
Abstract
After sequencing, it is common to screen ncRNA according to expression differences. But this may lose a lot of valuable information and there is currently no indicator to characterize the regulatory function and participation degree of ncRNA on transcriptome. Based on existing pathway enrichment methods, we developed a new algorithm to calculating the participation degree of ncRNA in transcriptome (PDNT). Here we analyzed multiple data sets, and differentially expressed genes (DEGs) were used for pathway enrichment analysis. The PDNT algorithm was used to calculate the Contribution value (C value) of each ncRNA based on its target genes and the pathways they participates in. The results showed that compared with ncRNAs screened by log2 fold change (FC) and p-value, those screened by C value regulated more DEGs in IPA canonical pathways, and their target DEGs were more concentrated in the core region of the protein–protein interaction (PPI) network. The ranking of disease critical ncRNAs increased integrally after sorting with C value. Collectively, we found that the PDNT algorithm provides a measure from another view compared with the log2FC and p-value and it may provide more clues to effectively evaluate ncRNA.
Similar content being viewed by others


Introduction
One of the most important applications of RNA sequencing is to compare the differences in the expression of the non-coding RNAs (ncRNAs). ncRNA refers to a kind of RNA that can be transcribed from the genome but not translated into proteins and can perform their biological functions at the RNA level, including rRNA, tRNA, snRNA, lncRNA, microRNA (miRNA) and others. They play important roles in normal development, physiology and disease1. miRNA and lncRNA are ncRNAs that have been widely studied and have been confirmed to have the strong regulatory ability on gene expression2,3,4,5,6. By direct or indirect means, a single miRNA or lncRNA can regulate hundreds of mRNAs.
High throughput sequencing is a common method for ncRNA research. People often select genes with high expression differences for follow-up function research9,10. In the traditional way, using log2 FC and p-value as thresholds to screen ncRNAs will obviously lose a lot of valuable information. In order to screen ncRNAs more scientifically, many analysis methods have been derived. There are many enrichment analysis methods and databases, such as GSEA11 IPA12, David13, Catmap14 and GlobalTest15. Their analytical methods have different priorities, but the general idea is the same, that is, to perform functional annotation on the RNA profile. But through these methods, we can only observe which genes and pathways are associated with ncRNAs. We do not have an indicator to measure the the regulatory function and participation degree of ncRNA on transcriptome expression. This lack will cause us to miss a lot of valuable information when we screen ncRNAs. Here, we developed an algorithm PDNT, through which we can get the contribution value (C value) of each ncRNA. C value is defined as a quantitative indicator of the participation degree of ncRNA in transcriptome. The algorithm is, (1) Enrich the pathways with DEGs in the dataset, and then use the −lg (p-value) of these pathways as the weighted phase; (2) Take the intersection of the target gene of ncRNA and DEGs, and calculate the proportion of this intersection in each pathway; (3) C value is equal to the weighted sum of these proportions. To verify the utility of the C value, we collected the existing sequencing results, including skeletal muscle denervation, Alzheimer's disease, prostate cancer, gastric cancer, and adipocyte differentiation. C57BL/6 mice were used as the model of skeletal muscle denervation, APP/PS1 mice as the model of Alzheimer's disease, prostate cancer, gastric cancer, and adipocyte differentiation samples were all from human16,17,18,19,20.
Our proposed algorithm PDNT takes into accounts the p-value for each enriched pathway and the proportion of ncRNA target genes in each pathway. We expect to quantify the participation degree of ncRNA in transcriptome, and to optimize the efficiency of screening ncRNA after high throughput sequencing.
Results
The C value of each DE ncRNA is equal to the sum of BP value, CC value, MF value and KEGG value
We calculated the C value of each DE miRNA in skeletal muscle denervation, prostate cancer, Alzheimer's disease and gastric cancer data sets respectively. In addition, we calculated the C value of each lncRNA in skeletal muscle denervation and adipocyte differentiation data sets. The details of these data were aggregated into a table (Table 1). The C values of each DE ncRNA based on biological process (BP), cellular component (CC), molecular function (MF) and KEGG analysis can be obtained, and we named these C values as BP value, CC value, MF value and KEGG value respectively. The total C value of each DE ncRNA was equal to the sum of BP value, CC value, MF value and KEGG value. The DE miRNAs were sorted with the total C value to obtain the 10 DE miRNAs with maximum C value, named as top10 C value miRNAs (Table 2). The top10 DE miRNAs with maximum absolute Log2 FC (top10 FC miRNAs), and the top10 DE miRNAs with minimum p-value (top10 p-value miRNAs), were obtained by sorting the DE miRNAs according to the absolute Log2 fold FC and p-value respectively (Supplementary Tables 1, 2). Similarly, DE lncRNAs were processed in the same way to obtain top5 C value lncRNAs, top5 FC lncRNAs, top5 p-value lncRNAs for adipocyte differentiation and top10 C value lncRNAs, top10 FC lncRNAs, top10 p-value lncRNAs for skeletal muscle denervation (Table 3, Supplementary Tables 3–6).
C value is superior to log2 FC and p-value in miRNA operation results
In each data set, the most significant enriched IPA canonical pathways were obtained by core analysis (Supplementary Table 7). We took the intersections of DEGs with the predicted target genes of top10 C value miRNAs, top10 FC miRNAs and top10 p-value miRNAs respectively, and then calculated the proportion of these intersections in the above pathways. It was found that the proportion of top10 C value miRNAs target genes was significantly larger than that of top10 FC miRNAs, top10 p-value miRNAs in most pathways (Fig. 1). We built several PPI networks based on DEGs, and calculated the degree of each node. The node with a larger degree had a darker color and was closer to the center. Then we divided these nodes into the core region (top 20% of degree), sub core region (top 20%-50% of degree) and noncore region (bottom 50% of degree) (Fig. 2a,e,i,m). In the PPI network, the predicted target genes of top10 C value miRNAs, top10 FC miRNAs and top10 p-value miRNAs were labeled in red (Fig. 2). It was found that the number of top10 C value miRNAs’ target genes in each region were larger than those of top10 FC miRNAs, and top10 p-value miRNAs, and the C value group are more concentrated in core region (Fig. 3) (Table 4).

Proportion of three groups in each IPA canonical pathway (a) Skeletal muscle denervation. (b) Prostate cancer. (c) Alzheimer's disease. (d) Gastric cancer. (FC group: the collection of the top10 FC miRNAs’ predictive target mRNAs; p-value group: the collection of the top10 p-value miRNAs’ predictive target mRNAs; C value group: the collection of the top10 C value miRNAs’ predictive target mRNAs). Picture drawn by Microsoft Excel.

Partition of PPI network and distribution of each group in PPI network. (a,e,i,m) PPI network of DEGs in the Skeletal muscle denervation dataset, Prostate cancer dataset, Alzheimer's disease dataset and Gastric cancer dataset. The degree of each node was calculated. The larger the degree of the node, the darker the color and the closer the position is to the center. The top 20% nodes are defined as core regions, the top 20%-50% nodes are defined as sub core regions, and the remaining nodes are noncore regions. (b,f,j,n) Distribution of FC group in PPI network. (c,g,k,o) Distribution of p-value group in PPI network. (d,h,l,p) Distribution of C value group in PPI network. Red is the selected node, blue is the unselected. Number of genes in core region, sub core region and noncore region of each group has been tagged. STRING v11.0 was used to generate protein interactions, and the resulting network was visualized using Cytoscape v3.7.2. (FC group: the collection of the top10 FC miRNAs’ predictive target mRNAs; p-value group: the collection of the top10 p-value miRNAs’ predictive target mRNAs; C value group: the collection of the top10 C value miRNAs’ predictive target mRNAs).

Statistics on the distribution of each group in the PPI network. (a) Skeletal muscle denervation. (b) Prostate cancer. (c) Alzheimer's disease. (d) Gastric cancer. The ratio of the number of genes in each group in different regions.
Based on extensive literature, we identified 14 skeletal muscle growth regulatory miRNAs, 6 Alzheimer’s disease associated miRNAs, 6 prostate cancer associated miRNAs, and 6 gastric cancer associated miRNAs and found that when DE miRNAs were sorted by C value, the sum of the ranks of these miRNAs was significantly smaller than that of the other two indexes, which means that these miRNAs sequences increased integrally (Fig. 4). When sorting by C value versus sorting by absolute Log2 FC/ p-value, most of the disease critical miRNAs ranked up (Fig. 4) (Supplementary Table 8).

After sorting with C value, the ranking of disease critical miRNAs increased integrally. (a) Skeletal muscle denervation. (b) Alzheimer's disease. (c) Prostate cancer. (d) Gastric cancer. Left: the sum of the ranks of disease critical miRNAs by the three indexes. Right: The number of mRNAs that rank up or down. (FC group: the collection of the top10 FC miRNAs’ predictive target mRNAs; p-value group: the collection of the top10 p-value miRNAs’ predictive target mRNAs; C value group: the collection of the top10 C value miRNAs’ predictive target mRNAs).
C value is superior to log2 FC and p-value in lncRNA operation results
In the skeletal muscle denervation data set, we calculated the proportion of the predicted target genes of top10 C value lncRNAs, top10 FC lncRNAs, and top10 p-value lncRNAs in the most enriched IPA canonical pathways respectively, and found that the proportion of the genes regulated by top10 C value lncRNAs was larger than that of top10 FC lncRNAs and top10 p-value lncRNAs (Fig. 5a). It was found that the number of top10 C value lncRNAs’ target genes in each region were larger than those of top10 FC lncRNAs, and top10 p-value lncRNAs and the C value group are more concentrated in the core region (Fig. 5b–e) (Table 5).

LncRNA operation results for skeletal muscle denervation data set (a) The ratio of predicted target genes to the total genes in IPA canonical pathways. The distribution of (b) top10 FC, (c) top10 p-value and (d) top10 C value lncRNAs’ predictive target mRNAs in the PPI network. Number of genes in core region, sub core region and noncore region of each group was tagged. (e) The ratio of the number of genes in each group in different regions. (FC group: the collection of the top10 FC lncRNAs’ predictive target mRNAs; p-value group: the collection of the top10 p-value lncRNAs’ predictive target mRNAs; C value group: the collection of the top10 C value lncRNAs’ predictive target mRNAs).
Since there are relatively few DE lncRNAs and DE mRNAs in the adipocyte differentiation data set, we take top5 C value lncRNAs, top5 FC lncRNAs, top5 p-value lncRNAs. The proportion of the genes regulated by top5 C value lncRNAs was larger than that of top5 FC lncRNAs and top5 p-value lncRNAs in enriched IPA canonical pathways (Fig. 6a). It was found that the number of top5 C value lncRNAs’ target genes in each region were larger than those of top5 FC lncRNAs, and top5 p-value lncRNAs and the C value group are more concentrated in the core region (Fig. 6b–e) (Table 5). And when DE lncRNAs were sorted by C value, the adipocyte differentiation associated lncRNAs sequences increased integrally than that of the other two indexes (Fig. 6f–g) (Supplementary Table 8).

LncRNA operation results for adipocyte differentiation data set (a) The ratio of predicted target genes to the total genes in IPA canonical pathways. The distribution of (b) top10 FC, (c) top10 p-value and (d) top10 C value lncRNAs’ predictive target mRNAs in the PPI network. Number of genes in core region, sub core region and noncore region of each group was tagged. (e) The ratio of the number of genes in each group in different regions. (f) The sum of the ranks of adipocyte differentiation associated lncRNAs by the three indexes. (g) The number of adipocyte differentiation associated lncRNAs that rank up or down. (FC group: the collection of the top5 FC lncRNAs’ predictive target mRNAs; p-value group: the collection of the top5 p-value lncRNAs’ predictive target mRNAs; C value group: the collection of the top5 C value lncRNAs’ predictive target mRNAs).
Efficiency comparison of different ncRNAs
Firstly, the results of IPA canonical pathways were analyzed, and the proportion of the C value group in the top10 pathways was calculated compared with the other two groups. We found that in miRNA data set, the efficiency of the C value group was improved by 61% compared with the FC group, and by 145% compared with the p-value group. In lncRNA data set, the C value group increased by 39% compared with the FC group, and by 78% compared with the p-value group (Table 6). Then, by analyzing the results of PPI network and calculating the ratio of the C value group in core region compared with the other two groups, we found that the C value group in miRNA data set increased by 10% compared with the FC group and by 18% compared with the p-value group. In lncRNA data set, the C value group increased by 85% compared with the FC group, and by 81% compared with the p-value group. In general, there is little difference between the results of miRNA and lncRNA, and a greater difference occurs between different data sets, which may be related to the quality of data sets (Table 7).
Discussion
After high-throughput sequencing, it is common to screen ncRNA according to expression differences. But this may lose a lot of valuable information and lead to biased results. Considering the strong regulatory function of ncRNA on gene expression, there is currently no indicator to characterize the regulatory function and participation degree of ncRNA on transcriptome expression to help us evaluate and screen ncRNA. Here we designed a new algorithm PDNT to calculate the Contribution value, which is defined as a quantitative indicator of the participation degree of ncRNA in transcriptome.
To test the superiority of C value, we compared it with absolute Log2 FC and p-value. Log 2 FC reflects the expression change of ncRNAs and p-value reflects how significant the change is. The two indexes of each DE RNA were obtained after the traditional whole transcriptome sequencing, and many follow-up studies have partially referenced Log2 FC and p-values in selecting the target gene9,10,16. We analyzed four microRNA data sets and two lncRNA data sets, and compared the C value with Log2 FC and p-value in each data set. First, we performed enrichment analysis on DEGs to obtain the most enriched IPA canonical pathways. We found that top C value ncRNAs targeted more genes in these pathways than FC and p-value groups, which may suggest that top C value ncRNAs have greater regulatory potential for enriched pathways. Further, we constructed a PPI network based on DEGs, partitioned the PPI by degree, and then observed the distribution of the three groups in different partitions. It was found that the number of target genes of top C value ncRNAs in each region was greater than that of the other two groups. At the same time, a larger proportion of target genes in the C value group were concentrated in the central region of the PPI. It suggests that the top C value ncRNA has a broader and more important influence on the PPI network than the other two groups. Finally, based on literature search, we obtained key ncRNAs that regulate various pathological/ physiological processes, and then tested the screening effect of the three indicators on these key ncRNAs in the datasets. It was found that using the C value to rank ncRNAs made the overall ranking of these key ncRNAs higher than the other two indicators. This suggests that ncRNAs screened with C values have a greater potential for regulating pathological/physiological processes.
In order to correct the bias caused by only considering expression differences when screening ncRNA, many analysis methods and databases have been derived, such as GSEA11 IPA12, David13, Catmap14 and GlobalTest15. Their analytical methods have different priorities, but the general idea is the same, that is, to perform functional annotation on the RNA profile. But through these methods, we can only observe which genes and pathways are associated with ncRNAs. We do not have a measure to evaluate the participation degree of ncRNA in transcriptome. This lack may result in our inability to assess the priority of two ncRNAs when their target genes are close in number. Or when the two ncRNA regulate similar pathways, we cannot judge their participation degree in the expression regulation of the transcriptome. The algorithm PDNT proposed in this study is based on these pathway analysis methods. We hope to make better use of the pathway enrichment results to evaluate ncRNA and we integrated more valuable information to optimize the screening efficiency of ncRNA. The limitation of this study is that we only calculated based on one pathway enrichment method. In the subsequent study, we will compare the differences between the results calculated based on different pathway enrichment methods, to provide more inspiration and help for related research.
Based on the above evidence, the PDNT is an efficient algorithm for calculating the participation degree of ncRNA in transcriptome based on pathway analysis. We found that the PDNT algorithm provides a measure from another view compared with the log2FC and p-value and it may provide more clues to effectively evaluate ncRNA.
Methods
Prediction of ncRNAs’ target mRNAs
MiRNA: MiRNAs target genes prediction software, miRanda-3.3a (http://www.microrna.org/) 22, uses a weighted dynamic programming algorithm to calculate the optimal sequence complementarity between a mature microRNA and a given mRNA. The main parameters are: -sc 140, -en -10, -scale 4, -strict -out.
LncRNA: The target genes of lncRNAs are predicted by co-expression analysis among samples. The Weighted Gene Correlation Network Analysis (http://www.r-project.org/) 23 was used to calculate Pearson correlation coefficients. The absolute value of the Pearson correlation coefficient ≥ 0.90, p-value < 0.01 and FDR < 0.01 was saved.
GO and KEGG pathway enrichment analysis
In this study, the screening criteria for DEG were p < 0.05 and absolute Log2 FC ≥ 1.
GO is a database established by Gene Ontology consortium (http://www.geneontology.org), which includes three parts: molecular function, biological process and cell composition. KEGG is based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.ad.jp/kegg/), Fisher exact test and × 2 test were used. Enrichment analysis of differentially expressed genes was performed using clusterProfiler R software package24, and gene length bias was corrected. The corrected p-value less than 0.05 was considered to be significantly enriched by differentially expressed genes.
C value mathematical model and its calculation
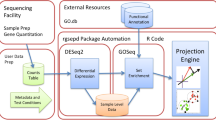
The C value of each DE ncRNA is calculated using the PDNT algorithm (Fig. 7):
p-value is the p-value of the pathway enriched by DEGs; Proportion refers to the proportion of the intersection between ncRNA target genes and DEGs in each pathway; n represents the number of pathways enriched by DEGs.

The operation and verification process of the PDNT algorithm.
Ingenuity pathway analysis (IPA) core analysis
IPA core analysis of DEGs (p < 0.05 and absolute Log2 FC ≥ 1) was performed using IPA (version 81,348,237, Qiagen), showing top10 canonical pathways according to p-value.
PPI network for DEGs
For each dataset, the STRING v.11.0 database was used to construct the PPI network based on DEGs. The images were then drawn by cytoscape3.72 (San Diego, CA, USA).
Retrieval and statistics of key miRNAs and lncRNAs
We searched PubMed (http://www.ncbi.nlm.nih.gov/pubmed) for miRNAs that play important roles in skeletal muscle denervation, Alzheimer's disease, prostate cancer and gastric cancer, respectively. The key words were "skeletal muscle AND microRNA", "Alzheimer's disease AND microRNA", "prostate cancer AND microRNA", and "gastric cancer AND microRNA". Next, we retrieved the lncRNAs that play an important role in skeletal muscle denervation and adipocyte differentiation. Keywords: "skeletal muscle AND lncRNA" and "adipocyte differentiation AND lncRNA". The results were shown in Table 8.
Data Analysis
The analysis platform is R 3.6.1 and the R package is clusterProfiler. The database is org.Mm.eg.db developed with the R package.
Abbreviations
- FC:
-
Fold change
- GSEA:
-
Gene set enrichment analysis
- IPA:
-
Ingenuity pathway analysis
- C value:
-
Contribution value
- DEG:
-
Differentially expressed gene
- PPI:
-
Protein–protein interaction
- ncRNA:
-
Non-coding RNA
- miRNA:
-
MicroRNA
- APP:
-
Amyloid beta precursor protein
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- BP:
-
Biological process
- CC:
-
Cellular component
- MF:
-
Molecular function
References
Mercer, T. R., Dinger, M. E. & Mattick, J. S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 10, 155–159. https://doi.org/10.1038/nrg2521 (2009).
He, L. & Hannon, G. J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 5, 522–531. https://doi.org/10.1038/nrg1379 (2004).
Bartel, D. P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116, 281–297. https://doi.org/10.1016/s0092-8674(04)00045-5 (2004).
Kornienko, A. E., Guenzl, P. M., Barlow, D. P. & Pauler, F. M. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 11, 59. https://doi.org/10.1186/1741-7007-11-59 (2013).
Beltran, M. et al. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev. 22, 756–769. https://doi.org/10.1101/gad.455708 (2008).
Aravin, A. A., Hannon, G. J. & Brennecke, J. The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science 318, 761–764. https://doi.org/10.1126/science.1146484 (2007).
Peng, H. et al. Pseudogene INTS6P1 regulates its cognate gene INTS6 through competitive binding of miR-17-5p in hepatocellular carcinoma. Oncotarget 6, 5666–5677. https://doi.org/10.18632/oncotarget.3290 (2015).
Wang, L. et al. Pseudogene OCT4-pg4 functions as a natural micro RNA sponge to regulate OCT4 expression by competing for miR-145 in hepatocellular carcinoma. Carcinogenesis 34, 1773–1781. https://doi.org/10.1093/carcin/bgt139 (2013).
Zhang, X. et al. Circular RNA circNRIP1 acts as a microRNA-149-5p sponge to promote gastric cancer progression via the AKT1/mTOR pathway. Mol. Cancer 18, 20. https://doi.org/10.1186/s12943-018-0935-5 (2019).
Bai, C., Gao, Y., Zhang, X., Yang, W. & Guan, W. MicroRNA-34c acts as a bidirectional switch in the maturation of insulin-producing cells derived from mesenchymal stem cells. Oncotarget 8, 106844–106857. https://doi.org/10.18632/oncotarget.21883 (2017).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. https://doi.org/10.1073/pnas.0506580102 (2005).
Kramer, A., Green, J., Pollard, J. Jr. & Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. https://doi.org/10.1093/bioinformatics/btt703 (2014).
Dennis, G. Jr. et al. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 4, P3 (2003).
Breslin, T., Eden, P. & Krogh, M. Comparing functional annotation analyses with Catmap. BMC Bioinf. 5, 193. https://doi.org/10.1186/1471-2105-5-193 (2004).
Goeman, J. J., van de Geer, S. A., de Kort, F. & van Houwelingen, H. C. A global test for groups of genes: Testing association with a clinical outcome. Bioinformatics 20, 93–99. https://doi.org/10.1093/bioinformatics/btg382 (2004).
Weng, J., Zhang, P., Yin, X. & Jiang, B. The whole transcriptome involved in denervated muscle atrophy following peripheral nerve injury. Front. Mol. Neurosci. 11, 69. https://doi.org/10.3389/fnmol.2018.00069 (2018).
Ma, N., Tie, C., Yu, B., Zhang, W. & Wan, J. Identifying lncRNA-miRNA-mRNA networks to investigate Alzheimer’s disease pathogenesis and therapy strategy. Aging (Albany NY) 12, 2897–2920. https://doi.org/10.18632/aging.102785 (2020).
Ye, Y., Li, S. L. & Wang, S. Y. Construction and analysis of mRNA, miRNA, lncRNA, and TF regulatory networks reveal the key genes associated with prostate cancer. PLoS ONE 13, e0198055. https://doi.org/10.1371/journal.pone.0198055 (2018).
Qi, M., Yu, B., Yu, H. & Li, F. Integrated analysis of a ceRNA network reveals potential prognostic lncRNAs in gastric cancer. Cancer Med. 9, 1798–1817. https://doi.org/10.1002/cam4.2760 (2020).
Chen, K., Xie, S. & Jin, W. Crucial lncRNAs associated with adipocyte differentiation from human adipose-derived stem cells based on co-expression and ceRNA network analyses. PeerJ 7, e7544. https://doi.org/10.7717/peerj.7544 (2019).
Wang, B. D. et al. Identification and functional validation of reciprocal microRNA-mRNA pairings in African American prostate cancer disparities. Clin. Cancer Res. 21, 4970–4984. https://doi.org/10.1158/1078-0432.CCR-14-1566 (2015).
Betel, D., Wilson, M., Gabow, A., Marks, D. S. & Sander, C. The microRNA.org resource: Targets and expression. Nucl. Acids Res. 36, 149–153. https://doi.org/10.1093/nar/gkm995 (2008).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinf. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Cheng, X. et al. MiR-204-5p regulates C2C12 myoblast differentiation by targeting MEF2C and ERRgamma. Biomed. Pharmacother. 101, 528–535. https://doi.org/10.1016/j.biopha.2018.02.096 (2018).
Feng, Y., Cao, J. H., Li, X. Y. & Zhao, S. H. Inhibition of miR-214 expression represses proliferation and differentiation of C2C12 myoblasts. Cell Biochem. Funct. 29, 378–383. https://doi.org/10.1002/cbf.1760 (2011).
Ge, G. et al. miR-10b-5p Regulates C2C12 Myoblasts Proliferation and Differentiation. Biosci. Biotechnol. Biochem. 83, 291–299. https://doi.org/10.1080/09168451.2018.1533805 (2019).
Gan, M. et al. miR-152 regulates the proliferation and differentiation of C2C12 myoblasts by targeting E2F3. In Vitro Cell Dev. Biol. Anim. 54, 304–310. https://doi.org/10.1007/s11626-017-0219-1 (2018).
Huang, Z., Chen, X., Yu, B., He, J. & Chen, D. MicroRNA-27a promotes myoblast proliferation by targeting myostatin. Biochem. Biophys. Res. Commun. 423, 265–269. https://doi.org/10.1016/j.bbrc.2012.05.106 (2012).
Liu, C. et al. miR-18a induces myotubes atrophy by down-regulating IgfI. Int. J. Biochem. Cell Biol. 90, 145–154. https://doi.org/10.1016/j.biocel.2017.07.020 (2017).
Mi, L. et al. MicroRNA-139-5p regulates C2C12 cell myogenesis through blocking Wnt/beta-catenin signaling pathway. Biochem. Cell Biol. 93, 8–15. https://doi.org/10.1139/bcb-2014-0079 (2015).
Qiu, H. et al. Regulatory axis of miR-195/497 and HMGA1-Id3 governs muscle cell proliferation and differentiation. Int. J. Biol. Sci. 13, 157–166. https://doi.org/10.7150/ijbs.17440 (2017).
Silva, W. J. et al. miR-29c improves skeletal muscle mass and function throughout myocyte proliferation and differentiation and by repressing atrophy-related genes. Acta Physiol. (Oxf) 226, 13278. https://doi.org/10.1111/apha.13278 (2019).
Tang, Z. et al. miR-34b modulates skeletal muscle cell proliferation and differentiation. J. Cell Biochem. 118, 4285–4295. https://doi.org/10.1002/jcb.26079 (2017).
Wang, H. et al. miR-22 regulates C2C12 myoblast proliferation and differentiation by targeting TGFBR1. Eur. J. Cell Biol. 97, 257–268. https://doi.org/10.1016/j.ejcb.2018.03.006 (2018).
Wang, M. et al. miRNA-34c inhibits myoblasts proliferation by targeting YY1. Cell Cycle 16, 1661–1672. https://doi.org/10.1080/15384101.2017.1281479 (2017).
Wei, X. et al. miR-378a-3p promotes differentiation and inhibits proliferation of myoblasts by targeting HDAC4 in skeletal muscle development. RNA Biol. 13, 1300–1309. https://doi.org/10.1080/15476286.2016.1239008 (2016).
Zhang, Z. et al. Identification of a novel miR-206-Notch3 pathway regulating mouse myoblasts proliferation. Gene 695, 57–64. https://doi.org/10.1016/j.gene.2019.01.045 (2019).
Endzelins, E. et al. Diagnostic, prognostic and predictive value of cell-free miRNAs in prostate cancer: A systematic review. Mol. Cancer 15, 41. https://doi.org/10.1186/s12943-016-0523-5 (2016).
Li, F. et al. MiR-218 impedes IL-6-induced prostate cancer cell proliferation and invasion via suppression of LGR4 expression. Oncol. Rep. 35, 2859–2865. https://doi.org/10.3892/or.2016.4663 (2016).
Song, Y., Hu, M., Zhang, J., Teng, Z. Q. & Chen, C. A novel mechanism of synaptic and cognitive impairments mediated via microRNA-30b in Alzheimer’s disease. EBioMedicine 39, 409–421. https://doi.org/10.1016/j.ebiom.2018.11.059 (2019).
Lei, X., Lei, L., Zhang, Z., Zhang, Z. & Cheng, Y. Downregulated miR-29c correlates with increased BACE1 expression in sporadic Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 8, 1565–1574 (2015).
Yao, X., Xian, X., Fang, M., Fan, S. & Li, W. Loss of miR-369 promotes tau phosphorylation by targeting the fyn and serine/threonine-protein kinase 2 signaling pathways in alzheimer’s disease mice. Front. Aging Neurosci. 11, 365. https://doi.org/10.3389/fnagi.2019.00365 (2019).
Swarbrick, S., Wragg, N., Ghosh, S. & Stolzing, A. Systematic review of miRNA as biomarkers in Alzheimer’s disease. Mol. Neurobiol. 56, 6156–6167. https://doi.org/10.1007/s12035-019-1500-y (2019).
Yu, B. et al. MiR-148a functions as a tumor suppressor by targeting CCK-BR via inactivating STAT3 and Akt in human gastric cancer. PLoS ONE 11, e0158961. https://doi.org/10.1371/journal.pone.0158961 (2016).
Li, X. et al. Involvement of miR-20a in promoting gastric cancer progression by targeting early growth response 2 (EGR2). Int. J. Mol. Sci. 14, 16226–16239. https://doi.org/10.3390/ijms140816226 (2013).
Chen, L. et al. MicroRNA-181b targets cAMP responsive element binding protein 1 in gastric adenocarcinomas. IUBMB Life 64, 628–635. https://doi.org/10.1002/iub.1030 (2012).
Wu, X. L. et al. MicroRNA-143 suppresses gastric cancer cell growth and induces apoptosis by targeting COX-2. World J. Gastroenterol. 19, 7758–7765. https://doi.org/10.3748/wjg.v19.i43.7758 (2013).
Tie, J. et al. MiR-218 inhibits invasion and metastasis of gastric cancer by targeting the Robo1 receptor. PLoS Genet. 6, 879. https://doi.org/10.1371/journal.pgen.1000879 (2010).
Zhang, Y., Han, T., Wei, G. & Wang, Y. Inhibition of microRNA-17/20a suppresses cell proliferation in gastric cancer by modulating UBE2C expression. Oncol. Rep. 33, 2529–2536. https://doi.org/10.3892/or.2015.3835 (2015).
Acknowledgements
This work was supported by the grants from National Natural Science Foundation of China (82072162 to X.Y. and 81971177 to B.J.), and Natural Science Foundation of Beijing, China (7192215 to X.Y.). We wish to thank all the members of the laboratory for the helpful discussion. Thank Mr. Zhang Guangze for providing technical guidance for this experiment.
Author information
Authors and Affiliations
Contributions
X.Y. designed the study. X.G. instructed all experiments and drafted the manuscript. S.W., B.J., Z.Q., J.D. and C.H. analysed the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gu, X., Wang, S., Jin, B. et al. A pathway analysis-based algorithm for calculating the participation degree of ncRNA in transcriptome. Sci Rep 12, 22654 (2022). https://doi.org/10.1038/s41598-022-27178-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-27178-8
- Springer Nature Limited