
Abstract
Serine proteases (SPs) and their homologs (SPHs) are among the best-characterized gene families. They are involved in several physiological processes, including digestion, embryonic development and immunity. In the current study, a total of 177 SPs-related genes were characterized in the genome of Ostrinia furnacalis. The activation site of SPs/SPHs and enzyme specificity of SPs were identified, and the findings showed that most of the SPs analyzed possessed trypsin substrate specificity. Several SPs/SPHs with similar simple gene structures had tandem repeat-like distributions on the scaffold, indicated that gene expansion has occurred in this large family. Furthermore, we constructed 30 RNA sequencing libraries including four with developmental stage and four middle larval stage tissues to study the transcript levels of these genes. Differentially upregulated and downregulated genes were obtained via data analysis. More than one-quarter of the genes were specifically identified as highly expressed in the midgut in compared to the other three tissues evaluated. In the current study, the domain structure, gene location and phylogenetic relationship of genes in O. furnacalis were explored. Orthologous comparisons of SPs/SPHs between model insects and O. furnacalis indicated their possible functions. This information provides a basis for understanding the functional roles of this large family.
Similar content being viewed by others
Introduction
Chymotrypsin-like serine proteases (SPs) and their homologs constitute a large gene family that plays essential roles in various insect physiological processes. SP-related peptides in inactive zymogen form are converted into their active forms by proteolytic cleavage of a particular peptide bond1,2. A specific catalytic triad comprising His57, Ser195, and Asp102 amino acid residues is harbored in the active center of SPs. Amino acid residues 189, 216 and 226 in chymotrypsin near the active site form the primary substrate-binding pocket, which is the predominant factor for determining SP substrate specificity3,4. This specific molecular recognition ability enables SPs to activate downstream enzymes, amplify signals, and form a cascade pathway. In addition to SPs, some members are enzymatically inactive owing to mutation of one or more catalytic residues. These proteins are known as serine protease homologs (SPHs) and are predicted to be cofactors of SPs. The roles played by SPs and SPHs have been extensively studied in invertebrates and found to range from the digestion of ingested proteins to immune defenses directed against various microbes and the establishment of embryo dorsoventral polarity5,6,7,8,9,10,11,12,13,14. SPs dominate the larval gut environment and contribute to approximately 95% of total digestive activity5. The functions of SPs and SPHs involved in epithelial morphogenesis of imaginal discs6,7 and somatic muscle attachment in embryos8,9 have been illustrated in Drosophila. The immune responses induced by SPs and SPHs have been reported in insect species10,11,12,13,14 and include melanotic encapsulation and induction of antimicrobial peptides.
Most SPs comprise fewer than 300 residues and harbor a single serine protease domain. These enzymes are mainly found in the gut and are involved in food digestion5. In addition, some SPs have disulfide-stabilized structures at the N-terminus; this structure is known as the clip domain and is formed by six conserved cycteine residues15,16. SPs and SPHs with clip domains are called CLIPs17. The roles played by CLIPs in the extracellular SP cascade, in which one SP activates the zymogen of another SP to trigger a rapid response, have been extensively explored in several insects18. In tobacco hornworm, HP6 activates HP8 to convert proSpatzle into Spatzle, thus inducing the synthesis of antimicrobial peptides (AMPs). Autoactivated HP14 activates proHP21, which further activates proPAP2 and PAP3 to induce proPO activation cascade. SPH1 and SPH2 are essential to this cascade, as they accelerate the production of high Mr active PO10,11,12. Holotrichia diomphalia PPAF-II and Tenebrio molitor SPH1 are involved in the sequential activation of proPOs to produce a melanization complex13,14. Some SPs present with other structural characteristics, such as a low-density lipoprotein receptor class A (LDLA) domain, Frizzled (Fz) domain, complement control protein (CCP) domain, or gastrulation defective (Gd) domain. These additional domains enable SPs to interact with other enzymes and thus participate in additional processes. For instance, Manduca sexta HP14 and Drosophila melanogaster modular serine protease (ModSP) have repeated LDLAs and CCP domain and, when initiated, receive signals from recognition receptors and activate proPAP in Toll pathway11,19. Limulus clotting factor C found in crayfish and horseshoe crab can form insoluble coagulin and plays a role in crosslinking20. Multidomain SPs, Nudel and Gd are involved in dorsal–ventral axis establishment21,22.
Advances in sequencing technology have increased the availability of genome data from different organisms. The number of SP-related genes in D. melanogaster, Apis mellifera, Bombyx mori, M. sexta, Anopheles gambiae and Pteromalus puparum has been identified at the genome level4,23,24,25,26,27. Sequencing technology has accelerated the identification of SP genes, in addition to the cloning and classification of single SPs. Orthologous comparison of these genes has enabled the prediction of the functional roles of newly discovered SPs/SPHs.
The Asian corn borer, O. furnacalis (Guenée), is an agricultural pest in Asia that causes significant damage in corn-producing countries with 20–80% yield losses28. The Asian corn borer has developed resistance to chemical and biotic pesticides, even in Bt crops28,29,30 making pest population control challenging. A previous study identified 13 CLIPs through transcriptome analysis31. The roles of four O. furnacalis SPs involved in the melanization pathway have been verified through biological experiments32,33,34. Although some SPs have been explored previously, the roles played by most SPs have not been elucidated. The roles of O. furnacalis SPs in digestion, development and immunity have not been fully elucidated. The O. furnacalis genome is currently available; thus, SP and SPH genes from O. furnacalis were characterized in the present study. Systematic analyses were conducted by determining catalytic characteristics, gene structure, and scaffold information; construction of evolution tree; and identifying expression patterns. This information enabled us to predict possible biological functions of O. furnacalis SP genes and provides a basis for further research.
Results
Overview of O. furnacalis SP and SPH sequences
Before the characterization of SP/SPH genes, we evaluated the completeness of the O. furnacalis genome by calculating the coverage for the set of single-copy orthologous genes in Arthropoda and Lepidoptera using BUSCO; the results showed genome coverage rates of 96.8% (981/1013) and 97.2% (5137/5286), respectively (Supplementary Figure S1). We also identified 971 and 5073 conserved single-copy Arthropoda and Lepidoptera genes in a BUSCO analysis of protein sequences, with 95.9% and 96.0% sequence similarity, respectively (Supplementary Figure S1).
Genomic evaluation of O. furnacalis indicated a total of 177 SP and SPH genes (Supplementary Table S1). The amino acid sequences of these SPs/SPHs are presented in Supplementary file in FASTA format. Twenty-eight SP/SPH genes were expressed as 2 to 4 isoforms, which were identified on the basis of structural gene annotation. The total number of SP-related genes was similar to that in Tribolium castaneum (175), and was significantly fewer than that in D. melanogaster (257) or A. gambiae (337) (Table 1). SPs and SPHs are synthesized as zymogens1. The amino acid at the P1 position of the activation site determines the enzyme specificity for SP/SPH catalysis. SPs/SPHs with Arg or Lys residue at P1 position are cleaved by trypsin-like proteases; SPs/SPHs containing Phe/Tyr/Leu residues at this position are substrates of chymotrypsin-like proteases, whereas SPs/SPHs with Ala/Gly/Val/Ile/Met/Ser residues at this position are activated by elastase-like proteases26. An analysis showed that 114 of 177 SPs/SPHs were activated by trypsin-like proteases, whereas 16 and 21 were presumably activated by chymotrypsin-like and elastase-like proteases, respectively. The residue at the P1 position was replaced by other amino acids in 21 SPs/SPHs, implying that they have unique features. The remaining 5 genes lacked residue at P1 position (Supplementary Table S1).
Notably, 43 of 177 SP-related genes were considered SPHs owing to the substitution of at least one conserved residue in the catalytic triad. These genes may encode cofactors for reactions. The remaining 134 SPs with enzymatic activity cleaved and activated downstream SPs/SPHs. An analysis showed that 80, 34 and 6 of the 134 SPs showed trypsin, chymotrypsin, and elastase substrate specificity (Supplementary Table S1).
Clip domain SP/SPH
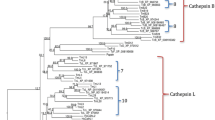
The clip domain can be sequentially activated to form a cascade pathway and play an important function in the immune response and embryonic development. An analysis revealed 29 cSP and 9 cSPH genes in the genome of O. furnacalis. Most CLIPs (30) carried a single clip domain, whereas 5 OfucSPs had 2 clip domains, and OfucSPH2, OfucSPH7, OfucSP9 had 3, 2, and 5 clip domains (Fig. 1). SP/SPH genes were assigned to four distinct clades based on the phylogenetic calculation results and were classified as CLIPAs to CLIPDs based on homologous serine protease-like genes that have been previously characterized (Fig. 2). The O. furnacalis genome included 8 CLIPAs, 11CLIPBs, 4CLIPCs and 15 CLIPDs. The total number of CLIPs in the Asian corn borer was within the median range compared with CLIPs in other related insect species. The ratio of O. furnacalis CLIPCs was the lowest among these species, whereas the number of O. furnacalis CLIPDs was highest among other species (Supplementary Table S2).

Domain organizations of SPs/SPHs with other structural elements in Ostrinia furnacalis. The schematic diagrams are not drawn to scale. CCP Sushi domain, also known as Sushi or SCR, CUB A domain identified in complement 1r/s, uegf, and bmp1, Fz Frizzled domain, Gd Gastrulation defective, LDLA LDLA-receptor class A domain, PD Serine protease domain, PLD serine protease-like domain, S Signal peptide, SR Scavenger receptor domain, TM Transmembrane region, TSP Thrombospondin.

Phylogenetic relationship of CLIPAs, CLIPBs, CLIPCs and CLIPDs in five insects. The serine protease-like domains of Ostrinia furnacalis (Ofu), Apis mellifera (Am), Manduca sexta (Ms), Drosophila melanogaster (Dm) and Tribolium castaneum (Tc) were aligned. The phylogenetic tree was constructed by IQ-TREE. SPs/SPHs from different species were marked with different color. Red spots at the nodes denoted bootstrap values were greater than 90 from 100 trials.
Based on the phylogenetic analysis, all OfucSPHs belonged to the CLIPA group except OfucSPH5, which formed clusters with cSPH genes in the CLIPB group (Fig. 2). OfucSP26 and OfucSP17 were orthologous to MsPAP1 and MsHP8, respectively. OfucSP10, 18, 19, and 24 clustered together with MsPAP3. OfucSP15 in the CLIPC group shared high amino acid sequence identity with MsHP6. There were 24 orthologous groups characterized by bioinformatics calculation, including 7 single copy orthologous sets (six in the CLIPD group and one in the CLIPA group) and 2 O. furnacalis specific orthologous sets (Supplementary Table S3).
SPs/SPHs with other domains
Eleven of the 177 SPs/SPHs with other domains were observed in the O. furnacalis genome. These domains included the LDLA domain, Fz domain, CCP domain, CUB domain, thrombospondin (TSP) domain, scavenger receptor Cys-rich (SR) domain and Gd domain (Fig. 1). The domain structure of O. furnacalis SP-like proteases was compared with that of proteases in fruit flies (Table 2). The findings showed that O. furnacalis SPs/SPHs were classified into 8 types of multidomain proteins, which was fewer than the number identified in D. melanogaster. OfuSP104 was orthologous to the Nudel gene that contains a serine protease-like domain between LDLA domains. OfuSP86 and OfuSP97 harbored Gd-like domains. OfuSP53/54/55 carried LDLAs followed by a CCP domain, and were probably ModSP-like proteases. OfuSP29 had Fz, LDLA and SR domains and shared domain similarity with Corin35.
Scaffold location of SPs/SPHs
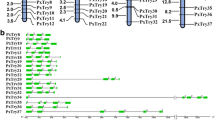
Gene duplication is common in the serine protease family. Gene duplication of 177 O. furnacalis SP and SPH genes was identified in the current study. Duplicated gene pairs were generated and are presented in Fig. 3. Based on this analysis, more than 40% (71/177) of the genes in the SP gene family underwent gene duplication (Fig. 3). Analysis showed that 9 SP/SPH genes on NW_021131465.1 were duplicated genes and formed the largest gene cluster. OfucSP5-8 formed a clade in the phylogenetic tree (Fig. 2), while they are also considered as duplicate genes and clustered in one scaffold.

Scaffold location of Ostrinia furnacalis SP and SPH genes. Scaffold name and gene names were shown on the left and right of the bar, respectively. The length of scaffold and the distance of two adjacent genes were scaled in figure. Adjacent genes underwent duplications were associated with red curve. Genes differentially upregulated in midgut were marked by symbols. “○”presented genes upregulated in midgut compared to genes in other three tissues; “□”presented genes upregulated in midgut compared to genes in either two of three tissues; “△”presented genes upregulated in midgut compared to genes in one of three tissue.
The expression patterns of SP/SPH genes
We performed deep RNA sequencing (RNA-seq) libraries of O. furnacalis with samples obtained from four developmental stages and from four different tissues to obtain the comprehensive expression profiles of O. furnacalis SPs and SPHs. Approximately 1.4 billion raw reads were obtained from 30 libraries, and approximately 43–51 million reads were generated per library. After removing the adaptor sequences, ambiguous reads and low-quality reads, approximately 1.36 billion clean reads were obtained. The average ratio of clean reads mapped to the reference genome was 83.48% (Supplementary Table S4).
A series of genome-wide expression profiling comparisons were conducted. Via pairwise comparison between samples at all these developmental stages and tissues, the number of differentially upregulated and downregulated genes were documented (Table 3). A Venn diagram shows the common or uniquely regulated genes at one stage or in one tissue (Figs. 4 and 5).

Venn diagram presented the number of upregulated or downregulated genes in embryo stage, newly hatched larval stage, middle larval stage, mature larval stage, pupal stage, and adult stage. The genes commonly upregulated or downregulated in one stage were listed. 1st instar larva, 3rd instar larva and 5th instar larva presented the newly hatched larvae, middle stage larvae and mature larvae.

Venn diagram presented the number of upregulated or downregulated genes in midgut, fat body, silk gland and hemolymph. The genes commonly upregulated or downregulated in one stage were listed.
There were more downregulated genes than upregulated genes between the embryo stage and other developmental stages. The results showed that 66, 56, 81, 76 and 77 SP/SPH genes were downregulated and 28, 27, 29, 20, and 27 SP/SPH genes were upregulated in the embryo stage compared to the newly hatched larval stage, middle larval stage, mature larval stage, pupal stage, and adult stage, respectively (Table 3). Among these genes, 31 SP/SPH genes were commonly differentially up- or downregulated in the embryo compared to the genes in the other four developmental stages, and approximately 50% (15/31) of these genes carried clip domains (Fig. 4). A comparative analysis of differentially expressed genes in the pupal stage revealed limited commonality of specifically expressed genes (only nine upregulated genes). Insight into gene expression profiles in the adult stage resulted in a total of 35 commonly and specifically highly expressed genes in comparison to genes expressed in other developmental stages, and none of these SP/SPH genes carried a clip domain (Fig. 4). Since there were few genes significantly expressed between either of the two larval stages, no common genes were identified at the middle- or mature-larval stage compared to the genes in the other four developmental stages, and only one commonly upregulated gene was identified in the newly hatched larval stage.
Analysis of gene expression patterns in different tissues showed that the number of SP/SPH genes that were differentially upregulated in the midgut was greater than the number of downregulated genes. Moreover, 46 commonly regulated genes and 7 genes shared with low expression were identified in the midgut (Fig. 5). All the upregulated genes formed simple gene structures (carrying only signal peptides and serine protease-like domains). There were 24, 21 and 42 SP/SPH genes highly expressed respectively in both the hemolymph and fat body; hemolymph and silk gland; hemolymph and midgut; the number of genes with low expression in these pairs was 48, 67 and 74, respectively (Table 3). In the fat body and silk gland, we only found common upregulated genes in comparison to genes in other three tissues (Fig. 5).
The expression patterns of SP/SPH genes in the midgut seemed to be correlated with their location in the scaffold. The genes highly expressed in the midgut are marked in Fig. 3, and a trend was found in the expression patterns, i.e., genes clustering on the same scaffold showed similar expression profiles. The transcript levels of SP and SPH genes on scaffold NW_021131465.1 were high in the midgut (Fig. 3). All these genes were single protease domain SPs/SPHs at amino acid residues 256–260 (Supplementary Table S1). Genes located on NW_021132648.1, NW_021130533.1 and NW_021134344.1 showed similar simple gene structures and expression profiles.
Discussion
A total of 177 SP/SPH genes were identified in O. furnacalis genome. Since the genome of O. furnacalis was sequenced by second-generation sequencing technology, not in combination with third-generation sequencing technology, the quality of O. furnacalis gene models was not as good as that in some model insects. The quality of gene models is partly reflected in the percentage of sequences with signal peptides. There are 141 of the 177 (80%) O. furnacalis SPs/SPHs possess signal peptides. The percentage of SPs/SPHs with signal peptide in O. furnacalis is lower than that in D. melanogaster (248/257) and A. gambiae (324/337), similar to that in Pteromalus puparum (152/183)36, but higher than that in Plutella xylostella (105/221) and T. molitor (142/200)37,38. With the cost of sequencing the genome decreased, we can improve O. furnacalis serine protease gene models by the use of the third-generation sequencing technology in the future work.
SPs are catalyzed into active enzymes that activate downstream SPs and SPHs. The findings showed that more than 60% (114/177) of SPs/SPHs were catalyzed by trypsin-like proteases. Most SPs exhibited trypsin substrate specificity (80/134). The majority of SPs/SPHs with trypsin-like properties were observed in insects such as D. melanogaster, A. gambiae and M. sexta4,26,27. Notably, the analysis showed a tandem repeat distribution in O. furnacalis SP/SPH genes (71/177). Genes adjacent to scaffold are essential for the expansion of the gene family, and unequal cross over may be an important mechanism for the generation of these clusters1. Gene duplication events have been reported in several insect species in this gene family4,23,24,26,27. An entire 11.1-kb region that contains 7 trypsin genes were amplified by PCR in A. gambiae39. Long segments harboring 5 trypsin genes also have been found in D. melanogaster and Drosophila erecta by PCR amplification40. We considered that gene duplication occurred in the serine protease family of O. furnacalis and some serine proteases were duplicated genes. In addition, an analysis of tandem repeat genes was based on the location information in the scaffold, not the chromosome, implying that some duplicated genes may have been left out.
The functions of SPs/SPHs were predicted by gene structures, construction of phylogenetic tree and determination of relative expression levels. Proteases in the CLIPB and CLIPC groups of several related species were included in the analysis. The findings showed that the CLIPC group comprised serine proteases upstream of SP cascades with members serving as activators of terminal proteases. Activated by CLIPCs, CLIPBs were cleaved and activated downstream proSatzles or proPOs15. An analysis revealed that the sequence of OfucSP17 showed high similarity with that of MsHP8, implying that OfucSP17 exhibits functions similar to those of MsHP8 and may play a role in the Toll pathway and induce the expression of AMP genes. OfucSP19 and OfucSP24, also known as O. furnacalis SP105 and OfPAP were components of the PPO activation system33,34. In addition, OfucSP19, OfucSP24 and MsPAP3 all comprised 2 clip domains. Similar sequence structures and functions of PAP-like genes in M. sexta and O. furnacalis indicate that these genes were involved in similar protease pathways. OfucSP10 and OfucSP18, formed a cluster with MsPAP3, OfucSP19 and OfucSP24 and harbored 2 clip domains. These findings suggested that OfucSP10 and OfucSP18 may exhibit functions similar to those of MsPAP3, OfucSP19 and OfucSP24 and may be involved in the melanization cascade.
An analysis showed OfucSP5-8 were different from other Clip D genes and displayed lineage-specific expansion. These four genes in this group showed similar expression patterns and were specifically upregulated in the middle and mature larval stages. The roles played by OfucSP5-8 should be explored further. Except 6 CLIPDs (OfucSP5-8, OfucSP12 and OfucSP20) considered as paralogous genes, the remaining 9 O. furnacalis CLIPDs were identified in 9 orthologous sets with CLIPDs from other four insects, indicating that most of CLIPDs were conserved.
In addition, several other genes that shared a high identity with known functional genes were identified through phylogenetic calculations. OfucSPH9 clustered with 5-clip SPHs in other species, indicating that the domain structure has been conserved and is similar across species. OfucSPH8 shared high identity with M. sexta SPH-2. MsSPH2 was a cofactor of PAP3 and has been implicated in mediating PO activity levels and can interact with immunlectine-2 by binding to lipopolysaccharide41,42. OfucSP2 presented high similarity with MsHP1a/b and clustered with TccSP52, DmcSP34 and AmcSP8. M. sexta proHP1 exerts its activity without proteolytic activation and can stimulate proHP6 to induce antimicrobial peptide synthesis and melanization43. These known functional serine protease-like proteins provide the potential function and characteristic of serine proteases in O. furnacalis.
Multidomain SPs can interact with other proteins through various structural modules to participate in more physiological processes4,16. SP cascades induce the dorsal–ventral axis reported in Drosophila. OfuSP104 is homologous to the Nudel gene and has been implicated in the processing of Gd. OfuSP86 and OfuSP97 were predicted to be Gd genes. These two genes play a role in cleaving and activating Snake, which subsequently processes Easter, and then cleaves Spatzle to form the active ligand for the Toll receptor22,44. The domain structures of OfuSP53/54/55 were similar to those of ModSP-like proteases, implying that they may interact with pattern recognition molecules and indirectly activate downstream proteins16.
Differentially expressed genes among different developmental stages and tissues were acquired and gave us comprehensive insight into their properties and functions. Approximately one-half of the genes highly expressed in embryos were CLIPs, including 3 CLIPBs and 3 CLIPDs. These genes may play essential roles in embryonic development. Hemolymph and fat bodies are important organs involved in immune defense. Five clip-domain SP genes and 3 clip-domain SPH genes, which are commonly highly expressed in the hemolymph compared to genes expressed in other tissues, may be involved in immune signaling pathways. More SPs/SPHs showed specifically high expression in the midgut in pairwise comparisons with genes in other tissues. Since serine-like proteases formed dominate population in digestive environment5, these midgut-specific upregulated genes may exert important roles in food digestion. An interesting phenomenon was that most of the tandem repeat SP/SPH genes with simple gene structures displayed similar upregulated expression profiles in the midgut. The common expression values were whether due to the multi-mapping of RNA-seq reads on homologous region of the genome, or the result of gene duplicated events, should be explored in further studies.
In summary, the present study classified the SP-related genes of O. furnacalis, and explored their gene structures, sequence characteristics, and expression patterns, thus providing a comprehensive understanding of O. furnacalis SP/SPH genes. Orthologous comparisons of SP/SPH genes between D. melanogaster, M. sexta and O. furnacalis provide a basis for functional studies on O. furnacalis SPs/SPHs.
Materials and methods
Insect rearing
Asian corn borers were reared under standard insectary conditions at 25 ± 1 °C and 75 ± 5% relative humidity with a photoperiod of 14 h: 10 h (light: darkness). Newly hatched larvae were fed with an artificial diet until they pupated, and adults were fed with 10% sucrose solution30.
Identification of O. furnacalis SP/SPH genes
The genome of O. furnacalis (accession number: ASM419383v1) was retrieved from NCBI. BUSCO was used to estimate genome assembly and for annotation completeness45. Arthropoda_odb10 and Lepidoptera_odb10 were chosen for performing this assessment. The genome file and GTF file of O. furnacalis were used to extract coding sequence regions as nucleotide sequences, and these sequences were batch translated into proteins with TBtools46. Serine protease domain sequences from known D. melanogaster and M. sexta serine protease sequences4,26 were used as queries for the BLAST search (E-value 1e-5). Identified genes were scanned by NCBI online BLASTP to remove sequences without serine protease domains. The remaining sequences were crosschecked with the O. furnacalis protein database via BLASTP to ensure that all the SP-like genes have been obtained. The identification of SPs and SPHs was based on the conserved His, Asp and Ser residues in catalytic triad residues (conserve region) of the serine protease domain. Sequences containing all three residues in TAAHC, DIAL and GDSGGP motifs were considered SPs. Sequences with amino acid substitution in one or more of these three residues were considered SPHs 23,26. The catalytic triad residues of these genes were manually checked, and severely incomplete sequences (sequence incomplete due to a missing catalytic triad residue in the serine protease domain) were removed. Fgenesh + was used for gene prediction to verify the accuracy of the assembly artifacts and annotation47. We retained only the longest sequence when one gene encoded more than one isoform. Finally, the amino acid sequences are listed in the Supplementary file in FASTA format. Signal peptides were predicted using SignalP 6.0 with default parameters. Pfam and ScanProsite were used to predict the domain structure of the protein sequences. Sequences with four cysteine residues and one cysteine doublet upstream of a serine protease domain were called clip SPs or SPHs. Residues 189, 216, and 226 in SPs determined the main specificity of the S1 pocket. SPs with Asp189, Gly216, and Glu/Ala/Ser226 played a role in the activation of trypsin-like proteases. SPs with Ser/Thr189, Gly216 and Gly/Ala/Ser226 residues conferred chymotrypsin-like protease activity. The SPs catalyzed elastase-like proteases when larger and nonpolar residues occur at position 216 or 2263,4,26. Three residues in the SP genes mentioned above were identified by sequence alignment, and the possible enzymatic activities of these SPs were predicted. Clip-domain serine protease-like sequences of O. furnacalis, D. melanogaster, T. castaneum, A. mellifera and M. sexta4,26 were obtained and used to characterize orthologs via OrthoFinder248.
Sequence alignment and phylogenetic analysis
SP/SPH protease-like domains of O. furnacalis were aligned with those of D. melanogaster, T. castaneum, A. mellifera and M. sexta sequences4,26. Amino acid sequence alignment were first analyzed by MAFFT and then trimmed by trimAI via default parameters49,50. ModelFinder51 implemented in IQ-TREE52 was used to seek the best substitution model according to the Bayesian Information Criterion (BIC). A phylogenetic tree was constructed by IQ-TREE with 1000 bootstrap replicates using the LG + I + G4 model suggested by ModelFinder. All the amino sequences used to construct phylogenetic tree has been listed in Supplementary file in FASTA format. The multiple sequence alignment of these serine protease-like sequences and the phylogenetic tree in newick format were also presented in Supplementary file.
Scaffold location and gene duplication of SP/SPH genes
Genomic location information of SP/SPHs of O. furnacalis was retrieved from the NCBI genome database, visualized using TBtools41, and then improved with Adobe Illustrator. MCScan with default parameters was used to analyze SP/SPH sequences and provide an integrated view of gene duplication53. Genes commonly differentially upregulated in the midgut in comparison with other tissues are marked with symbols.
Sample collection and RNA extraction
A graphic presentation of RNA-seq sample preparation is illustrated in Supplementary Figure S2. O. furnacalis embryos were collected less than 12 h post-oviposition. The 1st, 3rd and 5th instars of the Asian corn borer, which represent the newly hatched larvae, middle stage larvae and mature larvae, were harvested for subsequent experiments. Pupae and adults were grouped into females and males, and then, female and male samples were mixed at a ratio of 1:1. The samples were anesthetized at −70 °C for 8 min, washed three times in DEPC water, and then frozen in liquid nitrogen. Every 20 embryos or 20 1st instar larvae were pooled as one sample, and every six individuals in the other 4 insect stages were pooled as one sample.
The 3rd instar larvae were anesthetized on an ice plate for tissue extraction. The midgut, fat body and silk gland were isolated and transferred to 2-ml centrifuge tubes containing TriPure reagent. Twenty midguts, fat bodies or silk glands were pooled as one sample. Larvae and curved scissors were sterilized with alcohol-containing DEPC, and then bled onto Parafilm by cutting away the proleg. One hundred microliters of hemolymph from 20 larvae was homogenized using TriPure reagent. Three replicates of each tissue sample and three replicates of each developmental stage sample were obtained and processed for RNA extraction according to the manufacturer’s instructions.
Transcriptome sequencing and data analysis
Total RNA was used as input material for library preparation. Briefly, mRNA was purified from total RNA by using poly-T oligo-attached magnetic beads. First-strand cDNA was then synthesized using mRNA fragments as templates, followed by second-strand cDNA synthesis using DNA polymerase I and RNaseH. After the construction of the library, the library was tested to ensure its quality. Qualified libraries were sequenced by the Illumina NovaSeq 6000 to generate 150-bp paired-end short reads. Raw FASTQ format data were processed by fastp 0.19.7 with the parameter -q 5 -u 50 -n 15 -l 15054. In this step, clean data were obtained by removing reads containing adapters, reads containing N-bases and low-quality reads from the raw data. The Q20, Q30 and GC contents of the clean data were calculated. The reference genome and gene model annotation files of O. furnacalis were downloaded. The index of the reference genome was built using HISAT2 2.0.555, and paired-end clean reads were aligned to the reference genome using HISAT2 with the parameter -p 4 –dta -t –phred33. All the transcriptome data are available on the website of the NCBI GEO database (GEO accession: GSE197663).
Nucleotide sequences extracted from the genome of O. furnacalis were used as reference sequences, and the expression levels of these sequences were quantified by mapping the Illumina reads to the reference sequences with RSEM software using default parameters. Differential gene expression analysis was conducted using the DESeq2 1.16.156. DEGs with log2(ratio) ≥ 1 & q-value < 0.05 or log2(ratio) ≤ -1 & q-value < 0.05 were classified as significantly upregulated or downregulated genes57,58 (Supplementary Table S5).
Data availability
The RNA-seq datasets generated in this study have been submitted to Gene Expression Omnibus (GEO) database at the National Center for Biotechnology Information (NCBI) under accession no. GSE197663.
References
Ross, J., Jiang, H., Kanost, M. R. & Wang, Y. Serine proteases and their homologs in the Drosophila melanogaster genome: An initial analysis of sequence conservation and phylogenetic relationships. Gene 304, 117–131 (2003).
Rawlings, N. D. & Barrett, A. J. Evolutionary families of peptidases. Biochem. J. 290, 205–218 (1993).
Perona, J. J. & Craik, C. S. Structural basis of substrate-specificity in the serine proteases. Protein Sci. 4, 337–360 (1995).
Cao, X. et al. Sequence conservation, phylogenetic relationships, and expression profiles of nondigestive serine proteases and serine protease homologs in Manduca sexta. Insect Biochem. Molec. 62, 51–63 (2015).
Srinivasan, A., Giri, A. P. & Gupta, V. S. Structural and functional diversities in Lepidopteran serine proteases. Cel. Mol. Biol. Lett. 11, 132–154 (2006).
Appel, L. F. et al. The Drosophila stubble-stubbloid gene encodes an apparent transmembrane serine-protease required for epithelial morphogenesis. Proc. Natl. Acad. Sci. U. S. A. 90, 4937–5494 (1993).
Bayer, C. A., Halsell, S. R., Fristrom, J. W., Kiehart, D. P. & von Kalm, L. Genetic interactions between the RhoA and stubble-stubbloid loci suggest a role for a type II transmembrane serine protease in intracellular signaling during Drosophila imaginal disc morphogenesis. Genetics 165, 1417–1432 (2003).
Murugasuoei, B., Rodrigues, V., Yang, X. H. & Chia, W. Masquerade - a novel secreted serine protease-like molecule is required for somatic muscle attachment in the Drosophila embryo. Gene. Dev. 9, 139–154 (1995).
Huang, T. S. et al. A cell adhesion protein from the crayfish Pacifastacus leniusculus, a serine proteinase homologue similar to Drosophila Masquerade. J. Biol. Chem. 275, 9996–10001 (2000).
Gupta, S., Wang, Y. & Jiang, H. B. Manduca sexta prophenoloxidase (proPO) activation requires proPO-activating proteinase (PAP) and serine proteinase homologs (SPHs) simultaneously. Insect Biochem. Molec. 35, 241–248 (2005).
Wang, Y. & Jiang, H. B. Interaction of beta-1,3-glucan with its recognition protein activates hemolymph proteinase 14, an initiation enzyme of the prophenoloxidase activation system in Manduca sexta. J. Biol. Chem. 281, 9271–9278 (2006).
Takahashi, D., Garcia, B. L. & Kanost, M. R. Initiating protease with modular domains interacts with beta-glucan recognition protein to trigger innate immune response in insects. Proc. Natl. Acad. Sci. U. S. A. 112, 13856–13861 (2015).
Kim, M. S. et al. A new easter-type serine protease cleaves a masquerade-like protein during prophenoloxidase activation in Holotrichia diomphalia larvae. J. Biol. Chem. 277, 39999–40004 (2002).
Lee, K. Y. et al. A zymogen form of masquerade-like serine proteinase homologue is cleaved during pro-phenoloxidase activation by Ca2+ in Coleopteran and Tenebrio molitor larvae. Eur. J. Biochem. 269, 4375–4383 (2002).
Kanost, M. R. & Jiang, H. Clip-domain serine proteases as immune factors in insect hemolymph. Curr. Opin. Insect Sci. 11, 47–55 (2015).
Veillard, F., Troxler, L. & Reichhart, J.-M. Drosophila melanogaster clip-domain serine proteases: Structure, function and regulation. Biochimie 122, 255–269 (2016).
Waterhouse, R. M. et al. Evolutionary dynamics of immune-related genes and pathways in disease-vector mosquitoes. Science 316, 1738–1743 (2007).
Zou, Z., Shin, S. W., Alvarez, K. S., Kokoza, V. & Raikhell, A. S. Distinct melanization pathways in the mosquito Aedes aegypti. Immunity 32, 41–53 (2010).
Buchon, N. et al. A single modular serine protease integrates signals from pattern-recognition receptors upstream of the Drosophila Toll pathway. Proc. Natl. Acad. Sci. U. S. A. 106, 12442–12447 (2009).
Theopold, U., Li, D., Fabbri, M., Scherfer, C. & Schmidt, O. The coagulation of insect hemolymph. Cell. Mol. Life Sci. 59, 363–372 (2002).
Schneider, D. S., Jin, Y. S., Morisato, D. & Anderson, K. V. A processed form of the Spatzle protein defines dorsal-ventral polarity in the Drosophila embryo. Development 120, 1243–1250 (1994).
Cho, Y. S., Stevens, L. M. & Stein, D. Pipe-dependent ventral processing of Easter by Snake is the defining step in Drosophila embryo DV axis formation. Curr. Biol. 20, 1133–1137 (2010).
Yang, L. et al. The genomic and transcriptomic analyses of serine proteases and their homologs in an endoparasitoid, Pteromalus puparum. Dev. Comp. Immunol. 77, 56–68 (2017).
Zhao, P. et al. Genome-wide identification and expression analysis of serine proteases and homologs in the silkworm Bombyx mori. Bmc Genomics 11, 1 (2010).
Zou, Z., Lopez, D. L., Kanost, M. R., Evans, J. D. & Jiang, H. Comparative analysis of serine protease-related genes in the honey bee genome: possible involvement in embryonic development and innate immunity. Insect Mol. Biol. 15, 603–614 (2006).
Cao, X. & Jiang, H. Building a platform for predicting functions of serine protease-related proteins in Drosophila melanogaster and other insects. Insect Biochem. Molec. 103, 53 (2018).
Cao, X., Gulati, M. & Jiang, H. Serine protease-related proteins in the malaria mosquito, Anopheles gambiae. Insect Biochem. Molec. 88, 48–62 (2017).
Afidchao, M. M., Musters, C. J. M. & de Snoo, G. R. Asian corn borer (ACB) and non-ACB pests in GM corn (Zea mays L.) inthe Philippines. Pest Manag. Sci. 69, 792–801 (2013).
He, K. L. et al. Evaluation of transgenic Bt corn for resistance to the Asian corn borer (Lepidoptera : Pyralidae). J. Econ. Entomol. 96, 935–940 (2003).
Wang, Y. et al. Genetic basis of Cry1F-resistance in a laboratory selected Asian corn borer strain and its cross-resistance to other Bacillus thuringiensis Toxins. Plos One 11, e0161189 (2016).
Shen, D., Liu, Y., Zhou, F., Wang, G. & An, C. Identification of immunity-related genes in Ostrinia furnacalis against entomopathogenic fungi by RNA-seq analysis. Plos One 9, e86436 (2014).
Chu, Y. et al. Serine proteases SP1 and SP13 mediate the melanization response of Asian corn borer, Ostrinia furnacalis, against entomopathogenic fungus Beauveria bassiana. J. Invertebr. Pathol. 128, 64–72 (2015).
Chu, Y., Hong, F., Liu, Q. & An, C. Serine protease SP105 activates prophenoloxidase in Asian corn borer melanization, and is regulated by serpin-3. Sci. Rep. 7, 1 (2017).
Feng, C. et al. Clip domain prophenoloxidase activating protease is required for Ostrinia furnacalis Guenee to defend against bacterial infection. Dev. Comp. Immunol. 87, 204–215 (2018).
Yan, W., Wu, F. Y., Morser, J. & Wu, Q. Y. Corin, a transmembrane cardiac serine protease, acts as a pro-atrial natriuretic peptide-converting enzyme. Proc. Natl. Acad. Sci. U. S. A. 97, 8525–8529 (2000).
Lei, Y. et al. The genomic and transcriptomic analyses of serine proteases and their homologs in an endoparasitoid, Pteromalus puparum. Dev. Comp. Immunol. 77, 56 (2017).
Lin, H. et al. Genome-wide identification and expression profiling of serine proteases and homologs in the diamondback moth, Plutella xylostella (L.). BMC Genomic 16, 1 (2015).
Wu, C. et al. Identification and expression profiling of serine protease-related genes in Tenebrio molitor. Arch. Insect Biochem. 111, e21963 (2022).
Wu, D. D., Wang, G. D., Irwin, D. M. & Zhang, Y. P. A profound role for the expansion of trypsin-like serine protease family in the evolution of hematophagy in mosquito. Molecul. Biol. Evol. 26, 2333–2341 (2009).
Wang, S. J., Magoulas, C. & Hickey, D. Concerted evolution within a trypsin gene cluster in Drosophila. Molecul. Biol. Evol. 16, 1117–1124 (1999).
Wang, Y. & Jiang, H. B. Prophenoloxidase (proPO) activation in Manduca sexta: an analysis of molecular interactions among proPO, proPO-activating proteinase-3, and a cofactor. Insect Biochem. Molec. 34, 731–742 (2004).
Yu, X. Q., Jiang, H. B., Wang, Y. & Kanost, M. R. Nonproteolytic serine proteinase homologs are involved in prophenoloxidase activation in the tobacco hornworm, Manduca sexta. Insect Biochem. Molec. 33, 197–208 (2003).
He, Y., Wang, Y., Yang, F. & Jiang, H. Manduca sexta hemolymph protease-1, activated by an unconventional non-proteolytic mechanism, mediates immune responses. Insect Biochem. Molec. 84, 23–31 (2017).
LeMosy, E. K., Tan, Y. Q. & Hashimoto, C. Activation of a protease cascade involved in patterning the Drosophila embryo. Proc. Natl. Acad. Sci. U. S. A. 98, 5055–5060 (2001).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Chen, C. et al. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202 (2020).
Solovyev, V.V. Statistical approaches in Eukaryotic gene prediction. In (eds. Balding D., Cannings C., Bishop M.). Handbook of Statistical genetics, 3d edition, 1616 p (Wiley-Interscience, 2007).
Emms, D.M. and Kelly, S. OrthoFinder2: Fast and accurate phylogenomic orthology analysis from gene sequences. bioRxiv; doi.org/https://doi.org/10.1101/466201 (2018)
Capella-Gutierrez, S., Silla-Martinez, J. M. & Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Katoh, K. & Standley, D. M. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Kalyaanamoorthy, S., Bui Quang, M., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587 (2017).
Lam-Tung, N., Schmidt, H. A., von Haeseler, A. & Bui Quang, M. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Wang, Y. et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890 (2018).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907 (2019).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1 (2014).
Benjamini, Y. & Hochberg, Y. On the adaptive control of the false discovery fate in multiple testing with independent statistics. J. Edu. Behav. Stati. 25, 60–83 (2000).
Reiner, A., Yekutieli, D. & Benjamini, Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 19, 368–375 (2003).
Acknowledgements
The study is supported by National Natural Science Foundation of China (Grant no. 32001964, http://www.nsfc.gov.cn/).
Author information
Authors and Affiliations
Contributions
L.Y. and J.X. conceived the assay design; L.Y., X.X. and W.W. performed the experimental work; L.Y., X.C., C.P. and X.W. dealt with the data; and L.Y., X.W., and J.X. prepared the first draft of the article. All authors reviewed the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, L., Xu, X., wei, W. et al. Identification and gene expression analysis of serine proteases and their homologs in the Asian corn borer Ostrinia furnacalis. Sci Rep 13, 4766 (2023). https://doi.org/10.1038/s41598-023-31830-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-31830-2
- Springer Nature Limited