
Abstract
Hereditary glutathione reductase deficiency, caused by mutations of the GSR gene, is an autosomal recessive disorder characterized by decreased glutathione disulfide (GSSG) reduction activity and increased thermal instability. This study implemented computational analysis to screen the most likely mutation that might be associated with hereditary glutathione reductase deficiency and other diseases. Using ten online computational tools, the study revealed four nsSNPs among the 17 nsSNPs identified as most deleterious and disease associated. Structural analyses and evolutionary confirmation study of native and mutant GSR proteins using the HOPE project and ConSruf. HOPE revealed more flexibility in the native GSR structure than in the mutant structure. The mutation in GSR might be responsible for changes in the structural conformation and function of the GSR protein and might also play a significant role in inducing hereditary glutathione reductase deficiency. LD and haplotype studies of the gene revealed that the identified variations rs2978663 and rs8190955 may be responsible for obstructive heart defects (OHDs) and hereditary anemia, respectively. These interethnic differences in the frequencies of SNPs and haplotypes might help explain the unpredictability that has been reported in association studies and can contribute to predicting the pharmacokinetics and pharmacodynamics of drugs that make use of GSR.
Similar content being viewed by others
Introduction
Glutathione-disulfide reductase (GSR) protein is also known as Glutathione reductase (GR) enzyme encoded by the GSR gene in humans, which is located on chromosome 8p21 and consists of 13 exons1,2,3. Glutathione-disulfide reductase is a 522-amino acid protein that initiates the synthesis of mitochondrial and cytosolic GR. It is a member of the class-1 pyridine nucleotide-disulfide oxidoreductase family. Glutathione reductase is a homodimeric flavoprotein, a central enzyme of cellular antioxidant defense4, and reduces oxidized glutathione disulfide (GSSG) to the sulfhydryl form GSH, which works as a cellular antioxidant5,6.
Glutathione reductase catalyses the reaction (GSSG + NADPH + H + 2 (GSH) + NADP +) which is an important enzyme in this cellular system. Because it maintains the ratio of GSH/GSSG, it is involved in many cellular functions, including the activation of dormant cells7,8. GSH plays a key role in two ways: one maintaining the function and preventing oxidative stress in red blood cells4 and second clearing the electrophilic xenobiotics.
Hereditary glutathione reductase deficiency generally impairs cellular energy balance and increases the level of oxidative stress in red blood cells and has been related to hereditary hemolytic anemia9,10.
Familial deficiency of glutathione reductase in human blood cells has been reported by Loose et. al. and Roos et. al. in 1976 and 1979 respectively indicating the importance of GSH11,12. Non-synonymous single nucleotide polymorphisms (nsSNPs) occurring in the coding regions of gene that result in point mutations affect protein function and lead to pathogenic phenotypes13,14. nsSNP have the potential to alter the function of their protein, either directly or via disruption of structure15. To date, 18 mutations are reported in GSR gene which is responsible for glutathione reductase deficiency16,17,18. Mutations in GSR gene were reported in a northern Thailand population19 for the first time and subsequently in African Americans20, and Korea21 populations. It has been reported that the mutations in GSR gene cause glutathione reductase deficiency leading to reduced lifespan of red blood cells (RBCs)12.
Computational techniques has proven to be beneficial in determining the mutation and related effects effectively like finding out the non-significant SNPs from the significant SNPs that might produce more deleterious disease-associated consequences22. Incorporating the phenotypic changes along with the classical computational SNP prediction techniques have provided a high accuracy prediction level and helped in the classification of SNPs on the basis of their specific disease-associated consequences. This study examined and reports the most deleterious effect of nsSNPs reported in the Glutathione-disulfide reductase protein coding region. During this study SIFT23, Polyphen24, Mutation testers31, SNPs&Go28, SNAP229, and Provean tools30 were used to prioritize the deleterious disease-associated nsSNPs from the available SNP datasets obtained from the ClinVar25 database. In addition, a linkage disequilibrium (LD) and haplotype-based approach were used to examine GSR genes for linkage and association with other diseases. A high-resolution haplotype structure for the GSR gene and the association of the individual SNPs and haplotypes with the GSR gene in Han Chinese (CHB) individuals from the International Hapmap Project has been defined. The deleterious polymorphisms of gene and their impact on function of protein, association and linkage of selected gene polymorphisms of the glutathione S-reductase (GSR) with various defects/disorders like Obstructive Heart Defects (OHDs)) and hereditary anaemia in humans is also presented here. The findings of this study would be helpful in the development of personalized medicine.
Result
Mutation spread of GSR gene
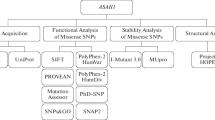
The SNPs present in the GSR gene were retrieved using the ClinVar database, and 18 various mutations were found in the gene. Among the coding regions of mutations identified, 17 were related to missense mutations, and 1 was related to nonsense mutations. An overview of the complete methodological approaches is summarized in a schematic diagram (Fig. 1).

Schematic diagram summarizing the study.
Identification of deleterious missense mutation
Six pathogenicity prediction web servers were used to predict the deleterious effect of nsSNPs. Six variants were observed to be damaged by using the SIFT tool and were further subjected to cross check by using five different tools (WHESS.db submodule PolyPhen-2 server, Mutation testers, SNPs&Go, SNAP2 and Provean tools). Out of a total of 17 nsSNPs identified, only four were predicted to be the most deleterious nsSNPs in all computational algorithms. The results are presented in Table 1.
Identification of nsSNPs on the domains of proteins
InterPro, a domain identification tool, predicts the domains and active sites of a protein through the functional analysis of protein families. It predicted four functional domains of GSR, which are pyridine nucleotide-disulfide oxidoreductase domains and FAD/NAD(P)-binding domains (65–390), and demonstrated that all 4 nsSNPs identified are positioned on these domains (Fig. S1).
Prediction of stability of the mutant protein
Protein stability was analysed by using the CUPSAT, I-Mutant-2 and DUET servers. The results revealed that four variants destabilized the GSR residue, namely, (V289A) rs151187899, (R233C) rs145851500, (A199T) rs141805635, and (R153C) rs8190955. The results are presented in Table 2.
Structural effect of point mutation on human GSR protein
Point mutation in genes has been studied by the Project HOPE server and the results revealed that the substitution of wild residues of R153C, R233C, A199T and V289A. These substitutions might be affecting the structure and function of the protein as the structure and physicochemical properties of different amino acids is diverse. For example in this case arginine residue is strong basic positively charged amino acid and has ability to form multiple hydrogen bonds and salt bridge in comparison to cysteine. Other mutations might be affecting the function and stability of the protein similarly (Table S2).
Evolutionary conservation analysis
The evolutionary conservancy of amino acid residues of the native GSR was examined by the ConSurf web server. It identified structural and functional residues of the 4 high-risk nsSNPs of the GSR protein using evolutionary conservation and solvent accessibility. We observed that R153 and V289 are exposed and functional, whereas residues A199 and R233 are buried and structural. All 2 of these residues are highly conserved (Table S1).
Study of LD and haplotype
LD (linkage disequilibrium) and haplotype were used to analyse the various genetic parameters of the GSR gene, and the genotype data of CHB (Han Chinese) were retrieved from the International Hapmap Project. Haplotype block reveals the combination of alleles at neighboring loci on the chromosome that may be transmitted together, and LD provides information on the measurement of the involvement of alleles (genetic marker) in a nonrandom mode.
Information about the predisposition of various diseases due to genetic variants has been evaluated. These parameters work as vital biomarkers for functional associations with a variety of diseases. The LD and haplotype study revealed an important block in the GSR gene, with three important SNPs having nonrandom associations, as represented in Fig. 2. Three critical SNPs out of five SNPs identified were rs3757918, rs8190955 and rs2978663 with minor allele frequencies T:C, C:T and A:G, respectively, with r2 ≥ 0.8, showing a high correlation between the loci (Table S3).

LD structure of GRS gene : (A) LD structure of maximum number of SNPs, (B) LD structure of GSR with minimum block size. (C) Haplotype block on GSR.
Furthermore, one of the haplotype blocks generated of the GSR gene involving 2 SNPs revealed that these variations may be responsible for obstructive heart defects ((rs2978663) OHDs) and hereditary anaemia (rs8190955) with different population frequencies. The TA haplotype was prominently found with a frequency of 0.533 in the studied population in comparison to haplotypes CG and CA.
Discussion
Glutathione-disulfide reductase is a flavoprotein involved in the glutathione redox cycle maintaining proper function and preventing oxidative stress in RBCs. The literature reveals that deregulation of glutathione-disulfide reductase protein leads to activation of dormant cells and deregulation of the cell cycle. Changes in the GSR protein during the glutathione antioxidant defence system play a vital role in executing its function, but any nsSNPs in the GSR gene lead to aberrant conformations, which in turn leads to glutathione reductase deficiency. Therefore, it becomes necessary to identify the effects of deleterious nsSNPs of GSR and their association with various diseases.
This study aimed to determine the most deleterious nsSNPs and their effects on the function of GSR proteins, characterize the haplotype structure of these genes and investigate markers associated with disease. The 17 molecular consequences found in the ClinVar database, were further subjected to analysis of six insilico SNP prediction tools, SIFT, Mutation Tester, Polyphen-2, SNP&GO, PROVEAN, SNAP2, and PredictSNP to identify significant deleterious nsSNPs. Among the identified 17 input non-synonymous SNP data-set, 4 nsSNPs (V289A, R233C, A199Tand R153C) were found to be “damaging” with score ranging between 0.5–1.000 and 0.990–1.000 in protein structure and function by SIFT and PolyPhen 2 tools respectively and the remaining two nsSNPs were characterized as benign (Table 1). These result were further confirmed for their pathogenicity of GSR gene using SNPs&GO, Mutation tester, SNAP2, and PROVEAN server. The results obtained supported the results of SIFT and PolyPhen2 i.e. V289A, R233C, A199T, R153C are potential deleterious nsSNPs (Table 1).
These four nsSNPs revealed different domains of the protein, where two nsSNPs were positioned in the pyridine nucleotide-disulfide oxidoreductase domain that interacts with the GSR association domain33. One nsSNP was located in the FAD/NAD-binding domain, which serves as the FAD/NAD-binding domain involved in oxidative metabolism of a variety of hydrocarbons (rubredoxin reductase, putidaredoxin reductase, terpredoxin reductase, ferredoxin-NAD + reductase components of benzene 1,2-dioxygenase, toluene 1,2-dioxygenase, chlorobenzene dioxygenase, biphenyl dioxygenase), NADH oxidase and NADH peroxidase1, 2, 3. The fourth nsSNP was present in the mitochondrial apoptosis-inducing factor, the C-terminal domain, which is crucial for cell apoptosis34.
The stability of the protein structure is crucial for the proper function of the protein. Alternations in the stability of proteins may cause misfolding and degradation of proteins. Therefore, to study the structural and functional activity of proteins, protein stability studies were carried out by using the CUPSAT and DUET and I-Mutant 2.0 servers. The result revealed that the same mutations (V289A, R233C, A199T, R153C) were responsible for affecting the stability of the GSR protein structure (Table 2). Furthermore, evolutionary conservancy of the GSR protein sequence is vital to determine whether a mutation has any negative effect on the host. Using the ConSurf server, it was observed that highly deleterious nsSNPs with high conservation scores were located in highly conserved regions, therefore increasing the risk of hereditary anaemia by altering the GSR protein sequence.
The Project Hope server revealed that these four highly risky nsSNPs negatively affect the structure of the GSR protein, among which two nsSNPs were structural and two nsSNPs were functional residues according to ConSurf. The server revealed that the wild-type residues of R153C and R233C are more hydrophobic than the mutant residues, and these variations in size and hydrophobicity disrupt the H-bond interactions with the adjacent molecules due to loss of hydrophobic interactions in the core of the protein. Arginine (Wild-type) residue is strong basic positively charged amino acid in nature and has ability to form multiple hydrogen bonds and salt bridge whereas cysteine due to their high reactivity of S–H group can possible distort the regular structure of protein by interacting with other reactive groups. The strong charge of the wild residue places it towards the outer hydrophilic surfaces of the proteins45,46. Thus it can be seen as arginine plays a crucial role in stability and flexibility of protein, as this types of interaction can be elucidated by opposite charge attraction, length and flexibility of side chain, and the potential to produce excellent hydrogen-bonding geometries with other biomolecules like nucleobases and phosphate groups.
A to T substitutions are likely to be the outcome of single nucleotide polymorphisms (SNPs), the most prevalent class of genetic variation among individuals. The human genome contains at least 11 million SNPs, about 1% of which are non-synonymous coding SNPs (nsSNPs). Most supposedly deleterious nsSNPs affect protein stability rather than functionality, underlying the importance of structural consequences caused by residue substitutions. Considering the local nature of the cross- sheet model, single substitutions can substantially change the tendency of the modified amino acid sequence to form -sheet aggregates. Threonine residue is reported to induce aggregation in proteins and supports the beta-sheet structures by unique effects on interaction with its surroundings including the consequent 3D structure, stability and dynamic behavior, in contrast to alanine which supports formation of α- helices. Among the amino acids (Thr, Leu, Phe, Trp, Ile, Val, Tyr residues) in proteins having strong tendency to form intra- or intermolecular β-sheets, threonine is the only polar residue in the list of β -sheet inducers, and should thus exert unique effects on interaction with its surroundings. The consequent three-dimensional structure, stability and dynamic behavior of such protein regions are all tightly linked to the process of aggregation47,48,49,50,51. Similarly valine also support the formation of β-sheet. Also valine has been proven important for the oxidation of the reduced flavin by molecular oxygen in choline oxidase and it has been reported that replacement of Val464 with alanine in the enzyme results in a twofold decrease in the limiting rate constant for flavin reduction (kred) and less than fivefold decrease in the equilibrium constant for formation of the enzyme–substrate complex (Kd)52. In the Val464Ala variant enzyme the substitution of the valine with an alanine has been reported to results in a 50-fold decrease in the bimolecular rate constant for reaction with oxygen, kcat/Koxygen. Further the authors have concluded that the presence of a nonpolar site is important for the oxidative half-reaction in which the enzyme-bound reduced flavin reacts with molecular oxygen to produce hydrogen peroxide and complete the catalytic cycle. It is proposed that the function of the nonpolar, aminoacyl side chain is to guide oxygen at the site where it subsequently will be activated to a superoxide species through electrostatic catalysis exerted by a positive charge53.
Linkage studies identified the 8p21 region as a susceptibility locus for obstructive heart defects (OHDs), hereditary anaemia. The LD pattern and haplotype structure for GSR in Han Chinese was characterized, and it revealed that there is LD across the GSR locus with little recombination. The rs3757918, rs8190955 and rs2978663 markers are loci enclosing a small part of the gene. Linkage disequilibrium has been reported between the common polymorphism found on GSR at positions 30619688 and 30627495. The analysis revealed that of the five nsSNPs identified, only three nsSNPs occurred and were linked in Han Chinese individuals. The results also indicated that only MAF (minor allele frequency) values of 0.467, 0.011 and 0.422 showed relatively strong linkage disequilibrium. The genotype of rs2978663 with the GSR gene increased the risk of occurrence of obstructive heart defects (OHDs). Right-sided and left-sided obstructive heart defects (OHDs) are subtypes of congenital heart defects in which the heart valves, arteries, or veins are abnormally narrow or blocked. Previous studies have suggested that the development of OHDs involves a complex interplay between genetic variants and maternal factors. Using data from 569 OHD case families and 1,644 control families enrolled in the National Birth Defects Prevention Study (NBDPS) between 1997 and 2008, we conducted an analysis to investigate the genetic effects of 877 single nucleotide polymorphisms (SNPs) in 60 candidate genes associated with the risk of OHDs and their interactions with maternal use of folic acid supplements and prepregnancy obesity. Applying log-linear models based on the hybrid design, we identified a SNP in the methylenetetrahydrofolate reductase (MTHFR) gene (C677T polymorphism) with a main genetic effect on the occurrence of OHDs. In addition, multiple SNPs in betaine-homocysteine methyltransferase (BHMT and BHMT2) were also identified to be associated with the occurrence of OHDs through significant main infant genetic effects and interaction effects with maternal use of folic acid supplements. We also identified multiple SNPs in glutamate-cysteine ligase, catalytic subunit (GCLC) and DNA (cytosine-5-)-methyltransferase 3 beta (DNMT3B) that were associated with an elevated risk of OHDs among obese women. Our findings suggested that the risk of OHDs was closely related to a combined effect of variations in genes in the folate, homocysteine, or glutathione/transsulfuration pathways, maternal use of folic acid supplements and prepregnancy. Obstructive heart defects associated with candidate genes, maternal obesity, and folic acid supplementation resolution in prepregnancy obese patients and maternal genotypes of SNPs in the GSR gene were associated with an increased risk of OHDs35. The genetic variant of GSR (rs8190955) was also found to be significantly associated with anaemia. This study demonstrates a potential connection between anaemia and oxidative stress, which could accelerate the production of ROS in addition to reducing the ability of the antioxidant defence system caused by SNPs of enzymes. LD and haplotype data should be useful in drug development and in understanding the genetic associations of GSR with adverse drug effects. These results provide an evidence of mutation leading to hereditary glutathione reductase deficiency and association with OHDs and can be further implemented for web lab studies.
Materials and method
Hereditary red blood cell enzymopathies of GSR gene-related information were collected from the database Online Mendelian Inheritance In Man (OMIM)13 and other reported literature. The dataset of chromosome number and position of the GSR gene in the human genome was collected from the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) only for missense variants25. Missense variants were chosen for further analysis because of their higher impact on the structure and function of proteins.
Identification of deleterious missense variants
Prediction of the deleterious effect of missense variants was performed by using seven different tools: Sorting Intolerant from Tolerant (SIFT)26 (http://sift.jcvi.org), Polymorphism Phenotypingv2 (PolyPhen-2)27 (http://genetics.bwh.harvard.edu/pph2/), SNP-GO28 (https://snps-and-go.biocomp.unibo.it/snps-and-go/), SNAP229, Provean30, predictor of human deleterious single nucleotide Polymorphisms (PhD-SNP)13,15and MUTATION TASTER31 (http://www.mutationtaster.org/) tools. The SIFT tool is a sequence homology-based tool; if the score is equal to or less than 0.05, the nsSNPs are considered deleterious nsSNPs. The WHESS.db module of the PolyPhen-2 server is a sequence and structure evolutionary conservation based on classifying the damaging effect of amino acid substitution; if the score lies between 0.801–1.00, then the nsSNPs are considered probably damaging. The PROVEAN server provides a pairwise sequence alignment (PSA) score and identifies nonsynonymous variants. Single nucleotide polymorphisms & Gene Ontology (SNPs&GO) and predictors of human deleterious single nucleotide polymorphisms (PhD-SNPs) are both support vector machine (SVM)-based tools used to predict evolutionary information, protein sequences and functions if the given mutation can be classified as disease related. Furthermore, mutation tester servers were used to evaluate the DNA sequence variants for disease-causing potential. Mutation tester scores that ranged from < 0.5 were considered disease-causing.
Identification of nsSNPs on the domains of GSR
The InterPro32 (https://www.ebi.ac.uk/interpro/) tool was used to identify the location of point mutations on the domains of glutathione-disulfide reductase protein, which can recognize motifs, active sites and domains of a protein.
Predicting mutation effect on protein stability
Protein stability analysis of mutant proteins was performed using three different tools: CUPSAT, the DUET server and I-Mutant233. CUPSAT was used to assess the effect of protein stability due to point mutation34. This tool uses structural environment-specific atom potential and torsion angle potentials to predict ΔΔG and the difference in the free energy between native and mutant proteins. Protein stability was also studied by using the DUET server35 and the I-Mutant 2.0 tool, which supports the vector machine SVM-based tool that predicts the change in protein stability upon single point mutation. The results obtained are in the form of stability of protein Gibbs free energy in the form of DDG values.
Analysing of GSR protein evolutionary conservation
To understand the evolutionary conservation of the amino acids in the protein sequence, ConSurf36 (https://consurf.tau.ac.il) was used to analyses the phylogenetic relationships between homologous sequences. Considered those nsSNPs of GSR that were found to be conserved for further analyses.
Prediction of the structural effect of nsSNPs on the human GSR protein
To identify the effect of the nsSNPs on the structure of the protein, HOPE (https://www3.cmbi.umcn.nl/hope) was used. HOPE is a web server that identifies the structural effects of point mutations in a protein sequence37. P00390 (UniProt-Accession Code of GSR) and the 4 SNPs were used individually as the input.
LD and haplotype block analysis
LD plays a key role in mapping complex disease or trail-associated genes. Haplotype block provides information on patterns of genetic variation that are associated with health and disease, and it can be used to examine stretches of DNA near the SNP cluster to identify the gene or genes responsible for causing the disease. Linkage disequilibrium (LD) is used for the study of population genetics for the nonrandom association of alleles at different loci38,39. The Haploview tool40 from the MIT/Harvard Broad Institute was used to study the genotype data for quantitative genetic parameters such as LD, and haplotype block data of Han Chinese (CHB) were retrieved from the International Hapmap Project. The data were visualized and analysed for any linkages and generation of LD and haplotype blocks.
Conclusion
GSR is a central enzyme in cellular antioxidant defence. The study of the functional SNPs of GSR provided significant insight into the deleterious effects of the nsSNPs identified in protein stability and cell functions. Two of the nsSNPs (rs2978663 and rs8190955) identified were also found to be associated with obstructive heart defects (OHDs) and hereditary anemia. It can be concluded that the LD and haplotype study unrevealed the relation of GSR with hereditary anaemia and OHD. Further that the understating of the oxidative stress pathways at molecular levels may be helpful in developing new interventions for the disease.
Abbreviations
- GSR:
-
Glutathione-disulfide reductase
- OHDs:
-
Obstructive heart defects
- nsSNPs:
-
Non-synonymous single nucleotide polymorphisms
References
George, D. L. & Francke, U. Gene dose effect: Regional mapping of human glutathione reductase on chromosome 8. Cytogenet. Cell Genet. 17, 282–286 (1976).
de la Chapelle, A. et al. Mapping of the gene for glutathione reductase on chromosome 8. Ann. Genet. 19, 253–256 (1976).
Kelner, M. J. & Montoya, M. A. Structural organization of the human glutathione reductase gene: Determination of correct cDNA sequence and identification of a mitochondrial leader sequence. Biochem. Biophys. Res. Commun. 269, 366–368 (2000).
Ho, H., Cheng, M. & Chiu, D. T. Glucose-6-phosphate dehydrogenase–From oxidative stress to cellular functions and degenerative diseases. Redox Rep. Commun. Free Radic. Res. 12, 109–118 (2007).
van Zwieten, R., Verhoeven, A. J. & Roos, D. Inborn defects in the antioxidant systems of human red blood cells. Free Radic. Biol. Med. 67, 377–386 (2014).
Pai, E. F. & Schulz, G. E. The catalytic mechanism of glutathione reductase as derived from x-ray diffraction analyses of reaction intermediates. J. Biol. Chem. 258(3), 1752–1757. https://doi.org/10.1016/S0021-9258(18)33050-3 (1983).
Kamerbeek, N. M. et al. Molecular basis of glutathione reductase deficiency in human blood cells. Blood 109(8), 3560–3566. https://doi.org/10.1182/blood-2006-08-042531 (2007).
Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta. 1830(5), 3217–3266. https://doi.org/10.1016/j.bbagen.2012.09.018 (2013).
Roos, D. et al. Protection of phagocytic leukocytes by endogenous glutathione: Studies in a family with glutathione reductase deficiency. Blood 53(5), 851–866. https://doi.org/10.1182/blood.V53.5.851.851 (1979).
Flatz, G. Population study of erythrocyte glutathione reductase activity. I. Stimulation of the enzyme by flavin adenine dinucleotide and by riboflavin supplementation. Humangenetik 11, 269–277 (1971).
Roos, D. et al. Protection of phagocytic leukocytes by endogenous glutathione: Studies in a family with glutathione reductase deficiency. Blood 53, 851–866 (1979).
Loos, H., Roos, D., Weening, R. & Houwerzijl, J. Familial deficiency of glutathione reductase in human blood cells. Blood 48, 53–62 (1976).
Kamaraj, B. & Purohit, R. Mutational analysis on membrane associated transporter protein (MATP) and their structural consequences in oculocutaeous albinism type 4 (OCA4)-A molecular dynamics approach. J. Cell. Biochem. 117, 2608–2619 (2016).
Kamaraj, B., Rajendran, V., Sethumadhavan, R., Kumar, C. V. & Purohit, R. Mutational analysis of FUS gene and its structural and functional role in amyotrophic lateral sclerosis 6. J. Biomol. Struct. Dyn. 33, 834–844 (2015).
Kumar, A. & Purohit, R. Use of long term molecular dynamics simulation in predicting cancer associated SNPs. PLoS Comput. Biol. 10, e1003318 (2014).
Kamerbeek, N. M. et al. Molecular basis of glutathione reductase deficiency in human blood cells. Blood 109, 3560–3566 (2007).
Nykamp, K. et al. Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med. Off. J. Am. Coll. Med. Genet. 19, 1105–1117 (2017).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Chang, J. C., van der Hoeven, L. H. & Haddox, C. H. Glutathione reductase in the red blood cells. Ann. Clin. Lab. Sci. 8, 23–29 (1978).
Gergely, P. et al. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 46, 175–190 (2002).
The Hereditary Hemolytic Anemia Working Party of the Korean Society of Hematology et al. Molecular diagnosis of hereditary spherocytosis by multi-gene target sequencing in Korea: matching with osmotic fragility test and presence of spherocyte. Orphanet J. Rare Dis. 14, 114 (2019).
Kumar, A. et al. Computational SNP analysis: Current approaches and future prospects. Cell Biochem. Biophys. 68, 233–239 (2014).
Kamaraj, B. & Purohit, R. Computational screening of disease-associated mutations in OCA2 gene. Cell Biochem. Biophys. 68, 97–109 (2014).
Kumar, A., Rajendran, V., Sethumadhavan, R. & Purohit, R. Evidence of colorectal cancer-associated mutation in MCAK: A computational report. Cell Biochem. Biophys. 67, 837–851 (2013).
Landrum, M. J. et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46(D1), D1062–D1067. https://doi.org/10.1093/nar/gkx1153 (2018).
Sim, N. L. et al. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40(Web Server issue), W452-457. https://doi.org/10.1093/nar/gks539 (2012).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 7, 7.20. https://doi.org/10.1002/0471142905.hg0720s76 (2013).
Capriotti, E. et al. WS-SNPs&GO: A web server for predicting the deleterious effect of human protein variants using functional annotation. BMC Genomics 14(Suppl 3), S6. https://doi.org/10.1186/1471-2164-14-S3-S6 (2013).
Hecht, M., Bromberg, Y. & Rost, B. Better prediction of functional effects for sequence variants. BMC Genomics 16(S8), S1. https://doi.org/10.1186/1471-2164-16-S8-S1 (2015).
Choi, Y. & Chan, A. P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinform. Oxf. Engl. 31(16), 2745–2747. https://doi.org/10.1093/bioinformatics/btv195 (2015).
Steinhaus, R. et al. MutationTaster2021. Nucleic Acids Res. 49(W1), W446–W451. https://doi.org/10.1093/nar/gkab266 (2021).
Apweiler, R. et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 29(1), 37–40. https://doi.org/10.1093/nar/29.1.37 (2001).
Capriotti, E., Fariselli, P. & Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 33(Web Server issue), W306-310. https://doi.org/10.1093/nar/gki375 (2005).
Parthiban, V., Gromiha, M. M. & Schomburg, D. CUPSAT: Prediction of protein stability upon point mutations. Nucleic Acids Res. 34(Web Server issue), W239–W242. https://doi.org/10.1093/nar/gkl190 (2006).
Pires, D. E. V., Ascher, D. B. & Blundell, T. L. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 42(Web Server issue), W314-319. https://doi.org/10.1093/nar/gku411 (2014).
Ashkenazy, H. et al. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44(W1), W344–W350. https://doi.org/10.1093/nar/gkw408 (2016).
Venselaar, H., Te Beek, T. A. H., Kuipers, R. K. P., Hekkelman, M. L. & Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 11, 548. https://doi.org/10.1186/1471-2105-11-548 (2010).
Blomhoff, A. et al. Linkage disequilibrium and haplotype blocks in the MHC vary in an HLA haplotype specific manner assessed mainly by DRB1*03 and DRB1*04 haplotypes. Genes Immun. 7(2), 130–140. https://doi.org/10.1038/sj.gene.6364272 (2006).
Long, J. R. et al. Patterns of linkage disequilibrium and haplotype distribution in disease candidate genes. BMC Genet. 5, 11. https://doi.org/10.1186/1471-2156-5-11 (2004).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 21(2), 263–265. https://doi.org/10.1093/bioinformatics/bth457 (2005).
Wang, T. et al. Pyridine nucleotide-disulphide oxidoreductase domain 2 (PYROXD2): Role in mitochondrial function. Mitochondrion 47, 114–124. https://doi.org/10.1016/j.mito.2019.05.007 (2019).
Daugas, E. et al. Apoptosis-inducing factor (AIF): A ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 476(3), 118–123. https://doi.org/10.1016/s0014-5793(00)01731-2 (2000).
Tang, X., National Birth Defects Prevention Study et al. Obstructive Heart Defects Associated with Candidate Genes, Maternal Obesity, and Folic Acid Supplementation.
Luscombe, N. M., Laskowski, R. A. & Thornton, J. M. Amino acid-base interactions: A three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 29, 2860–2874 (2001).
Hunter, T. Why nature chose phosphate to modify proteins. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 367, 2513–2516 (2012).
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011).
Chou, P. Y. & Fasman, G. D. Conformational parameters for amino acids in helical, β-sheet, and random coil regions calculated from proteins. Biochemistry 13, 211–222 (1974).
Frazer, K. A., Murray, S. S., Schork, N. J. & Topol, E. J. Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 10, 241–251 (2009).
Ramensky, V., Bork, P. & Sunyaev, S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 30, 3894–3900 (2002).
Chasman, D. & Adams, R. M. Predicting the functional consequences of non-synonymous single nucleotide polymorphisms: structure-based assessment of amino acid variation. J. Mol. Biol. 307, 683–706 (2001).
Podoly, E., Hanin, G. & Soreq, H. Alanine-to-threonine substitutions and amyloid diseases: Butyrylcholinesterase as a case study. Chem. Biol. Interact. 187, 64–71 (2010).
Finnegan, S. & Gadda, G. Substitution of an active site valine uncovers a kinetically slow equilibrium between competent and incompetent forms of choline oxidase. Biochemistry 47, 13850–13861 (2008).
Finnegan, S., Agniswamy, J., Weber, I. T. & Gadda, G. Role of valine 464 in the flavin oxidation reaction catalyzed by choline oxidase. Biochemistry 49, 2952–2961 (2010).
Acknowledgements
The authors are thankful to the Department of Biotechnology, Ministry of Science & Technology, Government of India for the Bioinformatics infrastructure facility at Jamia Hamdard under the BTISNet program.
Funding
One of the authors, Ms. Bharti Vyas, acknowledges the fellowship obtained from the Indian Council of Medical Research (ICMR), New Delhi.
Author information
Authors and Affiliations
Contributions
Conception or design of the work, Data collection, and Data analysis and interpretation was done by B.V. Critical revision of the article and final approval of the version to be published was done by Dr. M.A. R.B. was contributed to the Data collection. F.J.A. contributed to the analysis of the result. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vyas, B., Bhowmik, R., Akhter, M. et al. Identification, analysis of deleterious SNPs of the human GSR gene and their effects on the structure and functions of associated proteins and other diseases. Sci Rep 12, 5474 (2022). https://doi.org/10.1038/s41598-022-09295-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-09295-6
- Springer Nature Limited