
Abstract
Allogeneic hematopoietic stem cell transplants (allo-HSCTs) dramatically reduce HIV reservoirs in antiretroviral therapy (ART) suppressed individuals. However, the mechanism(s) responsible for these post-transplant viral reservoir declines are not fully understood. Therefore, we modeled allo-HSCT in ART-suppressed simian-human immunodeficiency virus (SHIV)-infected Mauritian cynomolgus macaques (MCMs) to illuminate factors contributing to transplant-induced viral reservoir decay. Thus, we infected four MCMs with CCR5-tropic SHIV162P3 and started them on ART 6–16 weeks post-infection (p.i.), maintaining continuous ART during myeloablative conditioning. To prevent graft-versus-host disease (GvHD), we transplanted allogeneic MHC-matched α/β T cell-depleted bone marrow cells and prophylactically treated the MCMs with cyclophosphamide and tacrolimus. The transplants produced ~ 85% whole blood donor chimerism without causing high-grade GvHD. Consequently, three MCMs had undetectable SHIV DNA in their blood post-transplant. However, SHIV-harboring cells persisted in various tissues, with detectable viral DNA in lymph nodes and tissues between 38 and 62 days post-transplant. Further, removing one MCM from ART at 63 days post-transplant resulted in SHIV rapidly rebounding within 7 days of treatment withdrawal. In conclusion, transplanting SHIV-infected MCMs with allogeneic MHC-matched α/β T cell-depleted bone marrow cells prevented high-grade GvHD and decreased SHIV-harboring cells in the blood post-transplant but did not eliminate viral reservoirs in tissues.
Similar content being viewed by others
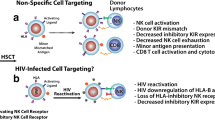


Allogeneic hematopoietic stem cell transplants (allo-HSCTs) are essential curative therapies for high-risk hematologic malignancies1,2,3, replacing recipient cells of hematopoietic origin with donor cells. In persons with HIV, allo-HSCTs also dramatically reduce viral reservoirs, enabling several transplant recipients to prevent or delay viral rebound after stopping antiretroviral therapy (ART)4,5,6,7,8,9,10. While allo-HSCTs are potent cellular immunotherapies, they are not scalable nor relevant to most people living with HIV without underlying hematologic cancers. Therefore, understanding the mechanism(s) of allo-HSCT-mediated reservoir decay can aid in designing safer and more efficacious treatment regimens. To this end, allo-HSCT components postulated to reduce viral reservoirs include pre-transplant conditioning regimens, protecting donor CD4+ T cells from infection with ART, graft-versus-host (GvH) responses, and donor HSCs lacking functional C-C chemokine receptor type 5 (CCR5)11,12.
To enter cells, HIV must sequentially bind to CD4 and a cellular coreceptor, primarily chemokine receptors CCR5 and CXCR4. These molecular interactions are disrupted in individuals homozygous for the CCR5∆32 gene variant (∆32/∆32), which prevents functional CCR5 from being expressed on cell surfaces, making ∆32/∆32 cells resistant to infection with CCR5-tropic HIV strains. The protective properties of ∆32/∆32 cells were employed in the hematologic cancer treatments of two men with HIV. In these men, transplanting ∆32/∆32 HSCs induced cancer remission and the rapid depletion of HIV reservoirs, allowing the “Berlin” and “London” patients to stop ART for at least 4 years without viral rebound5,6,8,13. Undoubtedly, the absence of CCR5 on donor cells prevented virus replication and the reestablishment of viral reservoirs post-transplant, supporting long-term HIV remission. However, additional transplant-related factors must have cleared pre-existing viral reservoirs.
Transplants with ∆32 heterozygous or CCR5 wild-type donors in ART-suppressed recipients provide further insights into eliminating latent HIV4,7,9,14,15. Notably, three CCR5 wild-type HSC recipients substantially reduced their viral reservoirs post-transplant and delayed HIV rebound up to 9 months after analytical treatment interruption (ATI)4,15. Mathematical modeling suggests that only hundreds to thousands of latently infected cells remained in these individuals at ART withdrawal16. Assuredly, maintaining ART during the peri-transplant period was an essential component of the allo-HSCT regimens, limiting HIV's spread from recipient cells to vulnerable donor cells17. Importantly, these allo-HSCTs also indicate that factors outside of donor genetics contribute to depleting viral reservoirs.
An attractive explanation for allo-HSCT-induced reservoir decay is the pre-transplant conditioning regimens used to reduce malignant cell burdens, create space for HSC engraftment, and ablate immune cells mediating graft rejections11,18. However, studies in humans and nonhuman primates (NHPs) indicate that pre-transplant conditioning regimens only transiently reduce viral reservoirs19,20,21,22,23,24, which are refilled by the homeostatic or antigen-driven proliferation of latently infected cells17,22. Thus, cytoreductive treatments alone are insufficient for eradicating HIV reservoirs, even when ART is maintained.
A more likely mechanism for allo-HSCT-mediated viral reservoir decay is the beneficial effects of GvH responses7,15,25. After allo-HSCTs, major histocompatibility complex (MHC)-matched donor T cells recognize minor alloantigens expressed by host leukocytes, eliminating malignant and HIV-infected cells simultaneously, thereby mediating graft-versus-tumor and graft-versus-viral reservoir (GvVR) effects, respectively26,27. But these GvH interactions can also result in life-threatening graft-versus-host disease (GvHD), a condition where donor cells attack non-hematopoietic origin cells or tissues. The primary cells mediating GvHD post-transplant are canonical T cells expressing αβ T cell receptors28,29, prompting the development of allo-HSCT regimens using cell preparations selectively depleted of αβ T cells30,31. Disentangling allo-HSCT's GvVR effects from GvHD may improve the safety and efficacy of cellular therapies targeting viral reservoirs.
In humans, variability in treatment regimens makes it challenging to investigate allo-HSCTs effects on HIV reservoirs. However, allo-HSCT treatment conditions can be standardized in NHP models of HIV infection. To this end, cynomolgus macaques (Macaca fascicularis) from the island of Mauritius are well-suited to model allo-HSCTs in people with HIV. Mauritian cynomolgus macaques (MCM) are descended from a small founder population32, resulting in limited genetic diversity, even at polymorphic loci. As a result, MCMs have only seven MHC haplotypes (termed M1-M7), facilitating the assembly of MHC-matched transplant pairs33,34,35. Moreover, SIV and simian-human immunodeficiency viruses (SHIVs) infections in MCMs recapitulate HIV replication and viral reservoir dynamics34,35,36,37, providing a platform for studying allo-HSCTs impact on viral reservoirs.
In this study, we modeled CCR5 wild-type allo-HSCTs in four SHIV-infected and ART-suppressed MCMs using our recently established TCRα/β-depleted MHC-matched bone marrow (BM) transplant model, which supports donor chimerism without GvHD38. As anticipated, the myeloablative conditioning regimen substantially depleted circulating leukocytes. As a result, we detected approximately 85% whole blood donor chimerism, with three MCMs having undetectable SHIV DNA in their peripheral blood mononuclear cells (PBMCs) post-transplant. However, SHIV-harboring cells persisted in various tissues, indicating that ART-suppression, allo-HSCs, and cytoreductive conditioning alone do not eliminate virally infected cells early after allo-HSCTs. Our findings suggest that extended ART treatments and GvH responses may be needed to eradicate viral reservoirs after allo-HSCTs.
Results
SHIV infection and ART suppression
We inoculated four MCMs (MCMs A-D) intravenously with SHIV162P3, a chimeric virus with a CCR5-tropic HIV envelope39. As anticipated, virus replication peaked by 3 weeks post-infection (p.i.) and declined thereafter (Fig. 1). To establish robust viral reservoirs, we allowed the SHIV infections to progress untreated for between 6 and 16 weeks p.i. before starting the MCMs on an ART regimen consisting of tenofovir, emtricitabine, and raltegravir. The ART regimen suppressed plasma viremia to undetectable levels within 3 weeks of initiation (< 100 viral RNA (vRNA) copy/eq per mL; Fig. 1). We maintained ART for the duration of the study, except where noted below, keeping plasma viremia undetectable, save for single viral blips in MCMs A and D.

Plasma viral loads after SHIV162P3 infection. Longitudinal plasma viral loads for the SHIV162P3-infected MCMs, with dashed lines indicating the qRT-PCR limit of detection (100 vRNA copy Eq/mL plasma). The gray boxes signify the timing of ART, the red arrows designate allo-HSCs infusions, and † indicate necropsies.
Allo-HSCT in SHIV-infected, ART-suppressed MCM
Our goal was to model allo-HSCTs with CCR5 wild-type cells in ART-suppressed people with HIV while minimizing the likelihood of severe GvHD. We collected BM aspirates from sex and MHC-matched MCM (Table 1) and depleted cells expressing α/β T-cell receptors (TCRs), removing T cells that are the primary driver of GvHD38. Approximately 1 month after starting ART, we delivered myeloablative doses of total body irradiation (TBI; two consecutive days of 5 Gray (Gy), 10 Gy total) and infused the MCMs with α/β T cell-depleted CD34+ HSCs (0.6–1.0 × 107/kg; Table 1). Post-transplant, we prophylactically treated the MCMs with cyclophosphamide and tacrolimus to prevent GvHD (Additional file 2: Table S1).
As expected, the myeloablative conditioning regimen rapidly depleted peripheral white blood cells (WBCs; Fig. 2a). However, the MCMs also developed chronic thrombocytopenia post-transplant (Fig. 2b), a common allo-HSCT complication40, which we treated with irradiated whole blood transfusions from SHIV-negative MCMs. Nevertheless, neutrophils reappeared in the peripheral blood of the MCMs by 21 days post-transplant, with absolute counts in MCM-C spiking above pre-transplant levels before rapidly declining (Fig. 2c). The remaining three MCMs had persistent neutropenia after their initial bursts of circulating neutrophils. Likewise, monocytes reemerged in the peripheral blood by 21 days post-transplant, with MCM-C again having the most robust recovery (Fig. 2d). The absolute monocyte counts were more variable than the neutrophil counts post-transplant, gradually declining in MCMs A and D, while rapidly increasing in MCMs B and C before their deaths.

Effect of allo-HSCTs on cell populations and platelets. Longitudinal absolute counts of (a) white blood cells, (b) platelets, (c) neutrophils, (d) monocytes, (e) lymphocytes, (f) CD4+ T cells, and (g) CD8+ T cells after allo-HSCTs. Counts are displayed as × 103/µL.
Lymphocytes recovered slowly after the allo-HSCTs (Fig. 2e). In MCM-C, lymphocytes reappeared in the peripheral blood approximately 3 weeks post-transplant and returned to near pre-transplant levels by day 38 post-transplant. Likewise, MCM-A exhibited a slow but steady increase in peripheral lymphocytes, reaching ~ 3500 cells/µL by day 71 post-transplant. In contrast, MCMs B and D had stunted lymphocyte recoveries, with absolute lymphocyte counts failing to reach 1000 cells/µL of blood by 5- and 7-weeks post-transplant, respectively. Similarly, CD4+ T cells failed to fully recover in the MCMs blood post-transplant (Fig. 2f). Finally, circulating CD8+ T cells increased in MCMs A, B, and D approximately 1-month post-transplant (Fig. 2g), fluctuating in MCM-A until its death. Meanwhile, CD8+ T cells poorly recovered in MCM-C, only reaching 86 CD8+ T cells/µL blood at 42 days post-transplant.
We measured post-transplant donor engraftment by deep sequencing single-nucleotide polymorphisms (SNPs) unique to the donor and recipient MCMs. Despite low absolute leukocyte counts, the allo-HSCT recipients exhibited greater than 85% whole blood donor chimerism by 3 weeks post-transplant, followed by modest declines to approximately 80% donor chimerism (Fig. 3a). Notably, MCM-A, the longest surviving transplant recipient, maintained 70–90% donor chimerism for 2 months until euthanasia. Additionally, we measured donor chimerism of circulating myeloid (monocytes) and lymphoid (B and T cells) lineages in MCMs A, C, and D at necropsy (cells from MCM-B were unavailable for analysis). The frequencies of donor-derived myeloid and lymphoid cells in MCMs C and D ranged between 65 and 93% of total cells (Fig. 3b), consistent with the whole blood chimerism results (Fig. 3a). In MCM-A, 78% of the B and T cells were of donor origin (Fig. 3b), similar to the 83% of donor cells detected in the whole blood. However, only 45% of the monocytes were of donor origin. Collectively, these results indicate that donor leukocytes replaced recipient cells early after transplant, with notable donor lymphocyte engraftment despite using α/β TCR-depleted grafts.

Donor chimerism after allo-HSCT. We determined donor chimerism levels post-transplant by sequencing SNPs differing between the donor and recipient. (a) Longitudinal percent donor chimerism in whole blood after allo-HSCT. (b) At necropsy, the percent donor chimerism among flow cytometry sorted PBMC monocytes (CD45midCD14+ lymphocytes), T cells (CD14-CD45hiCD3+ lymphocytes), and B cells (CD14-CD45hiCD20+).
Pre- and post-ART viral reservoirs
We designed our GvHD prophylaxis regimen of cyclophosphamide and tacrolimus to minimize high-grade GvHD. As anticipated, GvHD was limited post-transplant with only MCM-A exhibiting transient skin lesions (white flaking/dry skin) that resolved with minimal intervention. Additionally, MCMs C and D had diarrhea, which may have been a TBI side effect. Furthermore, the MCMs had no significant abnormalities in their liver functions or elevated total bilirubin post-transplant, indicating an absence of hepatic GvHD.
We measured cell-associated vDNA in PBMCs and inguinal lymph nodes to determine the effect of ART, myeloablative conditioning, and allo-HSCTs on viral reservoirs in the absence of severe GvHD (Fig. 4). Before starting ART, we detected more SHIV-harboring cells in the inguinal lymph nodes (range 743–2680 copy Eq/1 × 106 cells) than the PBMC (range 255–512 copy Eq/1 × 106 cells), consistent with lymph nodes being a primary viral reservoir41,42. Additionally, the lymph nodes of MCMs A and B contained more SHIV-infected cells than MCM-D, potentially due to these MCMs starting ART later in infection (14–16 weeks versus 6 weeks). Lastly, the number of SHIV-infected cells declined in the blood of all the MCMs between 36 and 64 days post-transplant, with MCMs A, B, and D having undetectable vDNA in their PBMCs (Fig. 4a).

SHIV DNA in allo-HSC transplanted MCMs. We measured copies of vDNA in (a) PBMCs and (b) peripheral lymph nodes before ART initiation, after ART but before allo-HSCTs, and post-transplant. (c) SHIV DNA levels in the tissues of MCMs B, C, and D at necropsy. Total SHIV DNA was measured by digital droplet PCR using SIV gag-specific primers and normalized to 106 cells by measuring macaque RNaseP p30, RPP30, in companion ddPCR assays. Post-transplant peripheral lymph node samples from MCM-B and MCM-D were unavailable for analysis.
Nevertheless, virally infected cells persisted in other tissues. MCMs-B, -C, and -D remained on ART until being necropsied, enabling a more thorough examination of post-transplant viral reservoirs. In MCM-C, we detected more vDNA in the inguinal lymph nodes at necropsy than in pre-transplant samples (1151 vs. 4807 vDNA copy Eq/million cells; Fig. 4b). This animal developed viral-induced colitis with ulcers post-transplant, potentially expanding latently infected cells or promoting localized virus replication, spreading SHIV-harboring cells into secondary lymphoid tissues. Nevertheless, we found vDNA outside of the secondary lymphoid tissues in the three MCMs, including the bone marrow, lung, and jejunum (Fig. 4c). Together these results demonstrate that viral reservoirs persist in various tissues early after allo-HSCTs without GvHD.
SHIV rapidly rebounds after removing ART
ATIs are used in HIV cure studies to evaluate treatment efficacy43,44. Therefore, we modeled ATI in MCM-A, an animal with a single viral blip but otherwise suppressed plasma viremia while on ART. One day before stopping ART, day 62 post-transplant, we collected blood and axillary lymph nodes to measure cell-associated SHIV. In these samples, we found no vDNA in the PBMCs but detected 271 vDNA copies/million cells in the lymph nodes, a slight decrease from pre-transplant inguinal lymph nodes (359 vDNA copies/million cells; Fig. 4b). Nevertheless, SHIV rebounded rapidly after removing ART (Fig. 1). Plasma viremia was 1 × 107 vRNA copy eq/mL 7 days after stopping treatment, roughly equivalent to peak acute viremia, and spiked to nearly 1 × 108 vRNA copy eq/mL plasma at 10 days post-ART. These results show that latent reservoirs can reactivate shortly after treatment interruption and initiate viral rebound early after allo-HSCTs.
Discussion
Allo-HSCT-associated HIV reservoir decay likely results from parallel processes protecting donor cells from infection and eliminating pre-existing latently infected cells10,11. Here, we employed the clinically relevant SHIV/MCM model to determine the effect of continuous ART and allo-HSCTs on viral reservoirs in the absence of high-grade GvHD. We recently reported that transplanting MCMs with MHC-matched TCRα/β+-depleted HSCs and prophylactically treating them with cyclophosphamide and tacrolimus prevents GvHD post-transplant38. Employing this regimen, only one SHIV-infected MCM had transient skin lesions post-transplant. These results contrast with previous NHP allo-HSCT studies where T-cell replete HSCs quickly induced GvHD in ART-suppressed SHIV-infected rhesus macaques45 and uninfected MCMs46.
Although we prevented GvHD, removing alloreactive TCRα/β+ T cells from the bone marrow grafts may have inhibited viral reservoir clearance. Still, our results agree with a previous study transplanting MHC-haploidentical HSCs into ART-suppressed SHIV-infected rhesus macaques that developed GvHD post-transplant49. Both studies observed reduced SHIV DNA levels in the blood and lymph nodes post-transplant, with SHIV reservoirs persisting in other tissues. Further, post-transplant follow-up was short in both studies, supporting the need for extended times on ART post-transplant to clear pre-existing latently infected cells.
Supplementing allo-HSCTs with cellular therapies may improve viral reservoir clearance47,48. Donor lymphocyte infusions (DLIs) can drive relapsing hematologic tumors into remission49. Conceivably, incorporating DLIs into allo-HSCT treatment plans could similarly augment GvVR responses25. However, since DLIs involve transferring polyclonal T cells, they also carry a significant risk of GvHD50. Therefore, targeted cellular immunotherapies maximizing GvVR effects while minimizing GvHD side effects are preferred. For instance, repurposing cancer adoptive cell therapies may boost GvVR activity51,52,53. HIV-specific chimeric antigen receptor (CAR) T- and NK-cell therapies are promising HIV cure strategies54 despite early iterations having limited impact on HIV reservoirs55,56,57,58. Alternatively, focusing T cells on minor histocompatibility antigens (mHAgs) exclusively expressed by host leukocytes may promote GvVR effects without causing GvHD59. We recently identified a mHAg in APOBEC3C that is expressed preferentially by MCM immune cells60. Hence, this epitope may serve as a model antigen for testing mHAg-targeted cellular immunotherapies in NHPs and examining their capacity to eradicate viral reservoirs.
In this study, we used the pathogenic SIV-HIV chimera SHIV162P3 to model HIV infections. This recombinant virus contains the env gene from the clade B isolate HIV-1 SF162 inserted into the SIVmac239 genomic backbone61. Importantly, SHIV162P3 exclusively uses CCR5 as a coreceptor, similar to most transmitted HIV-1 strains39,62. This coreceptor usage contrasts with common SIV strains, which can use alternate chemokine receptors and G-protein coupled receptors for cell entry63,64. However, SHIV162P3 replication is less robust than most SIV strains, resulting in lower chronic phase viral loads65,66. Indeed, MCM-A and -B spontaneously suppressed viremia to approximately 103 vRNA copies/mL plasma by 4 months post-infection. Nevertheless, SHIV162P3 established pre-transplant viral reservoirs in our MCMs consistent with previous SHIV/ SIV studies41,45,67,68.
Our study has several limitations. First, its small size and lack of a control group transplanted with TCR α/β-replete HSCs make it difficult to draw firm conclusions from the results. Second, ART was started 3–6 weeks before transplantation. While this provided sufficient time to suppress SHIV replication, it does not reflect the prolonged ART typical of HIV-infected allo-HSCT recipients4,5,8,14,15. Third, we could not determine whether virally infected cells were of donor or recipient origin post-transplant. A study by Colonna et al. exploited haploidentical allo-HSCTs in rhesus macaques to sort donor and recipient cells based on differential MHC expression, finding that most SHIV-infected cells were from the recipients early after transplantation45. However, our MHC-matched MCMs lacked unique surface markers to distinguish donor and recipient cells, prohibiting similar analyses. Lastly, we achieved poor donor cell engraftment, hindering post-transplant analyses and potentially limiting SHIV reservoir depletion. Consequently, we could not quantify donor chimerism in tissues or measure post-transplant antiviral and GvVR immune responses. The limited lymphocyte recovery also hampered our ability to longitudinally measure SHIV-infected cells post-transplant. Therefore, less toxic pre-transplant conditioning regimens, higher HSC doses, or T-cell replete HSCs may be necessary for sustaining donor cell engraftment. To this end, Burwitz et al. established an allo-HSCT model in MHC-matched MCMs that achieves durable post-transplant donor chimerism with reduced-intensity conditioning but has an increased risk of GvHD46. Thus, allo-HSCTs in NHPs will likely need to balance full donor engraftment with suppressing GvHD to dissect the mechanisms of GvVR effects.
Conclusions
In summary, MHC-matched bone marrow transplants in ART-suppressed SHIV-infected MCMs reduced, but did not eliminate, viral reservoirs early after transplantation. However, depleting TCRα/β T cells from the HSCs and post-transplant prophylaxis prevented high-grade GvHD. Lastly, our results indicate that this model has room for improvement because the treatment regimen was insufficient for robust donor cell engraftment, limiting lymphocyte recovery and making a detailed analysis of tissues and GvVR immune responses difficult.
Methods
Ethics statement and animal care
The staff at the Wisconsin National Primate Research Center (WNPRC) cared for the Cynomolgus macaques (Macaca fascicularis) according to the regulations and guidelines of the University of Wisconsin Institutional Animal Care and Use Committee, which approved this study (protocol g005424) following the recommendations of the Weatherall Report and the principles described in the National Research Council's Guide for the Care and Use of Laboratory Animals and in compliance with ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines. The MCMs were closely monitored for signs of stress or pain. In consultation with the WNPRC veterinarians, the MCMs were euthanized by anesthetizing with ketamine (≥ 15 mg/kg, intramuscular) followed by intravenous administration of sodium pentobarbital (> 50 mg/kg) when they developed an untreatable opportunistic infection, inappetence, and/or progressive decline in condition.
MHC typing
MHC genotyping was performed by Genetic Services at WNPRC as previously described69. Briefly, genomic DNA (gDNA) was isolated from whole blood or PBMCs and used as templates in polymerase chain reactions (PCRs) with primers flanking exon 2 of MHC class I (Mafa-A, Mafa-B, Mafi-I, and Mafa-E) and class II (Mafa-DRB, Mafa-DQA, Mafa-DQB, Mafa-DPA, and Mafa-DPB) loci. The PCR reactions were generated with a Fluidigm Access Array, which permitted all reactions to be multiplexed in a single experiment. After cleanup with AMPure beads (Beckman Coulter), the amplicons were sequenced on an Illumina MiSeq. The resulting sequences were mapped against a custom database of MCM class I and II sequences to assign one of seven MCM haplotypes (M1-M7).
SHIV infections and viral loads
We inoculated the MCMs intravenously with 500 median tissue culture infectious doses (TCID50) of SHIV162P339. Plasma viral loads were measured via quantitative real-time polymerase chain reactions (qRT-PCRs) as previously described70,71. Briefly, vRNA was isolated from plasma using the Maxwell Viral Total Nucleic Acid Purification kit (Promega) on a Maxwell 48 RSC instrument (Promega). Next, vRNA was reverse transcribed into complementary DNA (cDNA), amplified using the TaqMan Fast Virus 1-Step Master Mix qRT-PCR kit (Invitrogen) on a LightCycler 480 or LC96 instrument (Roche), and quantified by interpolation onto a standard curve of tenfold serial dilutions of an SIV gag in vitro transcript. The assay limit of detection was 100 vRNA copies/mL.
Antiretroviral treatment
We treated the SHIV-infected MCMs with a combination of reverse transcriptase inhibitors tenofovir (PMPA) and emtricitabine (FTC) and integrase inhibitor raltegravir (RAL). PMPA and FTC were pre-formulated at concentrations of 20 mg/mL and 40 mg/mL in water containing sodium hydroxide (NaOH) and administered subcutaneously once daily at 1 mL/kg of body weight. 100 mg of RAL was mixed twice daily with food72. Gilead Sciences kindly provided PMPA and FTC, and Merck kindly provided RAL through material transfer agreements.
Donor bone marrow collection and TCRα/β cell depletion
We prepared the HSCs as previously described38. Briefly, we collected BM from sex and MHC-matched MCMs (Table 1) by aspirating up to 5 mL from four sites, 20 mL total, and removed red blood cells using ACK lysis buffer (Thermo Fisher Scientific). Next, we washed the cells twice with phosphate-buffered saline and incubated them with anti-TCRα/β allophycocyanin (APC) antibodies (clone R73, BioLegend) for 20 min, followed by incubating with anti-APC microbeads (Miltenyi Biotec) for an additional 20 min at 4 °C. Finally, we passed the stained cells through two LS columns (Miltenyi Biotec) stacked on top of one another, collecting both negative and positive cell fractions, and cryopreserving them at 30 million/mL in serum-free expansion medium (SFEM; Stem Cell Technologies) containing 5% fetal bovine serum and 10% dimethylsulfoxide (DMSO) until further use.
Bone marrow transplants and care
First, we treated the MCMs with daily oral antibiotics trimethoprim (200 mg) and sulfamethoxazole (40 mg) to decontaminate the gut, starting 7 days pre-transplant (− 7) and continuing until day-1. Next, we started the MCMs on systemic bacterial prophylaxis by administering ceftriaxone (50 mg/kg) on day-2; MCM-D was switched to cefazolin (25 mg/kg twice daily) at day-1, and maintaining treatment until absolute neutrophil counts stabilized. On day-2, the MCMs received five separate 1 Gy fractions of TBI on two consecutive days (10 Gy total), sparing the lungs and eyes as previously described38. One day before transplantation, we thawed cryopreserved TCRα/β-depleted BM fractions at 37˚C and cultured them overnight in SFEM containing 100 ng/mL human stem cell factor (SCF, Peprotech), 100 ng/mL human Fms-related tyrosine kinase 3 (FLT3) ligand (Peprotech), and 50 ng/mL human thrombopoietin (TPO, Peprotech). On the day of transplantation, we resuspended the BM cells in 15 mL of Plasma-Lyte (Baxter), a nonpyrogenic isotonic solution, supplemented with 2% autologous serum and 5 U/mL heparin, and infused the cell suspension intravenously. Next, we prophylactically treated the MCMs for GvHD using cyclophosphamide (50 mg/kg) on days 4 and 5 post-transplant, followed by twice-daily tacrolimus (0.01 mg/kg) starting at day 5, maintaining tacrolimus serum levels between 5 and 15 ng/mL. To prevent fungal infections, we treated the MCMs with once-daily fluconazole (5 mg/kg) starting on day 0. Lastly, we treated MCMs C and D with oral eltrombopag (1.5 mg/kg) and N-acetyl l-cysteine (50 mg/kg), respectively, to support platelet engraftment. See Additional file 2: Table S1 for treatment details.
Measuring peripheral leukocyte populations and GvHD
Following transplantation, we collected peripheral blood from the MCMs twice per week for the first month, followed by bi-weekly blood draws for the next 2 months and monthly thereafter. We used an XS-1000i automated hematology analyzer (Sysmex) to measure platelets and leukocyte populations. In addition, GvHD was evaluated as previously described73.
We quantified CD4+ and CD8+ T cells by incubating 100 µL of whole blood with anti-CD8-Brilliant Violet 421 (clone SK1; BioLegend), anti-CD4-APC (clone L200; BD-Pharmingen), and anti-CD3-Alexa 700 (clone SP34; BD-Pharmingen) antibodies with Live/Dead Fixable Near-IR vital dye (ThermoFisher Scientific) for 30 min at 4˚C. Next, we lysed the red blood cells by adding 1 mL of FACS Lysing Solution (BD Biosciences) and incubated the samples for 10 min at room temperature. We washed the cells three times with FACS buffer (phosphate-buffered saline supplemented with 2% fetal calf serum), fixed with 2% paraformaldehyde, and ran the samples on a BD-LSR-II flow cytometer using FACSDiva software (Becton Dickinson). Finally, we analyzed the flow cytometry data using FlowJo software (Treestar), determining the absolute CD4+ and CD8+ T cell counts by multiplying the frequency of CD4+ or CD8+ T cells (singlets, live, CD3+, CD4+ or CD8+) by the white blood counts per microliter of blood from the matching complete blood counts (Additional file 1: Figure S1).
Lymphocyte isolation from tissues
We isolated lymphocytes from the blood, lymph nodes, spleen, and bone marrow as previously described74. Briefly, we isolated PBMCs from EDTA-anticoagulated blood by density gradient centrifugation using Ficoll-Paque PLUS (GE Healthcare), removing contaminating red blood cells with ACK lysis buffer. We collected peripheral lymph nodes before ART (MCM A and B: 77 days post-infection (dpi); MCM-C: unavailable; and MCM-D: 34 dpi), after ART but before allo-HSCT (MCM-A: 23 days post-ART; MCM-B: 24 days post-ART; MCM-C: 18 days post-ART; and MCM-D: 12 days post-ART). Additionally, we collected lymph nodes from MCM-A 62 days post-transplant but before ART cessation and the necropsies for MCM C and D, which occurred on days 46 and 39 post-transplant, respectively, while they were on ART. We were unable to collect lymph nodes at MCM-B's necropsy for analysis.
We isolated lymphocytes from the lymph nodes, spleen, and bone marrow by dicing the tissues with scalpels and forcing the cells through 100-µm cell strainers to remove connective tissues. Lymphocytes were enriched from the cell suspensions using Ficoll-Paque PLUS density gradient centrifugation.
Measuring donor chimerism
We identified SNPs that distinguished the donor and recipient MCMs using a panel of 12 SNPs (see Additional file 3:Table S2) and the rhAMP SNP Genotyping System (Integrated DNA Technologies) as described previously38,75, giving preference to homozygous/homozygous mismatches. We performed the rhAMP assays in triplicate using a LightCycler 96 Instrument (Roche Molecular Systems) in the endpoint analysis mode.
We quantified donor chimerism in whole PBMCs or immune subsets by sequencing the diagnostic SNPs with an Illumina MiSeq using previously described primer sets, PCR conditions, and analysis pipeline (see also Additional file 4:Table S3 and Additional file 5:Table S4)38. To isolate specific immune subsets, we stained PBMCs with anti-CD3 FITC (clone SP34; BD Pharmingen), anti-CD14 PerCP-Cy5.5 (clone M5E2; BioLegend), anti-CD20 APC (clone 2H7; BioLegend), anti-CD45 PE (clone D058-1283; BD Pharmingen), and Live/Dead Fixable Near-IR vital dye (ThermoFisher Scientific) and sorted into monocyte (singlets, live, lymphocytes, CD45mid, CD14+ cells), T lymphocyte (singlets, live, lymphocytes, CD14−, CD45hi, CD3+), or B lymphocyte (singlets, live, lymphocytes, CD14−, CD45hi, CD20+) populations using a BD FACSJazz cell sorter. We used between 179 and 61,251 cells for gDNA extraction and sequencing for each sorted population.
Quantifying cell-associated viral DNA
We extracted gDNA from tissues or PBMCs using the DNeasy Blood & Tissue Kit (Qiagen) per the manufacturer's instructions, except eluting the DNA twice with separate 75 µL of molecular grade water (150 µL total volume). We determined the concentration of DNA using a Nanodrop spectrophotometer (ThermoFisher Scientific), and we digested up to 3 µg of DNA with 3 µL of EcoRI-HF restriction enzyme (New England Biolabs) per 50 µL.
We performed two digital droplet PCR (ddPCR) reactions for each sample: one quantifying vDNA and the other determining total cell number. First, we quantified vDNA using the primers and probes for SIV gag and the cycling conditions described by Gama et al.76. Second, we normalized vDNA copies to cell numbers by quantifying the RNase P (RPP30) gene, as previously described77. We generated droplets and read the samples on a QX200 ddPCR system (Bio-Rad). We quantified the target genes in duplicate for each sample across at least two independent runs.
Data availability
The data that support the findings of this study are available within the paper and Supplementary files. The datasets generated during the study are available from the corresponding author upon reasonable request.
Abbreviations
- Allo-HSCTs:
-
Allogeneic hematopoietic stem cell transplants
- ART:
-
Antiretroviral therapy
- HIV:
-
Human immunodeficiency virus
- SHIV:
-
Simian-human immunodeficiency virus
- MCMs:
-
Mauritian cynomolgus macaques
- GvH:
-
Graft-versus-host
- GvHD:
-
Graft-versus-host disease
- GvVR:
-
Graft-versus-viral reservoir
- CCR5:
-
C-C chemokine receptor type 5
- CXCR4:
-
C-X-C chemokine receptor type 4
- CCR5∆32:
-
C-C chemokine receptor type 5 with 32 base-pair deletion
- MHC:
-
Major histocompatibility complex
- NHPs:
-
Nonhuman primates
- BM:
-
Bone marrow
- p.i.:
-
Post-infection
- Gy:
-
Gray
- WBCs:
-
White blood cells
- ATIs:
-
Analytical treatment interruptions
- DLIs:
-
Donor lymphocyte infusions
- CAR:
-
Chimeric antigen receptor
- mHAgs:
-
Minor histocompatibility antigens
- WNPRC:
-
Wisconsin national primate research center
- gDNA:
-
Genomic deoxyribonucleic acid
- PCRs:
-
Polymerase chain reactions
- Mafa:
-
Macaca fascicularis
- TCID50 :
-
Median tissue culture infectious doses
- qRT-PCRs:
-
Quantitative real-time polymerase chain reactions
- cDNA:
-
Complementary deoxyribonucleic acid
- PMPA:
-
Tenofovir (9-[9(R)-2-(phosphonomethoxy)propyl]adenine
- FTC:
-
Emtricitabine
- RAL:
-
Raltegravir
- SFEM:
-
Serum-free expansion medium
- TBI:
-
Total body irradiation
- SCF:
-
Stem cell factor
- FLT3:
-
Fms-related tyrosine kinase 3
- TPO:
-
Thrombopoietin
- PBMCs:
-
Peripheral blood mononuclear cells
- SNPs:
-
Single-nucleotide polymorphisms
- vDNA:
-
Viral deoxyribonucleic acids
- ddPCR:
-
Digital droplet polymerase chain reaction
- RPP30:
-
RNase P subunit p30
References
Dickinson, A. M. et al. Graft-versus-leukemia effect following hematopoietic stem cell transplantation for leukemia. Front. Immunol. 8, 496 (2017).
Falkenburg, J. H. & Warren, E. H. Graft versus leukemia reactivity after allogeneic stem cell transplantation. Biol. Blood Marrow Transplant. 17, S33–S38 (2011).
Warren, E. H. et al. Therapy of relapsed leukemia after allogeneic hematopoietic cell transplantation with T cells specific for minor histocompatibility antigens. Blood 115, 3869–3878 (2010).
Cummins, N. W. et al. Extensive virologic and immunologic characterization in an HIV-infected individual following allogeneic stem cell transplant and analytic cessation of antiretroviral therapy: A case study. PLoS Med. 14, e1002461 (2017).
Gupta, R. K. et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature 568, 244–248 (2019).
Gupta, R. K. et al. Evidence for HIV-1 cure after CCR5Δ32/Δ32 allogeneic haemopoietic stem-cell transplantation 30 months post analytical treatment interruption: A case report. Lancet HIV 7, e340–e347 (2020).
Henrich, T. J. et al. Long-term reduction in peripheral blood HIV type 1 reservoirs following reduced-intensity conditioning allogeneic stem cell transplantation. J. Infect. Dis. 207, 1694–1702 (2013).
Hutter, G. et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 360, 692–698 (2009).
Koelsch, K. K. et al. Impact of allogeneic hematopoietic stem cell transplantation on the HIV reservoir and immune response in 3 HIV-infected individuals. J. Acquir. Immune Defic. Syndr. 75, 328–337 (2017).
Prator, C. A., Donatelli, J. & Henrich, T. J. From Berlin to London: HIV-1 reservoir reduction following stem cell transplantation. Curr. HIV/AIDS Rep. 17, 385–393 (2020).
Smiley, S. T. et al. Progress toward curing HIV infections with hematopoietic stem cell transplantation. Clin. Infect. Dis. 60, 292–297 (2015).
Hutter, G. Stem cell transplantation in strategies for curing HIV/AIDS. AIDS Res. Ther. 13, 31 (2016).
Allers, K. et al. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood 117, 2791–2799 (2011).
Salgado, M. et al. Mechanisms that contribute to a profound reduction of the HIV-1 reservoir after allogeneic stem cell transplant. Ann. Intern. Med. 169, 674–683 (2018).
Henrich, T. J. et al. Antiretroviral-free HIV-1 remission and viral rebound after allogeneic stem cell transplantation: Report of 2 cases. Ann. Intern. Med. 161, 319–327 (2014).
Hill, A. L. et al. Real-time predictions of reservoir size and rebound time during antiretroviral therapy interruption trials for HIV. PLoS Pathog. 12, e1005535 (2016).
Eberhard, J. M. et al. Vulnerability to reservoir reseeding due to high immune activation after allogeneic hematopoietic stem cell transplantation in individuals with HIV-1. Sci. Transl. Med. 12, eaay9355 (2020).
Gyurkocza, B. & Sandmaier, B. M. Conditioning regimens for hematopoietic cell transplantation: One size does not fit all. Blood 124, 344–353 (2014).
Cillo, A. R. et al. Impact of chemotherapy for HIV-1 related lymphoma on residual viremia and cellular HIV-1 DNA in patients on suppressive antiretroviral therapy. PLoS ONE 9, e92118 (2014).
Cillo, A. R. et al. Plasma viremia and cellular HIV-1 DNA persist despite autologous hematopoietic stem cell transplantation for HIV-related lymphoma. J. Acquir. Immune Defic. Syndr. 63, 438–441 (2013).
Gabarre, J. et al. High-dose therapy plus autologous hematopoietic stem cell transplantation for human immunodeficiency virus (HIV)-related lymphoma: Results and impact on HIV disease. Haematologica 89, 1100–1108 (2004).
Henrich, T. J. et al. Human immunodeficiency virus type 1 persistence following systemic chemotherapy for malignancy. J. Infect. Dis. 216, 254–262 (2017).
Mavigner, M. et al. Persistence of virus reservoirs in ART-treated SHIV-infected rhesus macaques after autologous hematopoietic stem cell transplant. PLoS Pathog. 10, e1004406 (2014).
Simonelli, C. et al. Immune recovery after autologous stem cell transplantation is not different for HIV-infected versus HIV-uninfected patients with relapsed or refractory lymphoma. Clin. Infect. Dis. 50, 1672–1679 (2010).
Serrano, D. et al. Graft-versus-tumor effect after allogeneic stem cell transplantation in HIV-positive patients with high-risk hematologic malignancies. AIDS Res. Hum. Retrovir. 29, 1340–1345 (2013).
Griffioen, M., van Bergen, C. A. & Falkenburg, J. H. Autosomal minor histocompatibility antigens: how genetic variants create diversity in immune targets. Front. Immunol. 7, 100 (2016).
Warren, E. H. & Deeg, H. J. Dissecting graft-versus-leukemia from graft-versus-host-disease using novel strategies. Tissue Antigens 81, 183–193 (2013).
Huang, Y. et al. The role of alphabeta- and gammadelta-T cells in allogenic donor marrow on engraftment, chimerism, and graft-versus-host disease. Transplantation 72, 1907–1914 (2001).
Neipp, M. et al. T-cell depletion of allogeneic bone marrow using anti-alphabetaTCR monoclonal antibody: Prevention of graft-versus-host disease without affecting engraftment potential in rats. Exp. Hematol. 27, 860–867 (1999).
Or-Geva, N. & Reisner, Y. The evolution of T-cell depletion in haploidentical stem-cell transplantation. Br. J. Haematol. 172, 667–684 (2016).
Sahasrabudhe, K., Otto, M., Hematti, P. & Kenkre, V. TCR αβ+/CD19+ cell depletion in haploidentical hematopoietic allogeneic stem cell transplantation: A review of current data. Leuk. Lymphoma 60, 598–609 (2019).
Lawler, S. H., Sussman, R. W. & Taylor, L. L. Mitochondrial DNA of the Mauritian macaques (Macaca fascicularis): An example of the founder effect. Am. J. Phys. Anthropol. 96, 133–141 (1995).
Budde, M. L. et al. Characterization of Mauritian cynomolgus macaque major histocompatibility complex class I haplotypes by high-resolution pyrosequencing. Immunogenetics 62, 773–780 (2010).
O’Connor, S. L. et al. Comprehensive characterization of MHC class II haplotypes in Mauritian cynomolgus macaques. Immunogenetics 59, 449–462 (2007).
Wiseman, R. W. et al. Simian immunodeficiency virus SIVmac239 infection of major histocompatibility complex-identical cynomolgus macaques from Mauritius. J. Virol. 81, 349–361 (2007).
Antony, J. M. & MacDonald, K. S. A critical analysis of the cynomolgus macaque, Macaca fascicularis, as a model to test HIV-1/SIV vaccine efficacy. Vaccine 33, 3073–3083 (2015).
Burwitz, B. J. et al. Mauritian cynomolgus macaques share two exceptionally common major histocompatibility complex class I alleles that restrict simian immunodeficiency virus-specific CD8+ T cells. J. Virol. 83, 6011–6019 (2009).
D’Souza, S. S. et al. Transplantation of T-cell receptor α/β-depleted allogeneic bone marrow in nonhuman primates. Exp. Hematol. 93, 44–51 (2021).
Harouse, J. M. et al. Mucosal transmission and induction of simian AIDS by CCR5-specific simian/human immunodeficiency virus SHIV(SF162P3). J. Virol. 75, 1990–1995 (2001).
Bento, L., Canaro, M., Bastida, J. M. & Sampol, A. Thrombocytopenia and therapeutic strategies after allogeneic hematopoietic stem cell transplantation. J. Clin. Med. 11, 1364 (2022).
Cadena, A. M. et al. Persistence of viral RNA in lymph nodes in ART-suppressed SIV/SHIV-infected Rhesus Macaques. Nat. Commun. 12, 1474 (2021).
Wong, J. K. & Yukl, S. A. Tissue reservoirs of HIV. Curr. Opin. HIV AIDS 11, 362–370 (2016).
Julg, B. et al. Recommendations for analytical antiretroviral treatment interruptions in HIV research trials-report of a consensus meeting. Lancet HIV 6, e259–e268 (2019).
Lau, J. S. Y., Smith, M. Z., Lewin, S. R. & McMahon, J. H. Clinical trials of antiretroviral treatment interruption in HIV-infected individuals. AIDS 33, 773–791 (2019).
Colonna, L. et al. Evidence for persistence of the SHIV reservoir early after MHC haploidentical hematopoietic stem cell transplantation. Nat. Commun. 9, 4438 (2018).
Burwitz, B. J. et al. Allogeneic stem cell transplantation in fully MHC-matched Mauritian cynomolgus macaques recapitulates diverse human clinical outcomes. Nat. Commun. 8, 1418 (2017).
Ward, A. R., Mota, T. M. & Jones, R. B. Immunological approaches to HIV cure. Semin. Immunol. 51, 101412 (2020).
Zhou, Y., Maldini, C. R., Jadlowsky, J. & Riley, J. L. Challenges and opportunities of using adoptive T-cell therapy as part of an HIV cure strategy. J. Infect. Dis. 223, 38–45 (2021).
Kolb, H. J. et al. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood 86, 2041–2050 (1995).
Frey, N. V. & Porter, D. L. Graft-versus-host disease after donor leukocyte infusions: Presentation and management. Best Pract. Res. Clin. Haematol. 21, 205–222 (2008).
Mylvaganam, G., Yanez, A. G., Maus, M. & Walker, B. D. Toward T cell-mediated control or elimination of HIV reservoirs: Lessons from cancer immunology. Front. Immunol. 10, 2109 (2019).
Rust, B. J., Kiem, H. P. & Uldrick, T. S. CAR T-cell therapy for cancer and HIV through novel approaches to HIV-associated haematological malignancies. Lancet Haematol. 7, e690–e696 (2020).
Yang, H., Wallace, Z. & Dorrell, L. Therapeutic targeting of HIV reservoirs: How to give T cells a new direction. Front. Immunol. 9, 2861 (2018).
Mu, W., Carrillo, M. A. & Kitchen, S. G. Engineering CAR T cells to target the HIV reservoir. Front. Cell Infect. Microbiol. 10, 410 (2020).
Deeks, S. G. et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol. Ther. 5, 788–797 (2002).
Mitsuyasu, R. T. et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood 96, 785–793 (2000).
Scholler, J. et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 4, 132ra53 (2012).
Walker, R. E. et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood 96, 467–474 (2000).
Summers, C., Sheth, V. S. & Bleakley, M. Minor histocompatibility antigen-specific T cells. Front. Pediatr. 8, 284 (2020).
Weinfurter, J. T. et al. Identifying a minor histocompatibility antigen in mauritian cynomolgus macaques encoded by APOBEC3C. Front. Immunol. 11, 586251 (2020).
Luciw, P. A., Pratt-Lowe, E., Shaw, K. E., Levy, J. A. & Cheng-Mayer, C. Persistent infection of rhesus macaques with T-cell-line-tropic and macrophage-tropic clones of simian/human immunodeficiency viruses (SHIV). Proc. Natl. Acad. Sci. USA 92, 7490–7494 (1995).
Moore, J. P., Kitchen, S. G., Pugach, P. & Zack, J. A. The CCR5 and CXCR4 coreceptors–central to understanding the transmission and pathogenesis of human immunodeficiency virus type 1 infection. AIDS Res. Hum. Retrovir. 20, 111–126 (2004).
Sina, S. T., Ren, W. & Cheng-Mayer, C. Coreceptor use in nonhuman primate models of HIV infection. J. Transl. Med. 9(Suppl 1), S7 (2011).
Zhang, Y. et al. Use of inhibitors to evaluate coreceptor usage by simian and simian/human immunodeficiency viruses and human immunodeficiency virus type 2 in primary cells. J. Virol. 74, 6893–6910 (2000).
Pahar, B. et al. Control of viremia and maintenance of intestinal CD4(+) memory T cells in SHIV(162P3) infected macaques after pathogenic SIV(MAC251) challenge. Virology 387, 273–284 (2009).
Xu, H., Wang, X., Morici, L. A., Pahar, B. & Veazey, R. S. Early divergent host responses in SHIVsf162P3 and SIVmac251 infected macaques correlate with control of viremia. PLoS ONE 6, e17965 (2011).
Siddiqui, S. et al. Persistent viral reservoirs in lymphoid tissues in SIV-infected rhesus macaques of Chinese-origin on suppressive antiretroviral therapy. Viruses 11, E105 (2019).
Abreu, C. M. et al. Infectious virus persists in CD4+ T cells and macrophages in antiretroviral therapy-suppressed simian immunodeficiency virus-infected macaques. J. Virol. 93, e00065-19 (2019).
Wiseman, R. W. et al. Haplessly hoping: Macaque major histocompatibility complex made easy. ILAR J. 54, 196–210 (2013).
Cline, A. N., Bess, J. W., Piatak, M. Jr. & Lifson, J. D. Highly sensitive SIV plasma viral load assay: Practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J. Med. Primatol. 34, 303–312 (2005).
Friedrich, T. C. et al. Subdominant CD8+ T-cell responses are involved in durable control of AIDS virus replication. J. Virol. 81, 3465–3476 (2007).
Lewis, M. G. et al. Response of a simian immunodeficiency virus (SIVmac251) to raltegravir: A basis for a new treatment for simian AIDS and an animal model for studying lentiviral persistence during antiretroviral therapy. Retrovirology 7, 21 (2010).
Miller, W. P. et al. GVHD after haploidentical transplantation: A novel, MHC-defined rhesus macaque model identifies CD28- CD8+ T cells as a reservoir of breakthrough T-cell proliferation during costimulation blockade and sirolimus-based immunosuppression. Blood 116, 5403–5418 (2010).
Reynolds, M. R. et al. CD8+ T-lymphocyte response to major immunodominant epitopes after vaginal exposure to simian immunodeficiency virus: Too late and too little. J. Virol. 79, 9228–9235 (2005).
Maufort, J. P. et al. Major histocompatibility complex-matched arteries have similar patency to autologous arteries in a Mauritian cynomolgus macaque major histocompatibility complex-defined transplant model. J. Am. Heart Assoc. 8, e012135 (2019).
Gama, L. et al. Reactivation of simian immunodeficiency virus reservoirs in the brain of virally suppressed macaques. AIDS 31, 5–14 (2017).
Dyavar, S. R. et al. Normalization of cell associated antiretroviral drug concentrations with a novel RPP30 droplet digital PCR assay. Sci. Rep. 8, 3626 (2018).
Acknowledgements
We thank the WNPRC veterinary staff for their assistance.
Funding
This study was funded by the National Institutes of Health (NIH) via grants R24 OD021322 awarded to I.I.S., T.G., and M.R.R. Additional support was provided by the Office of Research Infrastructure Programs/ OD via grant P51 OD011106 awarded to the WNPRC at the University of Wisconsin-Madison, and by St. Baldrick's-Stand up to Cancer Dream Team Translational Research Grant SU2C-AACR-DT-27-17 to C.M.C and P.H., NIH/National Cancer Institute (NCI) Grant K08 CA174750 and NIH/NCI Grant R01 CA215461 to C.M.C.
Author information
Authors and Affiliations
Contributions
M.R.R., T. G., and I.I.S.: Conceptualization and study design; J.T.W and S.S.D.: coordinated the animal experiments; J.T.W.: processed samples and performed flow cytometry; S.S.D., K.S., and A.K.: prepared TCR α/β-depleted HSCs; L.M.M.: performed SHIV DNA measurements; S.B. and L.E.K.: performed chimerism analysis; J.C.: contributed to treatment design and handled animals; C.M.C. and P.H.: advised on the allo-HSCT platform, GvHD prophylaxis, and critically reviewed the manuscript; M.R.R.: wrote the manuscript. All authors revised the manuscript and approved its submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Weinfurter, J.T., D’Souza, S.S., Matschke, L.M. et al. Allogeneic MHC-matched T-cell receptor α/β-depleted bone marrow transplants in SHIV-infected, ART-suppressed Mauritian cynomolgus macaques. Sci Rep 12, 12345 (2022). https://doi.org/10.1038/s41598-022-16306-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-16306-z
- Springer Nature Limited