://doi.org/10.1111/j.1582-4934.2001.tb00172.x (2001).
Hu, J., Zhang, Z., Shen, W. J. & Azhar, S. Cellular cholesterol delivery, intracellular processing and utilization for biosynthesis of steroid hormones. Nutr. Metab. (Lond) 7, 47, doi:https://doi.org/10.1186/1743-7075-7-47 (2010).
Smals, A. G., Weusten, J. J., Benraad, T. J. & Kloppenborg, P. W. The HMG-CoA reductase inhibitor simvastatin suppresses human testicular testosterone synthesis in vitro by a selective inhibitory effect on 17-ketosteroid-oxidoreductase enzyme activity. J. Steroid Biochem. Mol. Biol. 38, 465–468. https://doi.org/10.1016/0960-0760(91)90333-z (1991).
Ference, B. A. et al. Mendelian randomization study of ACLY and cardiovascular disease. N. Engl. J. Med. 380, 1033–1042. https://doi.org/10.1056/NEJMoa1806747 (2019).
Lotta, L. A. et al. Association between low-density lipoprotein cholesterol-lowering genetic variants and risk of Type 2 diabetes: a meta-analysis. JAMA 316, 1383–1391. https://doi.org/10.1001/jama.2016.14568 (2016).
Walsh, E. I., Shaw, J. & Cherbuin, N. Trajectories of BMI change impact glucose and insulin metabolism. Nutr. Metab. Cardiovasc. Dis. 28, 243–251. https://doi.org/10.1016/j.numecd.2017.12.003 (2018).
Pasquali, R. et al. Insulin regulates testosterone and sex hormone-binding globulin concentrations in adult normal weight and obese men. J. Clin. Endocrinol. Metab. 80, 654–658. https://doi.org/10.1210/jcem.80.2.7852532 (1995).
Schooling, C. M. & Ng, J. C. M. Reproduction and longevity: A Mendelian randomization study of gonadotropin-releasing hormone and ischemic heart disease. SSM – Popul. Health 8, 100411. https://doi.org/10.1016/j.ssmph.2019.100411 (2019).
Clark, R. V. et al. Large divergence in testosterone concentrations between men and women: Frame of reference for elite athletes in sex-specific competition in sports, a narrative review. Clin. Endocrinol. (Oxf) 90, 15–22. https://doi.org/10.1111/cen.13840 (2019).
Zhao, J. V., Luo, S. & Schooling, C. M. Sex-specific Mendelian randomization study of genetically predicted insulin and cardiovascular events in the UK Biobank. Commun. Biol. 2, 332–332. https://doi.org/10.1038/s42003-019-0579-z (2019).
Eriksson, J. et al. Causal relationship between obesity and serum testosterone status in men: a bi-directional mendelian randomization analysis. PLoS ONE 12, e0176277. https://doi.org/10.1371/journal.pone.0176277 (2017).
Hernán, M. A. & Taubman, S. L. Does obesity shorten life? The importance of well-defined interventions to answer causal questions. Int. J. Obes. 32(Suppl 3), S8-14. https://doi.org/10.1038/ijo.2008.82(2008) (2005).
Bays, H. E., Chapman, R. H., Grandy, S. & Group, S. I. The relationship of body mass index to diabetes mellitus, hypertension and dyslipidaemia: comparison of data from two national surveys. Int. J. Clin. Pract. 61, 737-747, doi:https://doi.org/10.1111/j.1742-1241.2007.01336.x (2007)
Hu, X. et al. Exploring the causal pathway from body mass index to coronary heart disease: a network Mendelian randomization study. Ther. Adv. Chronic Dis. 11, 2040622320909040. https://doi.org/10.1177/2040622320909040 (2020).
Liao, J. K. & Laufs, U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 45, 89–118. https://doi.org/10.1146/annurev.pharmtox.45.120403.095748 (2005).
Lopez, P. M., Subramanian, S. V. & Schooling, C. M. Effect measure modification conceptualized using selection diagrams as mediation by mechanisms of varying population-level relevance. J. Clin. Epidemiol. 113, 123–128. https://doi.org/10.1016/j.jclinepi.2019.05.005 (2019).
Jenkins, D. A. et al. Estimating the causal effect of BMI on mortality risk in people with heart disease, diabetes and cancer using Mendelian randomization. Int. J. Cardiol. https://doi.org/10.1016/j.ijcard.2021.02.027 (2021).
Burgess, S. & Labrecque, J. A. Mendelian randomization with a binary exposure variable: interpretation and presentation of causal estimates. Eur. J. Epidemiol. 33, 947–952. https://doi.org/10.1007/s10654-018-0424-6 (2018).
Yarmolinsky, J. et al. Association between genetically proxied inhibition of HMG-CoA reductase and epithelial ovarian cancer. JAMA 323, 646–655. https://doi.org/10.1001/jama.2020.0150 (2020).
Ference, B. A. et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J. Am. Coll. Cardiol. 60, 2631–2639. https://doi.org/10.1016/j.jacc.2012.09.017 (2012).
Willer, C. J. et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 45, 1274–1283. https://doi.org/10.1038/ng.2797 (2013).
Zhou, W. et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341. https://doi.org/10.1038/s41588-018-0184-y (2018).
Nikpay, M. et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 47, 1121–1130. https://doi.org/10.1038/ng.3396 (2015).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37, 658–665. https://doi.org/10.1002/gepi.21758 (2013).
Sanderson, E., Davey Smith, G., Windmeijer, F., Bowden, J. An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int. J. Epidemiol. 48, 713–727, https://doi.org/10.1093/ije/dyy262 (2019).
Bowden, J., Davey Smith, G., Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525. https://doi.org/10.1093/ije/dyv080 (2015)
Burgess, S. & Thompson, S. G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 32, 377–389. https://doi.org/10.1007/s10654-017-0255-x (2017).
effect estimation and bias detection through Egger regression. Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments. Int. J. Epidemiol. 44, 512–525. https://doi.org/10.1093/ije/dyv080 (2015).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314, doi:https://doi.org/10.1002/gepi.21965 (2016).
Acknowledgements
We thank the UK Biobank for providing publicly available sex-specific summary data. We also thank Guoyi Yang for checking the study results.
Author information
Authors and Affiliations
Contributions
S.L. analyzed and interpreted the data and was a major contributor in writing the manuscript. C.M.S. designed and supervised the study and revised the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, S., Schooling, C.M. Investigating the effects of statins on ischemic heart disease allowing for effects on body mass index: a Mendelian randomization study. Sci Rep 12, 3478 (2022). https://doi.org/10.1038/s41598-022-07344-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-07344-8
- Springer Nature Limited