
Abstract
Missense mutation C694R in the RING domain of the LRSAM1 gene results in a dominantly inherited polyneuropathy, Charcot-Marie-Tooth disease type 2P (CMT2P). We have generated and characterized a Lrsam1C698R knock-in mouse model produced through CRISPR/Cas9 technology. Both heterozygous (Lrsam1+/C698R) and homozygous (Lrsam1C698/C698R) knock-in mice exhibited normal motor functions on behavioral tests as well as normal on nerve conduction studies. Axonal density and myelin thickness were not significantly different between mutants and wild-type mice by sciatic nerve morphometric analysis up to 17 months of age. In line with these normal findings, protein–protein interactions between mutant LRSAM1 and RNA-binding proteins (such as FUS and G3BP1) were still present in mouse cells, which differs from the disrupted interactions between these proteins in human CMT2P cells. However, after crush nerve injury, Lrsam1+/C698R mice had a mild, but statistically significant, reduced compound nerve action potential and conduction velocity during recovery. Therefore, C698R mutation results in a mild impaired nerve regeneration in mice. We speculate that repetitive nerve injuries may, at least partially, contribute to the slowly progressive axonal loss in CMT2P.
Similar content being viewed by others

Introduction
Charcot-Marie-Tooth disease (CMT) is a group of inherited peripheral nerve diseases with a collective prevalence of 1:2500, which are caused by monogenic mutations in more than 100 different human genes1. Patients with CMT typically present with sensory loss, muscle atrophy, weakness in distal limbs, and foot deformities. Autosomal dominantly inherited CMTs with de-/dysmyelination have been classified as CMT type-1, whereas autosomal dominant CMTs of axonal type are grouped as CMT type-2 (CMT2)2,3.
CMT2P is caused by autosomal dominant and recessive mutations in leucine rich repeat and sterile alpha motif 1 (LRSAM1) gene on human chromosome-94,5,6,7,8,9,10,11,12. Most patients with autosomal dominant CMT2P usually manifest a teenage or adult-onset, slowly progressive, length-dependent sensory-motor axonal polyneuropathy5,6,11,13.
LRSAM1 is an E3 ubiquitin ligase with a RING domain at its c-terminal. The RING domain is crucial for the ubiquitination function of LRSAM1. CMT mutations have been shown to disrupt its function13. CMT2P was initially found in multiple European families with frame-shift mutations, which altered a large portion of the RING amino acid sequence7,9,10. Of the 12 CMT2P-causing mutations, 11 are dominantly inherited and located in the catalytic RING domain13; which further supports the functional importance of the RING. Our group has described a missense mutation, Cys694Arg, that alters a highly conserved cysteine in the RING and is co-segregated with affected CMT2P members in an American family6. This conserved cysteine is required to maintain the structure of E3 ubiquitin ligase through its binding with two zinc ions4,6,13. We have further demonstrated that the C694R mutation altered the RNA-binding protein nuclear translocation likely by disrupting the protein–protein interaction between LRSAM1 and the RNA-binding proteins6, a potential mechanism in CMT2P pathogenesis.
To further explore this mechanism in vivo, we have generated a Lrsam1C698R knock-in mouse model, the amino acid substitution equivalent to the human C694R mutation. Herein, we present our initial characterization of the Lrsam1C698R mouse model. We did not observe axonal degeneration in the peripheral nerve in the naïve Lrsam1C698R knock-in mice; instead, we found a mild impairment of nerve repair in the mouse model.
Results
Generation of Lrsam1 C698R knock-in mouse
The missense mutation c.2080T > C (C694R) in human LRSAM1 that we described6 was introduced into the mouse genome through CRISPR/Cas9-mediated mutagenesis (Fig. 1A). Compared with the human sequence surrounding the c.2080T position, we found a C698R substitution in the mouse Lrsam1 gene equivalent to the human C694R mutation (Fig. 1A). Heterozygous (Lrsam1+/C698R) mice were crossed to obtain homozygous (Lrsam1C698R/C698R) mice. DNA sequencing revealed that the mutant mice carry both C698R and a silent C to A mutation 7 bp upstream to C698R mutation (Fig. 1B). Mutant mice were further verified by restriction fragment length polymorphism analysis to determine whether Lrsam1 knock-in occurs in both alleles or one allele (Fig. 1C). These knock-in mice did not appear to differ from Lrsam1+/+ mice in appearance and body weight (Fig. 1D,E).

Generation of Lrsam1C698R knock-in mouse. (A) CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated 9) targeting in mouse Lrsam1 locus. A single guide RNA sequence was underlined. The sequences of amino acid were shown under DNA sequences. The substituted nucleotides were shown in green (wild type) and red (mutation). The PAM (protospacer adjacent motif) sequence was shown in blue. HDR indicates homology-directed repair; ssDNA, single-strand DNA. Exons were represented by gray boxes. (B) The Lrsam1 knock-in allele was identified by sequencing the PCR amplicon surrounding the mutation site. The asterisk indicates the mutation site of knock-in of the Lrsam1 allele: 5′-TGC(Cys)-3′ > 5′-CGC(Arg)-3′. (C) Polymerase chain reaction (PCR) genotyping of Lrsam1C698R progeny. Mouse DNA segment around the C698 was amplified by the following primers and then digested with a restriction enzyme Mlsl recognizing tgg/cca. The PCR products were two bands at 285 bp and 215 bp for WT, one band at 499 bp for Lrsam1C698R. Lane 1 = DNA marker (M); Lane 2 = Lrsam1+/+ (WT/WT); Lane 3 = Lrsam1+/C698R (KI/WT); Lane 4 = Lrsam1C698R/C698R (KI/KI). (D) External physical analysis. 6-month-old WT/WT, KI/WT, and KI/KI littermates were placed next to a centimeter ruler. (E) At 14–16 months of age, we observed no significant difference in body weight between wild-type and mutant mice (P > 0.05, n = 5 in each group).
Behavioral studies showed no abnormality in Lrsam1 C698R mice
Rotarod and hindlimb clasping test assess mouse motor coordination and limb weakness. At 12 months of age, Lrsam1+/C698R and Lrsam1C698R/C698R mice did not show any abnormalities in hindlimb clasping or paw extension during tail suspension (Fig. 2A) when compared with Lrsam1+/+ mice. The fall latency from the rotating bas of Rotarod was not different among Lrsam1+/+, Lrsam1+/C698R and Lrsam1C698R/C698R mice measured at 6- and 8-months of age (Fig. 2B).

Behavioral analysis of Lrsam1C698R knockin mice. (A) Quantification of hindlimb clasping test at 12 months old. Animals were held at tails for 20 s, and time displaying hindlimb splaying was recorded. Data presented as the average time for hindlimb splaying. Average time on the hindlimb splaying was not different in Lrsam1+/C698R and Lrsam1C698R/C698R mice, compared to their Lrsam1+/+ littermates (P > 0.05, n = 6 for each genotyping group). (B) Rotarod performance showed the average time, in seconds (s), spent on the accelerating rotating rod of 6 and 8 months of age in Lrsam1+/C698R, Lrsam1C698R/C698R, and their Lrsam1+/+ littermates. The mutant mice showed no significant decline in the locomotor performance (P > 0.05, n = 6–9 for each genotyping group).
The nerve conduction study was not significantly different between mutants and wild-type mice
We measured compound muscle action potentials (CMAP) at the paw muscles and conduction velocities (CV) on the sciatic nerves. CMAPs and CVs were not significantly different between Lrsam1+/+, Lrsam1+/C698R and Lrsam1C698R/C698R mice up to 2.5 years of age (P > 0.05, Fig. 3A,B).

Nerve conduction studies (NCS) at 3, 5, 30-month-old WT and mutant mice. (A) NCS showed no significant difference in conduction velocity (CV) between Lrsam1+/+ (WT) and Lrsam1+/C698R (KI/WT) or Lrsam1C698R/C698R (KI/KI) mice tested in separate cohorts at 3, 6 and 30 months of age (P > 0.05, n = 18, 30, 13 for 3-month-old, 8, 11, 6 for 5-month-old, 7, 9, 3 for 30-month-old, Lrsam1+/+, Lrsam1+/C698R, Lrsam1C698R/C698R mice). (B) Compound muscle action potential (CMAP) was also normal in mutants.
Myelin and axon were morphologically normal in Lrsam1 C698R mice
We further studied mouse nerves morphologically. Axons and myelin were examined and quantified at 17 months of age in the sciatic nerves. Visual inspection on semithin sections did not find axonal degeneration or demyelination features, such as loss of axons, myelin debris, and clusters of regenerating axons (Fig. 4A). Myelinated nerve fibers on the cross-sections of the sciatic nerves were quantified using deep-learning-based analysis14 (Fig. 4B). There was no significant difference in axon diameter, fiber density, g-ratio, and myelin thickness in 17-month-old Lrsam1+/C698R and Lrsam1C698R/C698R sciatic nerves, compared with those in wild-type mice (Fig. 4C,F).

Morphometric analysis for Lrsam1C698R mouse sciatic nerve. (A) Semithin sections of sciatic nerves showed myelinated nerve fibers were morphologically similar between 17-month-old wild-type and Lrsam1+/C698R or Lrsam1C698R/C698R mice. Scale bars = 50 µm. (B) 40 × Bright Field Image (a) is processed by an ORS Dragonfly trained model (b), which produces a segmentation of myelin (c) and inner axonal areas (d). The software’s Region of Interest analysis tool is used to measure each fiber’s inner and outer areas (e). The results are exported into a spreadsheet, where myelin radii, thickness, and g-ratio are calculated (f). (C–F) Morphometric analysis showed that the axon radius, axon density, myelin thickness, and g-ratio (determined by dividing axonal radius by the outer fiber diameter) were not significantly different between 17-month-old wild-type and Lrsam1+/C698R or Lrsam1C698R/C698R sciatic nerves (P > 0.05, t-test, n = 5 per group, 3 males and 2 females each genotype).
Protein–protein interaction between LRSAM1 and RNA-binding proteins was not disrupted by the C698R mutation
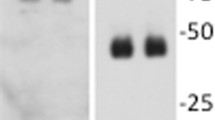
Our previous study in fibroblasts from patients with CMT2P showed that C694R mutation disrupts the protein–protein interactions between LRSAM1 and RNA-binding proteins (including FUS and G3BP1), leading to altered nuclear-cytoplasmic translocation of the RNA-binding proteins, a potential mechanism of neuronal degeneration in CMT2P6. To determine whether similar molecular abnormalities occur in mouse fibroblasts, we performed co-IP. We found that interactions were still present and not significantly different between FUS-mutant LRSAM1 and FUS-wild type LRSAM1 or between G3BP1-mutant LRSAM1 and G3BP1-wild type LRSAM1 (P > 0.05, Fig. 5A–D). There were no significant differences in nuclear / cytoplasmic ratios among LRSAM1, FUS and G3BP1 protein levels (P > 0.05, Fig. 5E–I). This finding differs from disrupted interactions between mutant LRSAM1 and RNA-binding proteins in human cells from patients with CMT2P.

C698R Lrsam1 mutation fails to disrupt interactions between LRSAM1 and RNA-binding proteins. (A) Cytoplasmic and nuclear protein fractions were extracted from Lrsam1+/+, Lrsam1+/C698R and Lrsam1C698R/C698R fibroblasts. Proteins were immunoprecipitated (IP) with anti-FUS antibody, and the precipitated proteins were immunobloted (IB) with anti-LRSAM1, anti-FUS antibody (IP lines). IgG was used as a IP negative control. Cell lysate was loaded as inputs. FUS antibody was able to pull down LRSAM1 in nuclear fractions of wild-type and C698R mutation fibroblasts. (B) Intensities of protein bands in co-IP experiments were measured using ImageJ (n = 3). Anti-LRSAM1 signals were normalized by either anti-FUS signals or anti-β-Actin signal. LRSAM1 levels displayed no significantly difference between wild-type and mutant cells (P > 0.05). Thus, C698R failed to disrupt the protein-protein interactions between LRSAM1 and FUS. (C) Cytoplasmic and nuclear protein fractions were separately used for IP with an anti-G3BP1 antibody and then blot with an anti-LRSAM1 and an anti-G3BP1 antibody. G3BP1 antibody was able to pull down LRSAM1 in nuclear fractions of both wild-type and C698R fibroblasts. (D) Intensities of LRSAM1 and G3BP1 protein bands were quantified using ImageJ. LRSAM1 levels were not significantly different between wild-type and mutant cells (P > 0.05). Thus, C698R mutation did not disrupt LRSAM1 interaction with G3BP1. (E) Cytoplasmic, nuclear proteins and whole cell were extracted from the mouse fibroblasts and examined by Western blot for levels of LRSAM1, FUS and G3BP1. HDAC3 served as a loading control of the nuclear extracts. β-actin was included as an overall loading control. (F–I) Histograms show the signal intensity of protein bands in arbitrary units after their normalization with the level of β-Actin. Each bar represents the mean of triplicate values. The C698R mutation did not significantly decrease the level of LRSAM1, FUS, G3BP1, or HDAC3 protein when compared with those in wild-type cells (P > 0.05).
Nerve regeneration was mildly impaired in Lrsam1 C698R nerves
Most patients with CMT2P are due to a heterozygous C694R mutation6. We thus examined nerve regeneration using crush-nerve injuries on Lrsam1+/C698R and Lrsam1+/+ mice that were age- and sex-matched. A baseline NCS found no difference in CMAP or CV between the two genotypic groups. After the nerve injury, NCS was done every 2–3 weeks. Sciatic nerves were dissected at 23 weeks after the second surgery or 43 weeks after the initial surgery respectively for morphometric analysis.
Lrsam1+/C698R mice had a statistically significant negative main effect on CMAP over the entire period (-0.516 mV), and during the second recovery period (− 0.606 mv) (Fig. 6A). The main effect of mutant on CV was statistically significant during the post-surgery periods: − 7.24 m/s over the whole time period, − 3.41 m/s during the first recovery, and − 7.25 m/s during the second recovery (Fig. 6B). These findings suggest an incomplete recovery of CMAP and CV after the injury15.

Nerve regeneration was slightly, but statistically significant, impaired in the Lrsam1C698R mice. (A) NCS was carried out before the surgery, 2 weeks post-surgery, and every 1–2 weeks afterward until 11 weeks. At 20 weeks, another baseline NCS was measured before the 2nd injury was done. NCS was then conducted every 2–3 weeks, until sciatic nerves were collected for morphometric analyses at 23 weeks, or 43 weeks after the initial surgery respectively. Baseline measurements showed no difference in CMAP between the genotypes (n = 15 for Lrsam1+/+ group, 12 for Lrsam1+/C698R group, P = 0.763). The linear mixed model showed a significant negative effect of Lrsam1+/C698R genotype on CMAP: − 0.516 mV over the whole period excluding the baseline, (P < 0.001), − 0.417 mV over the first postoperative period (P = 0.07), and − 0.606 mV over the second post-op period (P < 0.001). Time overall had a positive effect on CMAP, associated with 0.192 mV increase per time point in all animals (p < 0.001), 0.715 mV during the 1st post-op period (P < 0.001), 0.501 mV during the 2nd post-op period (P < 0.001). Interactions of time and genotype were not found to be significant. # = P < 0.001. (B) CV baseline measurements showed no significant difference between the genotypes n = 15 for Lrsam1+/+ group, 12 for Lrsam1+/C698R group, P = 0.961). A linear mixed model showed a significant negative effect of Lrsam1+/C698R genotype on CV: − 7.24 m/s over the whole time excluding the baseline, (P < 0.001), − 3.41 m/s over the first postoperative period (P < 0.001), and − 7.25 m/s over the second post-op period (P < 0.001). Time overall had a positive effect on CV, associated with 0.55 m/s increase per time point overall in all animals (P < 0.001), 1.868 m/s during the 1st post-op period (P < 0.001), 1.638 m/s during the 2nd post-op period (P < 0.001). Interactions of Genotype x Time were found to have a significant effect on CV and indicated a slowed rate of CV recovery in Lrsam1+/C698R mice during the whole time period (Lrsam1+/+ increased 0.641 mV (P < 0.001), Lrsam1+/C698R increased 0.437 mV (P < 0.001), interaction P < 0.001), over the first postoperative period (Lrsam1+/+ increase 2.078 mV (P < 0.001), Lrsam1+/C698R increased 1.628 mV (P < 0.001), interaction P < 0.001), and over the second post-op period (Lrsam1+/+ increased 1.906 mV (P < 0.001), Lrsam1+/C698R increased 1.294 mV (P < 0.001), interaction P < 0.001). # = p < 0.001. CV was significantly different between 5- and 20-weeks post-surgery. * = P < 0.001. (C–E) Sciatic nerves were collected from the surgery and control hindlimbs, embedded in Epon resin, sliced and imaged under a brightfield microscope at 40X magnification. Automatic segmentation of myelin was performed and refined in Dragonfly software. Results were analyzed using a mixed linear model and showed that crash nerve surgery was associated with an increase of axon density (+ 4758.8 fibers/mm2), a decrease of axon radius (− 0.74 µm), and a decrease of myelin thickness (− 0.30 µm), all statistically significant (P < 0.05). Genotype or interaction between genotype and surgery status were not found to be statistically significant. (F–G) Morphometric analysis of the nerve fibers revealed no significant difference in g-ratio and axon-diameter distribution in injured Lrsam1+/C698R nerves as compared to injured Lrsam1+/+ nerves (n = 6 per group, 4 males and 2 females each genotype). Ctrl = Control. Inj = crush injury.
Time had a statistically significant positive effect on CMAP and CV in both genotypes, indicating an effect of recovery process. More importantly, CV recovery showed a statistically significant genotype x time interaction over the two recovery periods, with Lrsam1+/+ recovering faster than Lrsam1+/C698R mice (Fig. 6A,B). In addition, Lrsam1+/C698R also showed a reduced rate of CV recovery as compared to Lrsam1+/+ mice.
Morphometric analysis showed that surgeries had main effects on axon density (an increase of 4758.8 fibers/mm2), a decrease of axon radius (− 0.74 µm), and a decrease of myelin thickness (− 0.30 µm), all statistically significant changes expected in regenerating nerves (P < 0.05). However, comparisons between Lrsam1+/+ and Lrsam1+/C698R nerve morphometrics, as well as the interaction of genotype and injury status were not significantly different (Fig. 6C–G).
Additionally, in homozygous C698R mice, CV and CMAP were not significantly different from that of heterozygous C698R mice after injury; but were significantly lower than that of wild-type mice (Supplementary Fig. 1A–B). Again, the morphometric analysis did not find a significant change in homozygous injured nerves, compared with heterozygous and wild-type injured nerves (Supplementary Fig. 1C–F).
Taken together, C698R Lrsam1 mutation only mildly impaired nerve function during regeneration.
Discussion
In this study, we have shown the production and initial characterization toward a new mouse model of CMT2P, where a knock-in of the C698R Lrsam1 mutation was introduced into the mouse genome. However, we observed no peripheral neuropathy in naïve Lrsam1+/C698R and Lrsam1C698R/C698R mice up to 2.5 years of age but did find a mildly impaired nerve function during regeneration after nerve injury. While the electrophysiological phenotype is subtle, it could contribute to the slowly progressive polyneuropathy in patients with CMT2P. As every person is exposed to many nerve injuries during their lifetime, we speculate that an accumulation of impaired nerve recoveries may eventually contribute to the polyneuropathy of CMT2P16,17,18.
This finding also brings up a challenging issue. The nerve abnormality in the knock-in mice was disproportionally subtle, comparing with the polyneuropathy in persons with CMT2P6. This mild phenotype is in line with absent disruption of protein–protein interactions between mutant LRSAM1 and RNA-binding proteins (Fig. 5). The exact reason for the loss of this disruption of protein–protein interaction by the mutation in mice is unclear at this point. However, there have been many precedents where mouse models could not completely recapitulate phenotypes in its human counterpart19. Perhaps, CMT2P needs to be modeled in larger animals.
Notice that we did not find an age-effect in the NCS of 30-month-old mice, while CV is known to decline over time20. This is not unexpected. Age-effect in CV is typically detectable in consecutive recordings in same animals. This was not possible in this study as tissues had to be collected at different ages; thus, our CV data in the 30-month-old mice were cross-sectional; thus may and may not show the expected age-effect. However, it would not affect the overall conclusion of this study. This was not likely a technical issue as the same NCS technique was used in our studies before and has detected age-effect21.
Another potential limitation relates to small n-numbers of some experimental groups. While our data do not provide evidence of obvious progressive neuropathy in mutant mice without the challenge of nerve injury, subtle alterations could be undetected due to the small n-numbers. However, impaired nerve regeneration after injury was reproduced in repeated experiments and also in a different experimental group (Fig S1), supporting the key conclusion of this study.
In summary, we have produced a knock-in mouse with C698R mutation in order to model CMT2P. While the phenotype is not robust, mild abnormality in nerve repair provides a helpful clue toward the slowly progressing polyneuropathy in CMT2P.
Methods
Animal generation
The Lrsam1C698R knock-in mice were generated on a C57Bl/6J background using the CRISPR/Cas9 technique. Briefly, single-cell zygotes from C57BL/6J mice were microinjected with mRNA encoding Cas9 and a guide sequence (5′-ACAGCAGCAGACGTGGCCAC-3′ at 20 ng/µl) to target the exon 26 of Lrsam1. A single stranded DNA oligo carrying the C698R mutation was co-injected at 20 ng/µl to promote homology-directed repair. The sequence of the oligo was (…ACGTCCGCTG…). The underlined "C" changes the first base of codon 698, creating the C to R substitution. Following implantation of the embryos into surrogate females, the microinjection resulted in 49 live-born mice. Fourteen of them carried idles consistent with non-homologous end joining (NHEJ) events. Two female offspring carried the C698R, as well as an upstream silent mutation. These mice were bred with wild-type C57BL/6 J males to establish the germline transmission of the mutation. The offspring were confirmed to carry both the C698R and the silent C to A mutation 7 bp upstream to C698R mutation. The silent mutation was likely a not-targeted change accidentally introduced by the genetic manipulation as we did not find this silent mutation in the progeny of Lrsam1+/+ mice. After germline transmission of the targeted variant allele, we used the congenic Lrsam1+/C698R mice derived from two founders to backcross with C57BL/6J mouse for at least 10 generations. A mixed mouse line from both founders were used for experiments. The experimental mice were obtained by intercrossing heterozygous mice (Lrsam1+/C698R) and were confirmed by both restriction fragment length polymorphism analysis and sequencing. The mice used in this study were age-matched and sex-mixed littermates. Mice were maintained on a standard 12 h light/12 h dark cycle, ad libitum access to food and water, and housed in plastic cages with standard rodent bedding. We did not observe any difference in mortality between genotypes.
Animal ethics statements
All animal experiments and procedures were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the Wayne State University and were performed in accordance with federal and university guidelines and regulations for the care and use of experimental vertebrate animals. Authors have complied with the ARRIVE guidelines for reporting.
Mouse PCR genotyping
DNA was extracted from ear clips by incubation in 100 μl DirectPCR Lysis buffer (Cat # 102T, Viagen) with 2 μl of 10 mg/ml proteinase K (Cat #P4850, Sigma) at 55 °C overnight. After deactivating the mixture at 85 °C for 45 min, the supernatant was used for genotyping by PCR. DNA segments around the C698 were amplified by the following primers: forward primer, 5′-CCA GGT AAG CAG TAC ACG CCT G-3′ and reverse primer 5′-GGT GAC AAA GGC CTA TGG CAG T-3′. PCR was programmed as follows: 94 °C, 5 min; (94 °C, 30 s; 60 °C, 1 min; 72 °C, 1 min) × 30 cycles; 72 °C, 6 min; 4 °C, 2 min. PCR products were digested with a restriction enzyme Mlsl recognizing tgg/cca at 37 °C for 3 h, deactivated at 65 °C for 20 min, and then fragments were imaged on an agarose gel. The wild-type (WT) allele produced two bands at 285 bp and 215 bp. Due to the silent mutation, the mutant allele only gave a band at 499 bp.
Rotarod test
Wild-type Lrsam1+/+ and Lrsam1C698R knock-in mice were placed on an accelerating (0.1 rpm/second) rotarod (Columbus Instruments, Columbus, OH) that progressed from 16 to 28 rpm over 2 min. If mice jumped off, turned around, or hung to the rod, they were given an additional attempt for the trial (up to a maximum of 3 attempts for any given trial). Once mice were in position, facing the correct direction, the device was turned on, and they walked until they lost balance and fell off or until the 2-min run-time ended. All mice were given 2 days of training prior to the test. Each mouse underwent 3 trials a day for 3 days following the training, with each trial separated by 30-min rest. The results were averaged for each animal. The cohorts of mice at 6- and 8-months of age were two separate groups; and were tested concurrently on the Rotarod. This was not a longitudinal test on the same group of mice at two different time points.
Hindlimb clasping test
Mice were held by the tail 30 cm above a tabletop, and video-recorded for 20 s. Abnormal postures such as hindlimb clasping and sustained straining of paws were recorded. Videos were analyzed by a researcher blinded to the genotype. The time span for clasping and paw straining was also calculated.
Nerve conduction studies (NCS)
NCS data were acquired as described21. Mice were anesthetized with isoflurane. CMAP was recorded from the intrinsic foot muscles using needle electrodes. Stimulation electrodes were placed in the mouse sciatic notch and at the ankle and recording was made on the paw. Supramaximal stimulations at each stimulation point evoked CMAP; and conduction velocity was calculated between the two stimulation points.
Sciatic nerve semithin section
This technique has been described previously22. Sciatic nerves were dissected and fixed in 4% paraformaldehyde and 2.5% glutaraldehyde mixture. Nerves embedded in Epon were sectioned to 1 µm thickness. The entire field of transverse sections of each nerve was imaged for morphometric analysis.
Nerve morphometrics
40X light images of mouse sciatic nerves were taken using a Leica THUNDER microscope. The segmentation model was developed based on a U-net architecture14. U-net architecture consists of interconnected expanding and contracting paths. This allows the model to localize image features, while retaining the original image detail. The model was implemented in ORS Dragonfly software, trained with ~ 150 mouse sciatic nerve images and their respective myelin segmentations. Extensive data augmentations of training were applied to the images, including image scaling, rotation, and other transformations. This approach allowed for more robust learning. The trained model was applied to the images to generate segmentations with isolated myelin and axons from the background. Each axon's inner and outer areas were measured. Then, each fiber’s diameter, myelin thickness, and g-ratio were calculated.
Primary mouse fibroblast culture
The primary fibroblasts were derived from toe skins of 2-month-old Lrsam1+/+,Lrsam1+/C698R and Lrsam1C698R/C698R mice. The tissue homogenate was incubated in DMEM high-glucose medium (Cat# 11995, Thermo Fisher Scientific) containing 20% FBS and 0.6 mg/ml collagenase II (Cat# LS004205, Worthington Biochemical) for 12 h at 37 °C incubator. Thereafter, cells were pelleted by centrifugation, washed with PBS and cultured in DMEM/10% FBS at 37 °C in a humidified atmosphere containing 5% CO2. Fibroblast outgrowth started at day 3–5. For the experiments, we used cells out of the first three passages of the primary culture.
Co-immunoprecipitation (co-IP) and immunoblotting
This technique has been described6. Whole-cell proteins were extracted using RIPA buffer (Cat# R0278, Sigma). Nuclear or cytoplasmic fractionation of mouse fibroblasts was isolated using NE-PER® Nuclear and Cytoplasmic Extraction Reagents (Cat#78833, Thermo Fisher Scientific). Cell lysates were incubated with primary antibodies overnight at 4 °C. Protein G agarose beads (Cat# 15920-010, Life technologies) were added for another 2 h incubation at 4 °C. Samples were eluted with Laemmli sample buffer, loaded into SDS-PAGE gels, and transferred to a PVDF membrane. The membranes were blotted with 5% milk and incubated overnight at 4 °C with primary antibodies and followed by secondary antibodies. The immune complexes were detected by the enhanced chemoilluminescence (Cat# NEL103001, Perkin Elmer). In some cases, the blots were stripped and re-probed with additional antibodies. Quantification of band intensity was performed using the ImageJ (http://rsbweb.nih.gov/ij/).
Sciatic nerve crush injury
While the mouse was under anesthesia with avertin (250 mg/kg, Cat #T48402, Sigma-Aldrich), a 1–2 cm incision just below the sciatic notch was made to expose the sciatic nerve. Forceps (Cat #11254-20, Fine Science Tools) were used to squeeze the nerve for 30 s; and turned 90 degrees to squeeze for another 30 s, which ensured that the whole nerve was crushed. Once the wound was clipped, Carprofen was administered over 3 days. NCS were performed immediately prior to the surgery and after the procedure to track the recovery of nerve function. The contralateral sciatic nerve was used as a sham control. The veterinarians and experimenters are always required to closely monitor the mouse's body weight after surgeries per IACUC protocol and found no significant weight loss. Mice among different experimental groups were evaluated simultaneously for NCS and tissue collections in nerve injury experiments.
Statistical analysis
The data were represented as the mean ± SD. P-values were obtained from the Student’s two-tailed t-test or repeated-measures ANOVA. For NCS data, we used a linear mixed model where time and genotype were coded as fixed effects on CMAP and CV. A linear mixed model was also chosen to analyze the crush nerve experiments as we were interested in determining effects on continuous outcome variables and dealing with clustered data (thousands of nerve fibers in each animal for morphometric analysis, as well as repeated measurements as part of NCS). Direct comparisons between genotypes were accomplished using a two-tailed t-test. We compared continuous variables between genotype/surgery status groups for morphometric analysis using another linear mixed model with genotype and surgical status as fixed effects. All statistical analysis was performed using SPSS version 27. P-value < 0.05 was used to determine significance.
Data availability
All data generated or analyzed in this study are provided in the Supplementary Information/Source data file. Additional details on protocols used in this study are available from the corresponding author on request. ORS Dragonfly myelin segmentation model is also available upon request.
References
Pipis, M., Rossor, A. M., Laura, M. & Reilly, M. M. Next-generation sequencing in Charcot-Marie-Tooth disease: Opportunities and challenges. Nat. Rev. Neurol. 15, 644–656. https://doi.org/10.1038/s41582-019-0254-5 (2019).
Morena, J., Gupta, A. & Hoyle, J. C. Charcot-Marie-tooth: From molecules to therapy. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20143419 (2019).
Li, J. Inherited neuropathies. Semin. Neurol. 32, 204–214. https://doi.org/10.1055/s-0032-1329198 (2012).
Guernsey, D. L. et al. Mutation in the gene encoding ubiquitin ligase LRSAM1 in patients with Charcot-Marie-Tooth disease. PLoS Genet. 6, 56. https://doi.org/10.1371/journal.pgen.1001081 (2010).
Hakonen, J. E. et al. LRSAM1-mediated ubiquitylation is disrupted in axonal Charcot-Marie-Tooth disease 2P. Hum. Mol. Genet. 26, 2034–2041. https://doi.org/10.1093/hmg/ddx089 (2017).
Hu, B., Arpag, S., Zuchner, S. & Li, J. A novel missense mutation of CMT2P alters transcription machinery. Ann. Neurol. 80, 834–845. https://doi.org/10.1002/ana.24776 (2016).
Nicolaou, P. et al. A novel LRSAM1 mutation is associated with autosomal dominant axonal Charcot-Marie-Tooth disease. Eur. J. Hum. Genet. 21, 190–194. https://doi.org/10.1038/ejhg.2012.146 (2013).
Peeters, K. et al. Charcot-Marie-Tooth disease type 2G redefined by a novel mutation in LRSAM1. Ann. Neurol. 80, 823–833. https://doi.org/10.1002/ana.24775 (2016).
Weterman, M. A. et al. A frameshift mutation in LRSAM1 is responsible for a dominant hereditary polyneuropathy. Hum. Mol. Genet. 21, 358–370. https://doi.org/10.1093/hmg/ddr471 (2012).
Engeholm, M. et al. A novel mutation in LRSAM1 causes axonal Charcot-Marie-Tooth disease with dominant inheritance. BMC Neurol. 14, 118. https://doi.org/10.1186/1471-2377-14-118 (2014).
Reilich, P. et al. Location matters—genotype-phenotype correlation in LRSAM1 mutations associated with rare Charcot-Marie-Tooth neuropathy CMT2P. Neuromuscul. Disord. 31, 123–133. https://doi.org/10.1016/j.nmd.2020.11.011 (2021).
Peretti, A. et al. LRSAM1 variants and founder effect in French families with ataxic form of Charcot-Marie-Tooth type 2. Eur. J. Hum. Genet. 27, 1406–1418. https://doi.org/10.1038/s41431-019-0403-8 (2019).
Palaima, P., Berciano, J., Peeters, K. & Jordanova, A. LRSAM1 and the RING domain: Charcot-Marie-Tooth disease and beyond. Orphanet. J. Rare Dis. 16, 74. https://doi.org/10.1186/s13023-020-01654-8 (2021).
Moiseev, D., Hu, B. & Li, J. Morphometric analysis of peripheral myelinated nerve fibers through deep learning. J. Peripher. Nerv. Syst. 24, 87–93. https://doi.org/10.1111/jns.12293 (2019).
Lim, E. F. et al. AlphaB-crystallin regulates remyelination after peripheral nerve injury. Proc. Natl. Acad. Sci. USA 114, E1707–E1716. https://doi.org/10.1073/pnas.1612136114 (2017).
Menorca, R. M., Fussell, T. S. & Elfar, J. C. Nerve physiology: mechanisms of injury and recovery. Hand Clin. 29, 317–330. https://doi.org/10.1016/j.hcl.2013.04.002 (2013).
Arthur-Farraj, P. & Coleman, M. P. Lessons from injury: How nerve injury studies reveal basic biological mechanisms and therapeutic opportunities for peripheral nerve diseases. Neurotherapeutics 18, 2200–2221. https://doi.org/10.1007/s13311-021-01125-3 (2021).
Auer-Grumbach, M. et al. Rare variants in MME, encoding metalloprotease neprilysin, are linked to late-onset autosomal-dominant axonal polyneuropathies. Am. J. Hum. Genet. 99, 607–623. https://doi.org/10.1016/j.ajhg.2016.07.008 (2016).
Juneja, M., Burns, J., Saporta, M. A. & Timmerman, V. Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development. J. Neurol. Neurosurg. Psychiatry 90, 58–67. https://doi.org/10.1136/jnnp-2018-318834 (2019).
Walsh, M. E. et al. Use of nerve conduction velocity to assess peripheral nerve health in aging mice. J. Gerontol. A Biol. Sci. Med. Sci. 70, 1312–1319. https://doi.org/10.1093/gerona/glu208 (2015).
Hu, B. et al. Tuning PAK activity to rescue abnormal myelin permeability in HNPP. PLoS Genet. 12, e1006290. https://doi.org/10.1371/journal.pgen.1006290 (2016).
Zhang, X. et al. Mutation of FIG4 causes a rapidly progressive, asymmetric neuronal degeneration. Brain 131, 1990–2001. https://doi.org/10.1093/brain/awn114 (2008).
Acknowledgements
The authors wish to thank Dr. Scott Millis for his assistance on statistics, Dr. Robert Burgess for his assistance in knock-in mouse production, and Perry Vera for his assistance in the Rotarod test. This research is supported by grants from the National Institute of Neurological Disorders and Stroke (R01NS115748) and the Department of Veterans Affairs (IBX003385A).
Author information
Authors and Affiliations
Contributions
D.M., Z.W., and D.L. acquisition and analysis of data. J.L. supervision, conception and design of the study; revising the manuscript. B.H. conception and design of the study; acquisition and analysis of data; and drafting the manuscript and figures.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moiseev, D., Wazir, Z., Liu, D. et al. C698R mutation in Lrsam1 gene impairs nerve regeneration in a CMT2P mouse model. Sci Rep 12, 12160 (2022). https://doi.org/10.1038/s41598-022-15902-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-15902-3
- Springer Nature Limited