
Abstract
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is a persistent environmental contaminant that induces the progression of steatosis to steatohepatitis with fibrosis in mice. Furthermore, TCDD reprograms hepatic metabolism by redirecting glycolytic intermediates while inhibiting lipid metabolism. Here, we examined the effect of TCDD on hepatic acetyl-coenzyme A (acetyl-CoA) and β-hydroxybutyrate levels as well as protein acetylation and β-hydroxybutyrylation. Acetyl-CoA is not only a central metabolite in multiple anabolic and catabolic pathways, but also a substrate used for posttranslational modification of proteins and a surrogate indicator of cellular energy status. Targeted metabolomic analysis revealed a dose-dependent decrease in hepatic acetyl-CoA levels coincident with the phosphorylation of pyruvate dehydrogenase (E1), and the induction of pyruvate dehydrogenase kinase 4 and pyruvate dehydrogenase phosphatase, while repressing ATP citrate lyase and short-chain acyl-CoA synthetase gene expression. In addition, TCDD dose-dependently reduced the levels of hepatic β-hydroxybutyrate and repressed ketone body biosynthesis gene expression. Moreover, levels of total hepatic protein acetylation and β-hydroxybutyrylation were reduced. AMPK phosphorylation was induced consistent with acetyl-CoA serving as a cellular energy status surrogate, yet subsequent targets associated with re-establishing energy homeostasis were not activated. Collectively, TCDD reduced hepatic acetyl-CoA and β-hydroxybutyrate levels eliciting starvation-like conditions despite normal levels of food intake.
Similar content being viewed by others


Introduction
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is the prototypical member of a class of persistent environmental contaminants termed polyhalogenated aromatic hydrocarbons, which include polychlorinated dibenzodioxins (PCDDs), dibenzofurans (PCDFs) and biphenyls (PCBs)1. A subset of these contaminants possesses lateral chlorines that induce a diverse spectrum of aryl hydrocarbon receptor (AHR)-mediated species-, sex-, tissue-, cell- and promoter-specific responses including the dose-dependent progression of hepatic steatosis to steatohepatitis with fibrosis. Moreover, TCDD and dioxin-like PCBs are classified as an International Agency for Research on Cancer (IARC) group 1 human carcinogen while the carcinogenicity of other toxic PCDDs and PCDFs in humans is equivocal2,3,4. TCDD and related compounds are non-genotoxic with most, if not all of their effects mediated by the AHR, a ligand-activated basic helix-loop-helix PER-ARNT-SIM transcription factor that is conserved in all vertebrate species5. It is activated by several structurally diverse chemicals, endogenous metabolites, microbial products, and natural compounds, although the physiological ligand is unknown. Following ligand binding and the dissociation of chaperone proteins, the AHR translocates to the nucleus and dimerizes with the AHR nuclear translocator (ARNT)6,7,8. In the proposed canonical mechanism, the heterodimer binds dioxin response elements (DREs; 5’-GCGTG-3’) throughout the genome and recruits multiple coactivators to elicit differential gene expression. However, several studies also report differential gene expression independent of DREs as well as alternate AHR binding partners suggesting alternative mechanisms of gene regulation7,9,10,11,12.
The emergence of transcriptomics and metabolomics provides the opportunity to comprehensively assess the effects of exogenous agents on gene expression and endogenous metabolite levels. Numerous studies have examined the consequences of PCDD, PCDF, or PCB exposure on gene expression and/or metabolite levels in diverse in vivo and in vitro models13,14,15,16,17,18,19,20. However, few have integrated transcriptomic and metabolomic datasets with complementary histopathology to distinguish adaptive events from key responses to elucidate causative mechanisms associated with adverse outcomes21,22,23,24,25. Despite decades of research establishing the central role of the AHR in mediating the effects of TCDD and related compounds, the mechanisms leading to toxicity remain poorly understood.
Acetyl-coenzyme A (acetyl-CoA) occupies a central position in multiple metabolic pathways. As a metabolite, it straddles carbohydrate, lipid, and amino acid catabolism, and can be used as a substrate for the synthesis of fatty acids, cholesterol, and ketone bodies. Acetyl-CoA is also used as a substrate for the posttranslational modification of proteins to regulate enzyme activity, protein stability, cellular location, and to remodel chromatin via histone acetylation to control gene expression, thus linking intermediate metabolism to cellular homeostasis26,27,28,29. Acetyl-CoA is not membrane permeable, therefore resulting in specific cellular pools each of which support distinct activities that are independently generated within mitochondrial, peroxisomal, endoplasmic reticulum, and nucleo-cytosolic compartments. For example, the mitochondrial pool is produced by the pyruvate dehydrogenase complex (PDC), fatty acid β-oxidation, and amino acid metabolism, while the nucleo-cytosolic pool is sourced from ATP-citrate lyase (ACLY), acyl-CoA synthetase short-chain family member 2 (ACSS2), and nuclear PDC30. Levels of protein acetylation are directly linked to acetyl-CoA levels that fluctuate depending on intra- and extra-cellular cues that also undergo circadian regulation. This dynamic regulation of acetyl-CoA metabolism not only affects global histone modifications but also synchronizes intermediate metabolism with feeding and active/rest cycles. Consequently, acetyl-CoA is not only a metabolic intermediate but also a surrogate indicator of nutritional status that coordinates metabolic reprogramming through epigenetic regulation and posttranslational modification to sustain survival, growth and proliferation during periods of starvation, nutrient availability and metabolic stress30,31. For instance, nutrient starvation causing the rapid depletion of acetyl-CoA triggers autophagy due to the activation of AMPK32. However, the dose-dependent effects of environmental contaminants, drugs, chemicals, or natural products on intermediate metabolism, and more specifically, acetyl-CoA levels, have not been examined and warrant further investigation.
Previous studies have reported decreased ATP levels in the liver and the induction of steatosis to steatohepatitis with fibrosis following treatment with TCDD and related compounds33,34,35,36,37,38. Moreover, TCDD dose-dependently reprogramed glucose metabolism by switching from PKM1 to PKM2 expression resulting in reduced glycolytic flux and the redirection of accumulating upstream intermediates to other pathways to support proliferation and/or reactive oxygen species (ROS) defenses39. In addition, the integration of transcriptomic and metabolomic data with chromatin immunoprecipitation analyses showed TCDD dose-dependently repressed fatty acid β-oxidation40. In this study, we further examined the effects of TCDD on pathways associated with acetyl-CoA metabolism to test the hypothesis that acetyl-CoA levels reduced by TCDD also affected ketone body synthesis, protein acetylation and β-hydroxybutyrylation, and AMPK activation. Our analysis found TCDD lowered hepatic acetyl-CoA and β-hydroxybutyrate levels. Accordingly, total hepatic protein acetylation and hydroxybutyrylation levels were reduced with increased levels of activated AMPK, suggesting the induction of a starvation-like phenotype in the liver despite unaffected levels of food intake. These results indicate that in addition to differential gene expression mediated by the AHR, TCDD can also elicit secondary effects by disrupting acetyl-CoA homeostasis.
Results
Gross morphology and histopathology
In agreement with previously reported findings, 30 μg/kg TCDD decreased terminal body weight by ~ 14% (Supplementary Fig. 1A), despite no significant change in daily food intake23. Absolute liver weights increased 12–30% between 0.3 and 30 μg/kg TCDD (Supplementary Fig. 1B), while relative liver weight dose-dependently increased 7–44% between 0.3 and 30 μg/kg TCDD (Supplementary Fig. 1C). Absolute and relative gonadal white adipose tissue weights have previously been reported to decrease 46% and 28%, respectively, at 30 μg/kg TCDD with no change in terminal brown adipose tissue weights41. Although a modest increase in serum ALT levels was observed following oral gavage with 30 µg/kg TCDD every 4 days for 28 days for a total of 7 treatments (Supplementary Fig. 1D), previous studies eliciting comparable effects exhibited no evidence of overt toxicity or body weight loss > 15%41,42.
Hepatic steatosis, immune cell infiltration, fibrosis, and bile duct proliferation have previously been reported to be dose-dependently induced following oral gavage with TCDD every 4 days for 28 days33. Specifically, there was evidence of hepatocyte vacuolization (fatty change) with minimal to slight hepatocyte necrosis at 3 μg/kg TCDD and immune cell infiltration after doses of ≥ 3 μg/kg TCDD after 28 days of exposure, with F4/80 staining confirming the presence of macrophages. At 30 μg/kg, bile duct proliferation was observed along with picro-Sirius red staining for collagen and inflammation surrounding the bile ducts (pericholangitis). Collectively, the gross morphology, histopathology, and ALT results suggest the effects on gene expression, protein levels and metabolite levels do not induce overt toxicity following oral gavage with TCDD every 4 days for 28 days.
MS analysis of acyl-CoA and CoA levels
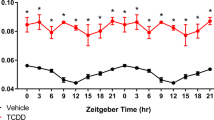
Previously reported untargeted metabolomics identified a dose-dependent decrease in hepatic acetyl-CoA levels40. These results were confirmed by targeted analysis with internal standards (Fig. 1A,B). Due to circadian regulation of carbohydrate, lipid, and protein metabolism, targeted analysis assessed samples collected in the morning (zeitgeber time [ZT] 0–3.5) and afternoon (ZT5.5–8.5) samples. Acetyl-CoA levels in controls (74.3 ± 6.4 nmol/g wet tissue; or 2.4 ± 0.21 nmol/mg total protein) did not change between morning and afternoon cohorts, and were comparable to previously reported levels43,44. TCDD lowered morning and afternoon acetyl-CoA levels 3.8- and 3.4-fold, respectively. In contrast, CoA levels dose-dependently increased 2.2-fold at lower TCDD doses but decreased 2.9-fold at 30 μg/kg TCDD in morning samples. Although TCDD did not affect afternoon CoA levels, the acetyl-CoA/CoA ratio exhibited a decreasing trend due to lowering acetyl-CoA levels (Fig. 1B).

Hepatic acetyl-CoA and coenzyme A (CoA) levels assessed in mice using targeted liquid chromatography tandem mass spectrometry. Mice were gavaged with TCDD (or sesame oil vehicle) between zeitgeber time (ZT) 0–1. Samples were collected at (A) ZT 0–3.5 h, or (B) ZT 5.5–8.5 h. Asterisk (*) denotes a significant change (p ≤ 0.05) as determined using a one-way ANOVA and Dunnett’s post-hoc analysis.
Glycolysis and PDH as a source of Acetyl-CoA
We next examined changes associated with carbohydrate catabolism, a primary source of acetyl-CoA (Fig. 2A). Figure 2 summarizes the TCDD-induced changes in the expression of glycolytic genes following oral gavage every 4 days for 28 days. The presence of a computationally identified putative DRE (pDRE) and AHR genomic binding (ChIPseq) data, as well as time course, diurnal regulated dose–response, and diurnal regulated RNAseq gene expression data are included in all heatmaps (Fig. 2B–E). Gene expression changes reported in the text represent the maximum fold-change determined in the diurnal regulated gene expression dataset with the corresponding ZT.

Glucose metabolism as a source of acetyl-CoA. Differential gene expression was assessed using RNA-seq. (A) The glycolysis pathway with regulated steps denoted with a double dagger (‡). (B) Computational identification of putative dioxin response elements (pDREs), and the detection of hepatic AhR genomic binding in ChIPseq analysis 2 h after oral gavage of 30 μg/kg TCDD. Genes are listed by the official symbol as designated in the mouse genome informatics (MGI) database. (C) Time-dependent hepatic expression of glycolysis-related genes (n = 3) following a single bolus gavage of 30 μg/kg TCDD. (D) Hepatic dose-dependent gene expression (n = 5) following oral gavage with TCDD every 4 days for 28 days. (E) Diurnal regulated gene expression denoted with a “Y”. An orange ‘X’ indicates oscillating gene expression was abolished following oral gavage with 30 μg/kg TCDD every 4 days for 28 days. ZT indicates the time of maximum induction/repression (P1(t) > 0.8). Counts represents the maximum number of raw reads for any treatment group. Low transcript counts (< 500 reads) are denoted in yellow with high transcript counts (> 10,000) denoted in pink. Differential gene expression with a posterior probability (P1(t)) > 0.80 are depicted with a black triangle in the upper right corner of the tile. Capillary electrophoresis was used to assess (F) phosphorylated pyruvate dehydrogenase (Ser300), (G) total pyruvate dehydrogenase, (H) ATP-citrate lyase, and (I) acyl-CoA synthetase short chain family member 2 protein levels in total lysate prepared from liver samples harvested between ZT0-3 (n = 3). (J) Hepatic levels of acetate, a precursor for acetyl-CoA, were assessed. Bar graphs denote the mean ± SEM. Significance (*p ≤ 0.05) was determined using a one-way ANOVA followed by Dunnett’s post-hoc analysis. The heatmap was created using R (v4.0.4). Plots were created using GraphPad Prism (v8.4.3). The biochemical reaction was created using Adobe Illustrator (v25.2).
Glucokinase (Gck), aldolase B (Aldob), liver/red blood cell pyruvate kinase (Pklr), and pyruvate dehydrogenase phosphatase catalytic subunit 2 (Pdp2) showed time and dose-dependent repression, while hexokinase 1, 2, and 3 (Hk1, 2, and 3), muscle pyruvate kinase (Pkm), and pyruvate dehydrogenase kinase 4 (Pdk4) were induced. Hk2 has recently been reported to be positively regulated by the AHR in human U2OS and 143B osteosarcoma cells, as well as human HCT116 colon cancer cell lines45. Glucose transporters GLUT2 (Slc2a2) and GLUT9 (Slc2a9), which are responsible for transporting glucose primarily in the liver46, were also dose-dependently repressed. Only Slc2a2, Slc2a9, Gck, Aldob, Pkm and Pdk4 exhibited AHR genomic enrichment 2 h after treatment with 30 μg/kg TCDD. TCDD and related polychlorinated biphenyls (PCB126 and 118) have been reported to specifically induce nuclear and cytosolic pyruvate kinase muscle isoform 2 (Pkm2)39,47,48. Nuclear PKM2 has been implicated in gene regulation while the cytosolic dimer is associated with the Warburg effect with lower catalytic activity compared to PKM1 and PKLR49,50. Following TCDD-induced PKM isoform switching, accumulating upstream glycolytic intermediates are redirected to the pentose phosphate pathway and serine/folate biosynthesis for biomass and NADPH production to support proliferation and the biosynthesis and recycling of glutathione, depending on the microenvironment51. Consequently, the reduced glycolytic flux following PKM2 induction and PKLR repression is consistent with decreased acetyl-CoA levels.
Acetyl-CoA levels are also affected by the activity of the pyruvate dehydrogenase complex (PDC). PDC is posttranslationally regulated by phosphorylation, acetylation, and succinylation52. The 131.1-fold induction of Pdk4 and the 4.8-fold repression of pyruvate dehydrogenase phosphatase 2 (Pdp2) suggest phosphorylation of the E1α subunit of pyruvate dehydrogenase (PDH) at Ser300. PDK4 is reported to primarily phosphorylate Ser293 and Ser300 sites of the PDH53, and was confirmed by capillary electrophoresis with the increase in phosphorylated PDH at 30 μg/kg TCDD that could not be attributed to overall higher total hepatic PDH levels (Fig. 2F,G). Moreover, estrogen-related receptor gamma (Esrrg), which induces PDK4 activity under hypoxic conditions54, was induced 3.9-fold. ACLY and ACSS2, which produce cytosolic pools of acetyl-CoA from citrate and acetate, were also repressed 4.3- and 8.8-fold, repression, respectively (Fig. 2H,I). Total hepatic acetate levels also decrease, but only at 30 μg/kg TCDD (Fig. 2J).
Coenzyme A biosynthesis
Acetyl-CoA biosynthesis is dependent on the availability of coenzyme A (CoA), an ubiquitous cofactor synthesized from the essential vitamin pantothenate55 (Fig. 3A). CoA is crucial to many metabolic pathways including the tricarboxylic acid and β-oxidation cycles. Biosynthesis begins with intestinal absorption of either dietary or microbial-derived pantothenate absorbed from the gut, after which it is transported to peripheral tissues and imported into cells via the sodium-dependent multivitamin transporter (Slc5a6; repressed 3.3-fold), or by passive diffusion55,56 (Fig. 3B–E). Pantothenate kinases (Pank1-4; repressed 2.8-, 1.5-, 2.9-, and 1.8-fold, respectively) phosphorylate pantothenic acid to yield 4′-phosphopantothenate which is then converted to 4′-phosphopantothenoylcysteine by 4′-phosphopantothenoylcysteine synthetase (Ppcs; repressed 1.6-fold), followed by decarboxylation mediated by 4′-phosphopantothenoylcysteine decarboxylase (Ppcdc; induced 2.1-fold) to yield 4′-phosphopantetheine. Bifunctional coenzyme A synthase (Coasy; repressed 1.5-fold) catalyzes the last two reactions to first yield dephospho-coenzyme A, and finally CoA. TCDD elicited a non-monotonic decrease in hepatic pantothenic acid levels that was only significant at 3 μg/kg TCDD (Fig. 3F). This increase in CoA (Fig. 1) cannot be attributed to CoA biosynthesis gene induction or increased hepatic levels of pantothenic acid (Fig. 3).

Biosynthesis of coenzyme A (CoA). Differential gene expression pertaining to CoA biosynthesis was assessed using RNA-seq. (A) The CoA biosynthesis pathway is shown with regulated steps denoted with a double dagger (‡). (B) Computational identification of putative dioxin response elements (pDREs), and the detection of hepatic AhR genomic binding in ChIPseq analysis 2 h after oral gavage of 30 μg/kg TCDD. Genes are listed by the official symbol as designated in the mouse genome informatics (MGI) database. (C) Time-dependent hepatic expression of glycolysis-related genes (n = 3) following a single bolus gavage of 30 μg/kg TCDD. (D) Hepatic dose-dependent gene expression (n = 5) following oral gavage with TCDD every 4 days for 28 days. (E) Diurnal regulated gene expression denoted with a “Y”. An orange ‘X’ indicates oscillating gene expression was abolished following oral gavage with 30 μg/kg TCDD every 4 days for 28 days. ZT indicates the time of maximum induction/repression (P1(t) > 0.8). Counts represent the maximum number of raw reads for any treatment group. Low transcript counts (< 500 reads) are denoted in yellow with high transcript counts (> 10,000) denoted in pink. Differential gene expression with a posterior probability (P1(t)) > 0.80 are depicted with a black triangle in the upper right corner of the tile. (F) Hepatic levels of pantothenic acid were assessed using target LC–MS/MS. Bar graphs denote the mean ± SEM. Significance (*p ≤ 0.05) was determined using a one-way ANOVA followed by Dunnett’s post-hoc analysis. The heatmap was created using R (v4.0.4). Plots were created using GraphPad Prism (v8.4.3). The biochemical reaction was created using Adobe Illustrator (v25.2).
Ketone body formation
In the fed state, cells can utilize acetyl-CoA for fatty acid biosynthesis, protein acetylation, cholesterol biosynthesis, and energy production via the TCA cycle30. Under fasting conditions, however, metabolic reprogramming shifts acetyl-CoA into mitochondrial oxidative catabolism to support the synthesis of either ATP or the easily transported ketone bodies, acetoacetate, β-hydroxybutyrate, and acetone30,57. Biosynthesis consumes two acetyl-CoAs for each ketone body (Fig. 4A), beginning with the formation of acetoacetyl-CoA via acyl-CoA:cholesterol acyltransferase (Acat1 and 2, repressed 3.1- and 4.3-fold, respectively)58,59. An additional acetyl-CoA undergoes condensation with acetoacetyl-CoA to form hydroxymethylglutaryl-CoA catalyzed by the rate-limiting hydroxymethylglutaryl (HMG)-CoA synthase (HMGCS). Hmgcs1 and 2 were repressed 10.4- and 1.9-fold, respectively, by TCDD (Fig. 4B–E). The HMG-CoA intermediate can then be metabolized into ketone bodies or shunted to the mevalonate pathway for cholesterol metabolism. However, TCDD repressed gene expression associated with de novo cholesterol biosynthesis14,60. Although HMG-CoA can be lysed to acetoacetate, HMG-CoA lyase (Hmgcl) was repressed 2.1-fold. In addition, TCDD repressed β-hydroxybutyrate dehydrogenase 1 (Bdh1) 3.2-fold thus limiting the oxidation of acetoacetate to β-hydroxybutyrate. β-Hydroxybutyrate dehydrogenase 2 (Bdh2) which catalyzes the reverse reaction and the formation of acetoacetate, was also repressed 106.4-fold. Although ketone bodies can be exported61, the induction of Slc16a6, the ketone body transporter, was negligible. Furthermore, following 30 μg/kg TCDD, serum β-hydroxybutyrate were reduced 2.5-fold, while hepatic β-hydroxybutyryl-CoA levels were undetectable (Fig. 4F,G). Overall, gene expression involved in ketone body biosynthesis was repressed, and for the most part, exhibited AHR genomic binding in the presence of a pDRE. TCDD also elicited a dramatic dose-dependent decrease in β-hydroxybutyrate levels in serum and hepatic extracts suggesting that decreased hepatic acetyl-CoA levels were not due to cholesterol nor ketone body biosynthesis.

Effects of TCDD on ketone body gene expression and levels. Differential gene expression pertaining to ketone body biosynthesis was assessed using RNA-seq. (A) The ketone body biosynthesis pathway with regulated steps denoted with a double dagger (‡). (B) Computational identification of putative dioxin response elements (pDREs), and the detection of hepatic AhR genomic binding in ChIPseq analysis 2 h after oral gavage of 30 μg/kg TCDD. Genes are listed by the official symbol as designated in the mouse genome informatics (MGI) database. (C) Time-dependent hepatic expression of glycolysis-related genes (n = 3) following a single bolus gavage of 30 μg/kg TCDD. (D) Hepatic dose-dependent gene expression (n = 5) following oral gavage with TCDD every 4 days for 28 days. (E) Diurnal regulated gene expression denoted with a “Y”. An orange ‘X’ indicates oscillating gene expression was abolished following oral gavage with 30 μg/kg TCDD every 4 days for 28 days. ZT indicates the time of maximum induction/repression (P1(t) > 0.8). Counts represent the maximum number of raw reads for any treatment group. Low transcript counts (< 500 reads) are denoted in yellow with high transcript counts (> 10,000) denoted in pink. Differential gene expression with a posterior probability (P1(t)) > 0.80 are depicted with a black triangle in the upper right corner of the tile. (F) Serum β-hydroxybutyrate and (G) hepatic β-hydroxybutyryl-CoA levels were assessed using a commercially available kit and targeted LC–MS/MS, respectively. Bar graphs denote the mean ± SEM. Significance (*p ≤ 0.05) was determined using a one-way ANOVA followed by Dunnett’s post-hoc analysis. The heatmap was created using R (v4.0.4). The biochemical reaction was created using Adobe Illustrator (v25.2).
Protein acetyl and β-Hydroxybutyryl posttranslational modifications
In addition to being metabolic intermediates, acetyl-CoA and β-hydroxybutyrate are also used as substrates for posttranslational modifications (PTMs). Approximately 90% of eukaryotic proteins undergo different types of reversible PTM, with moieties typically added onto lysine residues to regulate enzymatic activity, alter protein stability, and change protein localization and interactions with other proteins62,63. The level for protein acetylation and β-hydroxybutyrylation are dependent on the levels acetyl-CoA and β-hydroxybutyryl-CoA, respectively, that serve as source of the donor group64. β-Hydroxybutyrate is first activated to β-hydroxybutyryl-CoA by acyl-CoA short-chain synthetases such as ACSS263, which was repressed 9.1-fold in the present study. Consequently, capillary electrophoresis and Western blotting were used to investigate the effect of TCDD on the levels of total hepatic lysine-specific protein acetylation and β-hydroxybutyrylation (Fig. 5). TCDD dose-dependently decreased the level of total acetylated (Fig. 5A) and β-hydroxybutyrylated proteins in hepatic extracts prepared from treated mice. 135, 53, and 46 kDa protein(s) were dose-dependently decreased in acetylation (Fig. 5B–D), while β-hydroxybutyrylation was decreased in 28 to 155 kDa proteins (Fig. 5E–G). Reductions in the levels of acetylated and β-hydroxybutyrylated hepatic proteins are consistent with lower levels of acetyl-CoA and β-hydroxybutyrate.

(A) Total lysine-specific acetylated proteins were assessed in livers of mice (n = 3–5) by capillary electrophoresis. Mice were orally gavaged every 4 days for 28 days with 0.01, 0.03, 0.1, 0.3, 1, 3, 10 or 30 μg/kg TCDD prior to tissue collection. (B–D) Area under the three most prominent peaks for total proteins with acetylated lysine was quantified. (E,F) Total β-hydroxybutyrylated proteins were assessed by traditional Western blot (n = 3). Depicted is a representative Western blot of total β-hydroxybutyrylated proteins, as well as β-actin from total protein extracts. The full Western blots can be found in Supplementary Fig. 2. (G) The signal intensity of total β-hydroxybutyrylated proteins at various molecular weights for vehicle and 30 μg/kg treatment groups quantified using ImageJ as outlined in materials and methods. Bar graphs denote the mean ± SEM. Significance (*p ≤ 0.05) was determined using a one-way ANOVA followed by Dunnett’s post-hoc analysis. Plots were created using GraphPad Prism (v8.4.3).
AMPK activation
Cellular levels of acetyl-CoA provide a surrogate measure of the current metabolic state65. AMP-activated protein kinase (AMPK) senses energy status by monitoring changes in the AMP:ATP and ADP:ATP ratios66. Phosphorylation of the catalytic subunit of the trimeric AMPK protein is required for activation66. In the present study, TCDD dose-dependently decreased total hepatic AMPK, with a precipitous decrease at 30 μg/kg TCDD that corresponded with an increase in phosphorylated AMPK (P-AMPK) and the phosphorylated AMPK:total AMPK ratio (Fig. 6A–C). In response to low energy status, P-AMPK activates catabolic pathways to increase energy production. For example, in response to low energy levels, P-AMPK induces autophagy in an attempt to compensate for the lack of nutrients and provide substrates for catabolism. Autophagy can be independently regulated by P-AMPK following phosphorylation of the ULK complex that can then bind and phosphorylate autophagy related 14 (ATG14), a marker of cellular autophagy activity. Total and phosphorylated ATG14 levels (Fig. 6D,E) in hepatic extracts show that both ATG14 and phosphorylated ATG14 decreased following treatment with 30 μg/kg TCDD. Further investigation of P-AMPK activity on other known targets including acetyl-CoA carboxylase and RAPTOR were also equivocal suggesting P-AMPK was not activated despite phosphorylation.

Markers of AMPK activation. Mice (n = 3–5) were orally gavaged every 4 days for 28 days with TCDD prior to tissue collection. Protein levels were assessed in hepatic extracts using capillary immunoassay analysis. (A) Total AMPK and (B) phosphorylated AMPK (P-AMPK) and (C) the P-AMPK/AMP ratio two were assessed. Capillary immunoassay analysis of (D) total ATG14 and (E) phosphorylated ATG14 at serine 29 (P-ATG14 Ser29), a marker of P-AMPK activation. Bar graphs denote the mean ± SEM. Significance (*p ≤ 0.05) was determined using a one-way ANOVA followed by Dunnett’s post-hoc analysis. Plots were created using GraphPad Prism (v8.4.3).
Discussion
This is one of the first studies to examine acetyl-CoA levels in a model of TCDD-induced steatohepatitis with fibrosis. Acetyl-CoA is a central metabolite associated with several catabolic pathways including glycolysis, the tricarboxylic acid cycle, β-oxidation, and amino acid metabolism (e.g. lysine, valine, leucine and isoleucine), as well as a substrate used for the synthesis of fatty acids, cholesterol, and ketone bodies30,67. Previous studies have shown that TCDD dose-dependently caused metabolic reprograming affecting both glycolysis and fatty acid β-oxidation22,40. The present study further investigated TCDD-elicited metabolic reprogramming associated with acetyl-CoA metabolism. Our results indicate TCDD dose-dependently decreased the ratio of acetyl-CoA/CoA, an overall indicator of cellular energy status, and therefore additional enzymatic reactions and pathways associated with acetyl-CoA homeostasis were examined.
TCDD elicits a dose-dependent induction of pyruvate kinase isoform 2 (PKM2) which is reported to decrease glycolytic flux and shunt accumulating glycolytic intermediates to other pathways resulting in lower glucose-6-phosphate and fructose-6-phosphate levels22,39. Pyruvate, the end product of glycolysis, can then be oxidized to acetyl-CoA by the PDC composed of the (i) pyruvate dehydrogenase (E1), (ii) acetyl transferase (E2), and (iii) dihydrolipoyl dehydrogenase (E3) subunits. PDC is regulated by phosphorylation and dephosphorylation of the E1 subunit by pyruvate dehydrogenase kinases (PDKs) and pyruvate dehydrogenase phosphatases (PDPs), respectively. Specifically, E1 phosphorylation at Ser232, Ser293, and Ser300, inhibits PDC activity and reduces acetyl-CoA production68. TCDD dose-dependently induced Pdk4 and repressed Pdp2, while estrogen-related receptor γ (ERRγ) was induced which is associated with the induction of PDK4 in hepatoma cell lines under hypoxic conditions54. In addition, TCDD repressed expression of the highly expressed Acss2 and Acly, two important enzymes that contribute to the cytosolic acetyl-CoA pool. In previous studies, TCDD treatment increased hepatic levels of the TCA cycle intermediate oxaloacetate39. Increased oxalacetate levels may be due to depleted acetyl-CoA levels since both are required for the synthesis of the TCA cycle intermediate citrate. Collectively, metabolic reprogramming due to PKM2 induction, the inhibition of PDC following E1 phosphorylation, and the repression of Acss2 and Acly, two sources of acetyl-CoA produced from free acetate and citrate, respectively, are consistent with the overall reduction in acetyl-CoA. Missing in this analysis is an examination of the effects of TCDD on amino acid metabolism, another minor source of acetyl-CoA that is currently being investigated in a companion study.
Lower acetyl-CoA levels are indicators of insulin resistance, obesity, and cancer69,70. For example, squamous cell carcinomas and adenocarcinomas express high levels of PDK1 with evidence of PDC inactivation during metastasis71,72. Coincidentally, the International Agency for Research on Cancer (IARC) classifies TCDD as a human carcinogen3. Long-term treatment of rodents with TCDD leads to the development of tumors in multiple tissues including the liver, although the carcinogenic mechanism has not been resolved73. In humans, TCDD exposure has been associated with obesity, insulin resistance, and diabetes74,75,76. The doses used here take into consideration the relatively short duration of this study compared to the lifelong exposure of humans to diverse AhR ligands, the bioaccumulative nature of halogenated AhR ligands, and the differences in the half-life of TCDD between humans (1–11 years77,78) and mice (8–12 days79). The same dose levels and treatment regimen have also been used in previous studies and recently shown to approach steady-state levels14,22,41,80,81,82. Specifically, orally gavaging mice with 0.01 to 30 μg/kg TCDD every 4 days for 28 days resulted in mouse hepatic tissue levels that span human background serum concentrations reported in the United States, Germany, Spain, and the United Kingdom as well as serum levels in Viktor Yushchenko 4–39 months following intentional poisoning22. Collectively, this suggests that metabolic reprogramming resulting in reduced acetyl-CoA levels following persistent AhR activation by TCDD may have a role in the etiology of cancer and metabolic diseases such as insulin resistance and diabetes.
Few studies have assessed how liver disease progression affects acetyl-CoA and acetyl-CoA-derived intermediates. Studies examining fatty liver in mice on high-fat diets (HFD) have reported reduced pyruvate dehydrogenase complex activity, as well as Pdk2 and Pdk4 induction83. Lower β-hydroxybutyric acid in the serum for NAFLD patients also suggests fatty liver may impair ketogenesis84. Studies also report conflicting results regarding hepatic acetyl-CoA levels in HFD-induced NAFLD but this can likely be attributed to significant differences in models, HFDs and study duration85,86. Nevertheless, the possibility that TCDD does not act directly, and that hepatic fat accumulation reduces acetyl-CoA levels cannot be excluded given the results.
Under conditions of lower acetyl-CoA levels, intracellular ATP levels would also decrease and trigger the activation of AMPK, a ubiquitous sensor of cellular energy and nutrient status. AMPK monitors the AMP/ATP ratio, and is activated following phosphorylation to restore energy homeostasis by turning on catabolic pathways that provide substrates for ATP production while switching off biosynthetic pathways and other nonessential processes that consume energy87. Accordingly, TCDD dose-dependently increased the phosphorylated AMPK (P-AMPK, active form)/unphosphorylated AMPK (inactive form) ratio. However, known targets of P-AMPK such as acetyl-CoA carboxylase, a regulated enzyme in fatty acid biosynthesis, and downstream targets such as ATG14, a marker of autophagy, did not exhibit phosphorylation but were instead transcriptionally repressed by TCDD. Likewise, there was no evidence of RAPTOR phosphorylation that would induce the dissociation of mTOR from lysosomes to reduce biomass production in support of cell growth and proliferation. ATG14, a key regulator of autophagy, was also not phosphorylated and consequently, macromolecules were not available for catabolism to generate ATP. Overall, the lower acetyl-CoA/CoA ratio following TCDD treatment is consistent with the inability of P-AMPK to restore energy homeostasis. Further studies are needed to investigate why P-AMPK did not activate autophagy under conditions of low acetyl-CoA levels.
In addition to being a substrate for fatty acid and cholesterol synthesis which are repressed by TCDD14,88, acetyl-CoA can be used as a substrate for hepatic ketone body production during starvation or low circulating glucose levels30. The present study showed that after 6 h of fasting, hepatic and serum levels of β-hydroxybutyrate were dose-dependently decreased by TCDD, consistent with lower levels of acetyl-CoA production. Furthermore, acetyl-CoA and β-hydroxybutyrate-CoA can be used as substrates for the reversible posttranslational modification of proteins that can affect protein structure, enzymatic activity, cellular location, and protein–protein interactions89,90. Protein acetylation and β-hydroxybutyrylation PTMs are particularly important in gluconeogenesis, glycolysis, the TCA and urea cycles, glycogen metabolism, and fatty acid metabolism. Consequently, these intermediates can exert a signaling function that links metabolism and metabolite levels to gene expression63,90,91. In the present study, TCDD dose-dependently decreased total hepatic protein acetylation and β-hydroxybutyrylation PTMs which has been shown to correlate with acetyl-CoA and β-hydroxybutyrate levels92. This extends the potential effects of TCDD beyond direct AHR-mediated effects on gene expression to later indirect consequences due the disruption of protein PTM.
In summary, TCDD elicited dose-dependent hepatic metabolic reprogramming by direct AHR-mediated action on gene expression, while also causing later indirect effects by altering protein acetylation and β-hydroxybutyrylation. Specifically, the present study presents further evidence of TCDD-mediated metabolic reprograming events that contribute to an energy crisis as demonstrated by the reduced levels of intracellular acetyl-CoA and the induction of P-AMPK. We have previously shown that exposure to TCDD in mice results in the metabolic inhibition of glycolysis and fatty acid β-oxidation, pathways that replenish acetyl-CoA levels when cell energy stores are low. The present study provides evidence that TCDD impeded glycolysis not only due to PKM isoform switching, but also through the inactivation of the PDC, the gateway between glycolysis and the TCA cycle. Further investigation is necessary to elucidate the paradoxical energy dysregulation induced by TCDD.
Materials and methods
Animal treatment
Mice were housed and treated as previously described40. Briefly, postnatal day (PND) 25 male C57BL/6 mice, obtained from Charles River Laboratories (Kingston, NY), were house in Innovive Innocages (San Diego, CA) containing ALPHA-dri bedding (Shepherd Specialty Papers, Chicago, IL). Cages were housed in a 12 h/12 h light/dark cycle and at 23 °C environment with 30–40% humidity. Harlan Teklad 22/5 Rodent Diet 8940 (Madison, WI) and Aquavive water (Innovive) were provided ad libitum. A TCDD stock was prepared as previously described40. On PND28, mice were orally gavaged at the start of the light cycle (zeitgeber [ZT] 0) with 0.1 ml sesame oil vehicle (Sigma-Aldrich, St. Louis, MO) or 0.03, 0.1, 0.3, 1, 3, 10, and 30 μg/kg body weight TCDD every 4 days for 28 days for a total of 7 treatments. This dosing regimen was selected to approach steady state levels given the 8–12 day half-life of TCDD in mice93. Comparable treatment has been used in previous studies14,22,23,25,40,81,94. Following 28 days, mice were weighed and euthanized. Serum and liver tissues were collected and immediately flash-frozen in liquid nitrogen and stored at -80 °C. This study was conducted in accordance with relevant guidelines and regulations. All animal procedures were approved by the Michigan State University (MSU) Institutional Animal Care and Use Committee (IACUC; PROTO201800043) and meet the ARRIVE guidelines.
Liquid chromatography tandem mass spectrometry
Previously published untargeted liquid chromatography tandem mass spectrometry data was used to assess hepatic β-hydroxybutyryl-CoA levels40. Dataset was accessed through the NIH Metabolomics Workbench (ST001379). Targeted acetyl-CoA and coenzyme A samples were measured on a Xevo G2-XS QTof attached to a Waters UPLC (Waters, Cambridge, Massachusetts, United States). The liquid chromatography mobile phases, gradient flow rates, and columns were used as previously published23,40. Mass spectra were acquired using negative-mode electrospray ionization run in MSE continuum mode. The metabolite raw signals were quantified by retention time and accurate mass using MassLynx Version 4.2 (Waters). Acetyl-CoA levels were determined by measuring the response (unlabeled acetyl-CoA signal: 13C2-acetyl-CoA signal) in each sample. Concentration was determined by a 5-point calibration curve containing unlabeled acetyl-CoA (0.005–5 µM) and 13C2-acetyl-CoA at a constant 2 µM. Since isotopic labeled standards of CoA were unavailable, the standard addition method was used to measure CoA concentrations and correct for matrix effects. Briefly, CoA raw signal was measured in the samples used for acetyl-CoA analysis and in the sample with 1 µM unlabeled CoA standard added. To account for signal carry-over, CoA raw signal was corrected with the average % carry-over. The average % carry-over was determined by averaging % carry-over signal of CoA in the blanks and the sample run on the instrument prior to the blank. For all samples, the raw CoA signal was corrected for carry-over by the following equation: (corrected raw CoA signal) = (raw CoA signal)—(the average % carry-over) X (raw CoA signal in sample run prior on the instrument).
Clinical chemistry and hepatic acetate quantitation
β-Hydroxybutyrate levels in undiluted serum samples were assessed using a commercially available kit (Sigma-Aldrich) according to manufacturer’s protocol. Similarly, acetate levels were assessed liver samples (~ 40 mg) using a commercially available kit (Sigma-Aldrich) according to manufacturer’s protocol. An Infinite M200 plate reader (Tecan, Durham, North Carolina) was used to assay all replicates.
Protein extraction and quantification
Frozen liver tissues (~ 50 mg) were homogenized in RIPA buffer with protease inhibitors (Sigma-Aldrich) using a Polytron PT2100 homogenizer (Kinematica, Lucerne, Switzerland) followed by sonication on ice. Samples were centrifuged, after which the supernatant was collected, and protein concentration measured using a bovine serum albumin standard curve and a bicinchoninic acid (BCA) assay (Sigma-Aldrich).
Capillary electrophoresis protein analysis
The WES capillary electrophoresis system (ProteinSimple, San Jose, CA) was used following standard manufacturer protocols to assess protein levels on total liver lysates. Compass for SW (v4.0.0; ProteinSimple) was used to assess the area under each peak using the Gaussian fit method. The following antibodies and dilutions were used from the respective manufacturers: PDH (1:50; #2784; Cell Signaling, Danvers, MA); PDH p-Ser300 (1:50; AP1064; Sigma-Aldrich); ACLY (1:65; #4332; Cell Signaling); Total Acetylated Lysine (1:65; #9441; Cell Signaling); AMPK (1:50; #2603; Cell Signaling); AMPK p-Thr172 (1:50; #2535; Cell Signaling); ATG14 (1:50; PD026MS; MBL International, Woburn, MA); ATG14 p-Ser29 (1:50; #92340; Cell Signaling).
Western blotting
Protein samples (20 μg) from total liver lysates were resolved via 10% SDS-PAGE gels (Bio-Rad, San Diego, CA, USA) and transferred to nitrocellulose membranes (GE Healthcare, Chicago, IL) using the Mini Trans-Blot Cell Unit (BioRad) by wet electroblotting (100 V, 45 min). The membranes were then blocked with 5% nonfat milk (in Tris-buffered saline [TBS] + 0.01% Tween) for 1 h and incubated with primary antibodies: anti-β-hydroxybutyryllysine (1:1000; PTM-1201; PTM Biolabs, China) or anti-β-actin (1:1000; #4970; Cell Signaling) overnight at 4 °C. Blots were visualized using horseradish peroxidase (HRP)-linked secondary antibodies (1:3,000; Cell Signaling Technology) and an ECL kit (Millipore Corporation, Billerica, MA). Membranes were scanned on a Sapphire Biomolecular Imager (Azure Biosystem, Dublin, CA). Protein density values were assessed and calculated using ImageJ software (version 1.47; National Institutes of Health, Bethesda, MD). The expression for the protein of interest was standardized to β-actin levels.
Protein localization data
Protein subcellular localization data were acquired using COMPARTMENTS95 as previously described40. Subcellular localizations are listed using the following abbreviations unless otherwise noted: cytosol (C), endoplasmic reticulum (ER), extracellular space (ES), Golgi apparatus (GA), lipid droplet (LD), lysosome (L), mitochondrion (M), mitochondrial outer membrane (OMM), mitochondrial inner membrane (IMM), nucleus (N), peroxisome (P), and plasma membrane (PM).
Data availability
Hepatic RNA-seq data sets were previously published23,41,96. Differentially expressed genes (|fold-change|≥ 1.5 and posterior probability (P1(t)) ≥ 0.8) were determined by empirical Bayes analysis97. Time course (GSE109863), dose response (GSE203302), and diurnal rhythmicity (GSE119780) sequencing data are available at the Gene Expression Omnibus. Diurnal rhythmicity was determined using JTK_CYCLE as previously described23. AHR ChIP-seq (GSE97634) and computationally identified putative dioxin response elements (pDREs, https://doi.org/10.7910/DVN/JASCVZ) data were previously published41. ChIP-seq analysis used a false discovery rate (FDR) ≤ 0.05. pDREs were considered functional with a matrix similarity score (MSS) ≥ 0.856 and associated with genes when located 10 kb upstream of the transcription start site (TSS) to the transcription end site (TES). Raw metabolomic data were previously published40 and can be access through the NIH Metabolomics Workbench (ST001379).
References
Safe, S. Polychlorinated biphenyls (PCBs), dibenzo-p-dioxins (PCDDs), dibenzofurans (PCDFs), and related compounds: Environmental and mechanistic considerations which support the development of toxic equivalency factors (TEFs). Crit. Rev. Toxicol. 21, 51–88. https://doi.org/10.3109/10408449009089873 (1990).
Polychlorinated Dibenzo-Para-Dioxins and Polychlorinated Dibenzofurans. IARC Monogr Eval Carcinog Risks Hum 69, 1-631 (1997).
Polychlorinated Biphenyls and Polybrominated Biphenyls. IARC Monogr Eval Carcinog Risks Hum 107, 9-500 (2016).
McGregor, D. B., Partensky, C., Wilbourn, J. & Rice, J. M. An IARC evaluation of polychlorinated dibenzo-p-dioxins and polychlorinated dibenzofurans as risk factors in human carcinogenesis. Environ. Health Perspect. 106(Suppl 2), 755–760. https://doi.org/10.1289/ehp.98106755 (1998).
Avilla, M. N., Malecki, K. M. C., Hahn, M. E., Wilson, R. H. & Bradfield, C. A. The Ah receptor: Adaptive metabolism, ligand diversity, and the xenokine model. Chem. Res. Toxicol. 33, 860–879. https://doi.org/10.1021/acs.chemrestox.9b00476 (2020).
Denison, M. S. & Nagy, S. R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 43, 309–334. https://doi.org/10.1146/annurev.pharmtox.43.100901.135828 (2003).
Denison, M. S., Soshilov, A. A., He, G., DeGroot, D. E. & Zhao, B. Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 124, 1–22. https://doi.org/10.1093/toxsci/kfr218 (2011).
Hankinson, O. The aryl hydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol. 35, 307–340. https://doi.org/10.1146/annurev.pa.35.040195.001515 (1995).
Beischlag, T. V., Luis Morales, J., Hollingshead, B. D. & Perdew, G. H. The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot. Gene Expr. 18, 207–250. https://doi.org/10.1615/critreveukargeneexpr.v18.i3.20 (2008).
Dere, E., Lo, R., Celius, T., Matthews, J. & Zacharewski, T. R. Integration of genome-wide computation DRE search, AhR ChIP-chip and gene expression analyses of TCDD-elicited responses in the mouse liver. BMC Genom. 12, 365. https://doi.org/10.1186/1471-2164-12-365 (2011).
Huang, G. & Elferink, C. J. A novel nonconsensus xenobiotic response element capable of mediating aryl hydrocarbon receptor-dependent gene expression. Mol. Pharmacol. 81, 338–347. https://doi.org/10.1124/mol.111.075952 (2012).
Tanos, R. et al. Aryl hydrocarbon receptor regulates the cholesterol biosynthetic pathway in a dioxin response element-independent manner. Hepatology 55, 1994–2004. https://doi.org/10.1002/hep.25571 (2012).
Kopec, A. K. et al. Automated dose-response analysis and comparative toxicogenomic evaluation of the hepatic effects elicited by TCDD, TCDF, and PCB126 in C57BL/6 mice. Toxicol. Sci. 118, 286–297. https://doi.org/10.1093/toxsci/kfq236 (2010).
Nault, R., Fader, K. A., Lydic, T. A. & Zacharewski, T. R. Lipidomic evaluation of aryl hydrocarbon receptor-mediated hepatic steatosis in male and female mice elicited by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Chem. Res. Toxicol. 30, 1060–1075. https://doi.org/10.1021/acs.chemrestox.6b00430 (2017).
Liu, Q. et al. A quantitative HILIC-MS/MS assay of the metabolic response of Huh-7 Cells exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Metabolites 9, 118. https://doi.org/10.3390/metabo9060118 (2019).
Carlson, E. A. et al. Divergent transcriptomic responses to aryl hydrocarbon receptor agonists between rat and human primary hepatocytes. Toxicol. Sci. 112, 257–272. https://doi.org/10.1093/toxsci/kfp200 (2009).
Ruiz-Aracama, A. et al. An untargeted multi-technique metabolomics approach to studying intracellular metabolites of HepG2 cells exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. BMC Genom. 12, 251. https://doi.org/10.1186/1471-2164-12-251 (2011).
Vezina, C. M., Walker, N. J. & Olson, J. R. Subchronic exposure to TCDD, PeCDF, PCB126, and PCB153: Effect on hepatic gene expression. Environ. Health Perspect. 112, 1636–1644. https://doi.org/10.1289/txg.7253 (2004).
Rowlands, J. C. et al. Transcriptional profiles induced by the Aryl Hydrocarbon Receptor agonists 2,3,7,8-tetrachlorodibenzo-p-dioxin, 2,3,7,8-tetrachlorodibenzofuran and 2,3,4,7,8-pentachlorodibenzofuran in primary rat hepatocytes. Chemosphere 85, 232–238. https://doi.org/10.1016/j.chemosphere.2011.06.026 (2011).
Jennen, D. G. et al. Comparison of HepG2 and HepaRG by whole-genome gene expression analysis for the purpose of chemical hazard identification. Toxicol. Sci. 115, 66–79. https://doi.org/10.1093/toxsci/kfq026 (2010).
Nault, R., Colbry, D., Brandenberger, C., Harkema, J. R. & Zacharewski, T. R. Development of a computational high-throughput tool for the quantitative examination of dose-dependent histological features. Toxicol. Pathol. 43, 366–375. https://doi.org/10.1177/0192623314544379 (2015).
Nault, R. et al. Dose-dependent metabolic reprogramming and differential gene expression in TCDD-elicited hepatic fibrosis. Toxicol. Sci. 154, 253–266. https://doi.org/10.1093/toxsci/kfw163 (2016).
Fader, K. A., Nault, R., Doskey, C. M., Fling, R. R. & Zacharewski, T. R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin abolishes circadian regulation of hepatic metabolic activity in mice. Sci. Rep. 9, 6514. https://doi.org/10.1038/s41598-019-42760-3 (2019).
Forgacs, A. L. et al. Comparative metabolomic and genomic analyses of TCDD-elicited metabolic disruption in mouse and rat liver. Toxicol. Sci. 125, 41–55. https://doi.org/10.1093/toxsci/kfr262 (2012).
Matsubara, T. et al. Metabolomics identifies an inflammatory cascade involved in dioxin- and diet-induced steatohepatitis. Cell Metab. 16, 634–644. https://doi.org/10.1016/j.cmet.2012.10.006 (2012).
Kaelin, W. G. Jr. & McKnight, S. L. Influence of metabolism on epigenetics and disease. Cell 153, 56–69. https://doi.org/10.1016/j.cell.2013.03.004 (2013).
Menzies, K. J., Zhang, H., Katsyuba, E. & Auwerx, J. Protein acetylation in metabolism - metabolites and cofactors. Nat. Rev. Endocrinol. 12, 43–60. https://doi.org/10.1038/nrendo.2015.181 (2016).
Wang, Q. et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007. https://doi.org/10.1126/science.1179687 (2010).
Zhao, S. et al. Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004. https://doi.org/10.1126/science.1179689 (2010).
Pietrocola, F., Galluzzi, L., Bravo-San Pedro, J. M., Madeo, F. & Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 21, 805–821. https://doi.org/10.1016/j.cmet.2015.05.014 (2015).
Sivanand, S., Viney, I. & Wellen, K. E. Spatiotemporal control of Acetyl-CoA metabolism in chromatin regulation. Trends Biochem. Sci. 43, 61–74. https://doi.org/10.1016/j.tibs.2017.11.004 (2018).
Marino, G. et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol. Cell 53, 710–725. https://doi.org/10.1016/j.molcel.2014.01.016 (2014).
Fader, K. A. et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-elicited effects on bile acid homeostasis: Alterations in biosynthesis, enterohepatic circulation, and microbial metabolism. Sci. Rep. 7, 5921. https://doi.org/10.1038/s41598-017-05656-8 (2017).
Cave, M. et al. Polychlorinated biphenyls, lead, and mercury are associated with liver disease in American adults: NHANES 2003–2004. Environ. Health Perspect. 118, 1735–1742. https://doi.org/10.1289/ehp.1002720 (2010).
Zheng, S. et al. Effects of environmental contaminants in water resources on nonalcoholic fatty liver disease. Environ. Int. 154, 106555. https://doi.org/10.1016/j.envint.2021.106555 (2021).
Kumar, J. et al. Persistent organic pollutants and liver dysfunction biomarkers in a population-based human sample of men and women. Environ. Res. 134, 251–256. https://doi.org/10.1016/j.envres.2014.07.023 (2014).
Shertzer, H. G. et al. TCDD decreases ATP levels and increases reactive oxygen production through changes in mitochondrial F0F1-ATP synthase and ubiquinone. Toxicol. Appl. Pharmacol. 217, 363–374. https://doi.org/10.1016/j.taap.2006.09.014 (2006).
Boverhof, D. R. et al. Temporal and dose-dependent hepatic gene expression patterns in mice provide new insights into TCDD-Mediated hepatotoxicity. Toxicol. Sci. 85, 1048–1063. https://doi.org/10.1093/toxsci/kfi162 (2005).
Nault, R. et al. Pyruvate kinase isoform switching and hepatic metabolic reprogramming by the environmental contaminant 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 149, 358–371. https://doi.org/10.1093/toxsci/kfv245 (2016).
Cholico, G. N. et al. Thioesterase induction by 2,3,7,8-tetrachlorodibenzo-p-dioxin results in a futile cycle that inhibits hepatic beta-oxidation. Sci. Rep. 11, 15689. https://doi.org/10.1038/s41598-021-95214-0 (2021).
Fader, K. A. et al. Convergence of hepcidin deficiency, systemic iron overloading, heme accumulation, and REV-ERBalpha/beta activation in aryl hydrocarbon receptor-elicited hepatotoxicity. Toxicol. Appl. Pharmacol. 321, 1–17. https://doi.org/10.1016/j.taap.2017.02.006 (2017).
Nault, R. et al. From the cover: Coagulation-driven hepatic fibrosis requires protease activated receptor-1 (PAR-1) in a mouse model of TCDD-elicited steatohepatitis. Toxicol Sci 154, 381–391. https://doi.org/10.1093/toxsci/kfw175 (2016).
Chow, J. D. et al. Genetic inhibition of hepatic acetyl-CoA carboxylase activity increases liver fat and alters global protein acetylation. Mol Metab 3, 419–431. https://doi.org/10.1016/j.molmet.2014.02.004 (2014).
Perry, R. J. et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 160, 745–758. https://doi.org/10.1016/j.cell.2015.01.012 (2015).
Watzky, M. et al. Hexokinase 2 is a transcriptional target and a positive modulator of AHR signalling. Nucleic Acids Res. 50, 5545–5564. https://doi.org/10.1093/nar/gkac360 (2022).
Mueckler, M. & Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med. 34, 121–138. https://doi.org/10.1016/j.mam.2012.07.001 (2013).
Liang, W., Zhang, Y., Song, L. & Li, Z. 2,3’4,4’,5-Pentachlorobiphenyl induces hepatocellular carcinoma cell proliferation through pyruvate kinase M2-dependent glycolysis. Toxicol. Lett. 313, 108–119. https://doi.org/10.1016/j.toxlet.2019.06.006 (2019).
Song, L., Guo, L. & Li, Z. Molecular mechanisms of 3,3’4,4’,5-pentachlorobiphenyl-induced epithelial-mesenchymal transition in human hepatocellular carcinoma cells. Toxicol. Appl. Pharmacol. 322, 75–88. https://doi.org/10.1016/j.taap.2017.03.003 (2017).
Lee, J., Kim, H. K., Han, Y. M. & Kim, J. Pyruvate kinase isozyme type M2 (PKM2) interacts and cooperates with Oct-4 in regulating transcription. Int. J. Biochem. Cell Biol. 40, 1043–1054. https://doi.org/10.1016/j.biocel.2007.11.009 (2008).
Mazurek, S., Boschek, C. B., Hugo, F. & Eigenbrodt, E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 15, 300–308. https://doi.org/10.1016/j.semcancer.2005.04.009 (2005).
Gorrini, C., Harris, I. S. & Mak, T. W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947. https://doi.org/10.1038/nrd4002 (2013).
Park, S. et al. Role of the pyruvate dehydrogenase complex in metabolic remodeling: Differential pyruvate dehydrogenase complex functions in metabolism. Diabetes Metab. J. 42, 270–281. https://doi.org/10.4093/dmj.2018.0101 (2018).
Rardin, M. J., Wiley, S. E., Naviaux, R. K., Murphy, A. N. & Dixon, J. E. Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal. Biochem. 389, 157–164. https://doi.org/10.1016/j.ab.2009.03.040 (2009).
Lee, J. H. et al. Hypoxia induces PDK4 gene expression through induction of the orphan nuclear receptor ERRgamma. PLoS One 7, e46324. https://doi.org/10.1371/journal.pone.0046324 (2012).
Miller, J. W. & Rucker, R. B. Present Knowledge in Nutrition 375–390 (Wiley, 2012).
Leonardi, R., Zhang, Y. M., Rock, C. O. & Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 44, 125–153. https://doi.org/10.1016/j.plipres.2005.04.001 (2005).
Cahill, G. F. Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 26, 1–22. https://doi.org/10.1146/annurev.nutr.26.061505.111258 (2006).
Haapalainen, A. M. et al. Crystallographic and kinetic studies of human mitochondrial acetoacetyl-CoA thiolase: The importance of potassium and chloride ions for its structure and function. Biochemistry 46, 4305–4321. https://doi.org/10.1021/bi6026192 (2007).
Song, X. Q. et al. Molecular cloning and nucleotide sequence of complementary DNA for human hepatic cytosolic acetoacetyl-coenzyme A thiolase. Biochem. Biophys. Res. Commun. 201, 478–485. https://doi.org/10.1006/bbrc.1994.1726 (1994).
Sato, S. et al. Low-dose dioxins alter gene expression related to cholesterol biosynthesis, lipogenesis, and glucose metabolism through the aryl hydrocarbon receptor-mediated pathway in mouse liver. Toxicol. Appl. Pharmacol. 229, 10–19. https://doi.org/10.1016/j.taap.2007.12.029 (2008).
Hugo, S. E. et al. A monocarboxylate transporter required for hepatocyte secretion of ketone bodies during fasting. Genes Dev. 26, 282–293. https://doi.org/10.1101/gad.180968.111 (2012).
Xia, C., Tao, Y., Li, M., Che, T. & Qu, J. Protein acetylation and deacetylation: An important regulatory modification in gene transcription (Review). Exp. Ther. Med. 20, 2923–2940. https://doi.org/10.3892/etm.2020.9073 (2020).
Huang, H. et al. The regulatory enzymes and protein substrates for the lysine beta-hydroxybutyrylation pathway. Sci. Adv. 7, eabe2771. https://doi.org/10.1126/sciadv.abe2771 (2021).
Chypre, M., Zaidi, N. & Smans, K. ATP-citrate lyase: A mini-review. Biochem. Biophys. Res. Commun. 422, 1–4. https://doi.org/10.1016/j.bbrc.2012.04.144 (2012).
Shi, L. & Tu, B. P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 33, 125–131. https://doi.org/10.1016/j.ceb.2015.02.003 (2015).
Garcia, D. & Shaw, R. J. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 66, 789–800. https://doi.org/10.1016/j.molcel.2017.05.032 (2017).
Cai, L. & Tu, B. P. On acetyl-CoA as a gauge of cellular metabolic state. Cold Spring Harb. Symp. Quant. Biol. 76, 195–202. https://doi.org/10.1101/sqb.2011.76.010769 (2011).
Patel, M. S., Nemeria, N. S., Furey, W. & Jordan, F. The pyruvate dehydrogenase complexes: Structure-based function and regulation. J. Biol. Chem. 289, 16615–16623. https://doi.org/10.1074/jbc.R114.563148 (2014).
Jeoung, N. H. & Harris, R. A. Pyruvate dehydrogenase kinase-4 deficiency lowers blood glucose and improves glucose tolerance in diet-induced obese mice. Am. J. Physiol. Endocrinol. Metab. 295, E46-54. https://doi.org/10.1152/ajpendo.00536.2007 (2008).
McFate, T. et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J. Biol. Chem. 283, 22700–22708. https://doi.org/10.1074/jbc.M801765200 (2008).
Koukourakis, M. I., Giatromanolaki, A., Bougioukas, G. & Sivridis, E. Lung cancer: A comparative study of metabolism related protein expression in cancer cells and tumor associated stroma. Cancer Biol. Ther. 6, 1476–1479. https://doi.org/10.4161/cbt.6.9.4635 (2007).
Koukourakis, M. I. et al. Pyruvate dehydrogenase and pyruvate dehydrogenase kinase expression in non small cell lung cancer and tumor-associated stroma. Neoplasia 7, 1–6. https://doi.org/10.1593/neo.04373 (2005).
Knerr, S. & Schrenk, D. Carcinogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in experimental models. Mol. Nutr. Food Res. 50, 897–907. https://doi.org/10.1002/mnfr.200600006 (2006).
Chang, J.-W., Chen, H.-L., Su, H.-J. & Lee, C.-C. Abdominal obesity and insulin resistance in people exposed to moderate-to-high levels of dioxin. PLOS ONE 11, e0145818. https://doi.org/10.1371/journal.pone.0145818 (2016).
Roh, E. et al. Serum aryl hydrocarbon receptor ligand activity is associated with insulin resistance and resulting type 2 diabetes. Acta Diabetol. 52, 489–495. https://doi.org/10.1007/s00592-014-0674-z (2015).
Warner, M. et al. Diabetes, metabolic syndrome, and obesity in relation to serum dioxin concentrations: The Seveso women’s health study. Environ. Health Perspect. 121, 906–911. https://doi.org/10.1289/ehp.1206113 (2013).
Sorg, O. et al. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) poisoning in Victor Yushchenko: Identification and measurement of TCDD metabolites. Lancet 374, 1179–1185. https://doi.org/10.1016/S0140-6736(09)60912-0 (2009).
Wolfe, W. H. et al. Determinants of TCDD half-life in veterans of operation ranch hand. J. Toxicol. Environ. Health 41, 481–488. https://doi.org/10.1080/15287399409531858 (1994).
Gasiewicz, T. A., Geiger, L. E., Rucci, G. & Neal, R. A. Distribution, excretion, and metabolism of 2,3,7,8-tetrachlorodibenzo-p-dioxin in C57BL/6J, DBA/2J, and B6D2F1/J mice. Drug Metab. Dispos. 11, 397–403 (1983).
Lee, J. H. et al. A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 139, 653–663. https://doi.org/10.1053/j.gastro.2010.03.033 (2010).
Angrish, M. M., Dominici, C. Y. & Zacharewski, T. R. TCDD-elicited effects on liver, serum, and adipose lipid composition in C57BL/6 mice. Toxicol. Sci. 131, 108–115. https://doi.org/10.1093/toxsci/kfs277 (2013).
Liu, R., Zacharewski, T. R., Conolly, R. B. & Zhang, Q. A physiologically based pharmacokinetic (PBPK) modeling framework for mixtures of dioxin-like compounds. Toxics 10, 700. https://doi.org/10.3390/toxics10110700 (2022).
Lu, Q. et al. Metabolic changes of hepatocytes in NAFLD. Front. Physiol. 12, 710420. https://doi.org/10.3389/fphys.2021.710420 (2021).
Fletcher, J. A. et al. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight https://doi.org/10.1172/jci.insight.127737 (2019).
Luo, G., Xiang, L. & Xiao, L. Acetyl-CoA deficiency is involved in the regulation of iron overload on lipid metabolism in apolipoprotein E knockout mice. Molecules 27, 4966. https://doi.org/10.3390/molecules27154966 (2022).
Carrer, A. et al. Impact of a high-fat diet on tissue acyl-CoA and histone acetylation levels. J. Biol. Chem. 292, 3312–3322. https://doi.org/10.1074/jbc.m116.750620 (2017).
Hardie, D. G. AMPK: Positive and negative regulation, and its role in whole-body energy homeostasis. Curr. Opin. Cell Biol. 33, 1–7. https://doi.org/10.1016/j.ceb.2014.09.004 (2015).
Lakshman, M. R., Campbell, B. S., Chirtel, S. J. & Ekarohita, N. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on de novo fatty acid and cholesterol synthesis in the rat. Lipids 23, 904–906. https://doi.org/10.1007/BF02536213 (1988).
Narita, T., Weinert, B. T. & Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 20, 156–174. https://doi.org/10.1038/s41580-018-0081-3 (2019).
Koronowski, K. B. et al. Ketogenesis impact on liver metabolism revealed by proteomics of lysine beta-hydroxybutyrylation. Cell Rep. 36, 109487. https://doi.org/10.1016/j.celrep.2021.109487 (2021).
Alleyn, M., Breitzig, M., Lockey, R. & Kolliputi, N. The dawn of succinylation: A posttranslational modification. Am. J. Physiol. Cell Physiol. 314, C228–C232. https://doi.org/10.1152/ajpcell.00148.2017 (2018).
Xie, Z. et al. Metabolic regulation of gene expression by histone lysine beta-hydroxybutyrylation. Mol. Cell 62, 194–206. https://doi.org/10.1016/j.molcel.2016.03.036 (2016).
Birnbaum, L. S. Distribution and excretion of 2,3,7,8-tetrachlorodibenzo-p-dioxin in congenic strains of mice which differ at the Ah locus. Drug Metab. Dispos. 14, 34–40 (1986).
Lee, J. H. et al. A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 139, 653–663. https://doi.org/10.1053/j.gastro.2010.03.033 (2010).
Binder, J. X. et al. COMPARTMENTS: Unification and visualization of protein subcellular localization evidence. Database (Oxford) 2014, bau012. https://doi.org/10.1093/database/bau012 (2014).
Nault, R., Doskey, C. M., Fader, K. A., Rockwell, C. E. & Zacharewski, T. Comparison of hepatic NRF2 and aryl hydrocarbon receptor binding in 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated mice demonstrates NRF2-independent PKM2 induction. Mol. Pharmacol. 94, 876–884. https://doi.org/10.1124/mol.118.112144 (2018).
Eckel, J. E., Gennings, C., Chinchilli, V. M., Burgoon, L. D. & Zacharewski, T. R. Empirical bayes gene screening tool for time-course or dose-response microarray data. J. Biopharm. Stat. 14, 647–670. https://doi.org/10.1081/BIP-200025656 (2004).
Acknowledgements
This project was supported by the National Institute of Environmental Health Sciences (NIEHS) Superfund Research Program [NIEHS SRP P42ES004911] and the NIEHS Research Project Grant Program [NIEHS R01ES029541] to TRZ. TRZ was partially supported by AgBioResearch at Michigan State University. GNC and RRF were supported by NIEHS Multidisciplinary Training in Environmental Toxicology [T32ES007255]. KAF was supported by the Canadian Institutes of Health Research Doctoral Foreign Study Award [DFS-140386].
Author information
Authors and Affiliations
Contributions
G.N.C. and T.R.Z. designed the project. R.R.F., K.A.F., N.A.Z. and R.N. performed the animal work. G.N.C., K.O., W.J.S., R.R.F., N.A.Z. and K.A.F. performed the experiments. R.R.F. developed the LC–MS workflow for targeted metabolomics. G.N.C. compiled all the data and produced the figures and tables. G.N.C. and T.R.Z. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cholico, G.N., Orlowska, K., Fling, R.R. et al. Consequences of reprogramming acetyl-CoA metabolism by 2,3,7,8-tetrachlorodibenzo-p-dioxin in the mouse liver. Sci Rep 13, 4138 (2023). https://doi.org/10.1038/s41598-023-31087-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-31087-9
- Springer Nature Limited