
Abstract
Neuroinflammation is a hallmark of frontotemporal dementia (FTD), a heterogeneous group of proteinopathies characterized by the progressive degeneration of the frontal and temporal lobes. It is marked by microglial activation and subsequent cytokine release. Although cytokine levels in FTD brain and CSF have been examined, the number of cytokines measured in each study is limited and knowledge on cytokine concentrations in FTD serum is scarce. Here, we assessed 48 cytokines in FTD serum and brain. The aim was to determine common cytokine dysregulation pathways in serum and brain in FTD. Blood samples and brain tissue samples from the superior frontal cortex (SFC) were collected from individuals diagnosed with behavioral variant FTD (bvFTD) and healthy controls, and 48 cytokines were measured using a multiplex immunological assay. The data were evaluated by principal component factor analysis to determine the contribution from different components of the variance in the cohort. Levels of a number of cytokines were altered in serum and SFC in bvFTD compared to controls, with increases in GRO-α and IL-18 in both serum and SFC. These changes could be associated with NLRP3 inflammasome activation or the NFκB pathway, which activates NLRP3. The results suggest the possible importance of the NLRP3 inflammasome in FTD. An improved understanding of the role of inflammasomes in FTD could provide valuable insights into the pathogenesis, diagnosis and treatment of FTD.
Similar content being viewed by others


Introduction
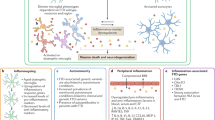
Neuroinflammation is recognized as a hallmark for neurodegenerative diseases including frontotemporal dementia (FTD), a heterogeneous group of neurodegenerative clinical syndromes characterized by progressive behavioral and/or language changes and associated cognitive deficits1. There are three clinical subtypes of FTD: behavioral variant FTD (bvFTD), nonfluent variant primary progressive aphasia and semantic variant primary progressive aphasia, with bvFTD being the most common2. Neuropathologically, FTD is categorized based on the pathological cellular inclusions, with tau and Tar-DNA binding protein-43 (TDP-43) being the most prevalent, and both of which are known to trigger neuroinflammation3,4. Neuroinflammation is primarily mediated by activated astrocytes and microglia, the resident immune cells of the central nervous system. Microglia are mainly responsible for maintaining homeostasis and mediating host defense against pathogens and toxic protein aggregates. These stimuli trigger the activation of microglia, which releases pro-inflammatory cytokines, chemokines and reactive oxygen species (ROS). While early studies referred to four primary features of neuroinflammation—microglial activation, increased cytokines/chemokines, recruitment of peripheral immune cells and local tissue damage—the definition for this term has since broadened to cover most immune processes in the nervous system5. Physiologically, consequences of neuroinflammation include elevated ROS/oxidative stress, neuronal cell death, impaired phagocytosis and autophagy, mitochondrial dysfunction and protein aggregation6,7,8,9, all of which are known to contribute to the pathogenesis of neurodegenerative diseases10,11,12,13.
Neuroinflammation in FTD brain is evident by the presence of activated microglia in disease-affected regions, as shown by immunohistochemistry14,15,16,17,18. Indeed, the presence of activated microglia in the brain of FTD patients have been confirmed in vivo by positron emission topography (PET) using inflammation markers C-PK11195 or C-PBR28 that bind to the 18-kDa translocator protein (TPO) of activated microglia16,19,20,21,22. Furthermore, these studies showed a positive correlation between neuroinflammation and protein aggregations16 and disease progression21 across the FTD spectrum and in semantic dementia, respectively. Of note, PET imaging of seven cases of familial FTD with mutations in the three most common FTD causal genes, C9ORF72, MAPT and GRN, all showed in vivo inflammation, suggesting neuroinflammation is a part of the pathophysiology of familial FTD22. These three genes, which account for 35–65% of familial FTD cases23, and other FTD causal genes24 are all implicated in neuroinflammation25,26,27,28. Interestingly, the brain of MAPT mutation carriers show microglial activation in disease-affected regions prior to the development of protein aggregation and atrophy28, while the leukocytes from symptomatic GRN mutation carriers have increased expression of inflammatory genes compared to those of healthy controls29. In addition, the FTD risk gene TREM2 has been reported to attenuate neuroinflammation30,31 and is used as a biomarker for microglial activation32.
Microglial activation leads to the release of cytokines that amplify and modulate the innate immune response to a foreign pathogen through binding to specific receptors and activating signaling cascade pathways to alter gene expression33,34. This allows cells to communicate with one another and orchestrate complex multicellular processes35. Alterations in brain and CSF cytokines levels in neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis and FTD have been reported36,37,38,39,40,41,42,43,44,45,46,47. Symptomatic FTD patients with GRN mutations have altered levels of pro-inflammatory cytokines in the serum and CSF48,49. Monocyte chemoattractant protein-1 (MCP-1), interferon-γ-inducible protein 10 (IP-10), IL-6, IL-11, IL-12 and IL-15, tumor necrosis factor β (TNFβ) and leukemia inhibitory factor (LIF)46,49,50,51,52,53 are all altered in the CSF or blood of FTD patients. Of note, the cytokines interleukins IL-1β and IL-18 are crucial for the NLRP3 inflammasome-mediated inflammatory responses54. NLRP3 inflammasomes are multi-protein complex comprised of a sensor (NLRP3), an adaptor (ASC) and an effector (caspase-1). The complex cleaves the pro-inflammatory interleukins IL-1β and IL-18, leading to their activation and release, resulting in a plethora of inflammatory responses including pyroptosis. NLRP3 inflammation activation have recently been implicated in driving the tau pathology55. The relationship between NLRP3 inflammasome and tau appears to be reciprocal. Activation of NLRP3 inflammasome can be caused by tau seeds in primary microglia56 in FTD brain with tauopathy55. Inflammasome activation is also known to regulate TDP-43 expression57. In addition, amyloid-β and α-synuclein were reported to induce NLRP3 inflammasome activation in Alzheimer’s58,59 and Parkinson’s60 disease, respectively.
Considering the importance of cytokines in neuroinflammation and neurodegenerative diseases, an improved understanding of their different roles in FTD could provide insights into the pathogenesis of FTD and other degenerative diseases. Currently, while there are numerous reports on cytokine changes in FTD, most of these analyses were performed on CSF, with little published examining cytokine changes in serum24 or in the brain, and only a small number of cytokines were measured. Furthermore, no direct comparisons were made between serum and brain cytokine levels; this would enable the identification of common cytokines that are altered in both the brain/CSF and serum in FTD. This is of particular significance as such cytokines could act as reliable neuroinflammation biomarkers that is detectable in the serum, thus providing a non-invasive means to assess neuroinflammation. In this current study, 48 cytokines were measured in the blood, using a multiplex assay, and were compared to those in the brain in bvFTD and healthy controls. The data were then evaluated by principal component factor analysis to understand the contribution from different components of the variance in our cohort. The primary aim was to determine whether cytokine dysregulation is evident in the serum and brain in FTD, and to identify common cytokines that are altered in both blood and the brain. The second aim was to examine whether any of these affected cytokines had similar functional roles that could lead to the discovery of hitherto unknown neuroinflammatory pathways that contribute to the pathogenesis of FTD.
Materials and methods
Participant blood serum
Individuals diagnosed with sporadic bvFTD and healthy controls were recruited from FRONTIER, the frontotemporal dementia clinical research group previously at Neuroscience Research Australia and now at the University of Sydney Brain and Mind Centre, and from a panel of healthy study volunteers61 with no neurological (i.e. no evidence of cognitive or motor impairment) or psychiatric disorders. The study was approved by the University of New South Wales human ethics committee (approval number: HC12573). All methods were carried out in accordance with the relevant guidelines and regulations. Blood samples were obtained following written informed consent from the participant and/or primary carer. All patients and controls underwent a neurological examination, a comprehensive cognitive assessment and structural brain MRI, and met current consensus diagnostic criteria for probable bvFTD62, as previously described61. Ten bvFTD cases (6 male, 4 female) and 10 controls (4 male, 6 female) were used in this study (Table 1). The mean age of the two groups at recruitment were 66.9 and 75.9 years, respectively. Two blood samples were collected from each person 12-months apart (i.e. Year-1 and Year-2), i.e. 40 samples in total. Blood samples (9 mL) were collected in tubes (BD Vacutainer SST II Advance Tube #367958), and serum prepared by centrifugation at 3500 rpm for 10 min at 4 ℃, which was then aliquoted and stored at −80 ℃ until use.
Participant brain tissues
A different cohort of bvFTD patients and controls (Table 2) was used for the brain tissue analysis. Fresh-frozen post-mortem brain tissue samples were obtained with consent from the Sydney Brain Bank at Neuroscience Research Australia and NSW Brain Tissue Resource Centre at the University of Sydney (both brain banks ethically approved through their institutions to collect, characterize and bank brain tissue for research purposes). Ethics approval for this tissue study was from the University of New South Wales Human Research Ethics (approval number: HC15789). All brain donors underwent standardized assessments in life and standardized neuropathological examination, and met current consensus diagnostic criteria for sporadic bvFTD with TDP-43 pathology63,64 or no significant neuropathology (controls)65,66. The Sydney Brain Bank collects brain tissue from brain donors participating in the FRONTIER brain donor program approved through the South Eastern Sydney Local Health District Human Research Ethics (approval number: HREC 10/092) and so the bvFTD cases with brain tissue were clinically assessed via the same procedures as indicated for the patient blood serum. Tissue samples from the superior frontal cortex were collected from ten bvFTD cases (5 male, 5 female)67 and 11 controls (5 male, 6 female)68. The mean age of the two groups were 72.9 and 79.5 years, respectively. Pathological severity of FTD was also assessed within the bvFTD group which could be split further into early stage 1 (N = 5) versus later stage disease (N = 5)69, reflecting the average disease durations of these two subgroups (mean ± standard deviation for stage 1 disease duration of 2.7 ± 1.5 years versus stage 2/3 disease durations of 8 ± 4 years).
Protein fraction extraction from human brain tissues
The TBS fractions, which contain the cytosolic proteins, were used in cytokine analysis. The TBS fractions were extracted from the superior frontal cortex as previously described68. Briefly, tissue (100 mg) was suspended in TBS homogenization buffer (20 mM Tris, 150 mM NaCl, pH 7.4, 5 mM EDTA, 0.02% sodium azide) containing protease inhibitor cocktail (Roche). The samples were then homogenized using Qiagen tissue lyser (30 Hz cycles, 3 × 30 s) and centrifuged for 1 h (100,000×g at 4 °C). The resultant supernatant then becomes the TBS fraction. Samples were stored at −80 °C until analysis.
Cytokine assay
A total of 48 cytokines including interleukins, chemokines, colony stimulating factors, growth factors, interferons, growth factors and tumor necrosis factor were measured in this study. Cytokine concentrations were measured in serum and human brain tissue TBS extracts using the Pro Human Cytokine Screening Panel 48-plex assay (Bio-Rad, Hercules, California, USA) and 5-point standard curve consisting of S3, S4, S5, S6, S7 standards and a blank, which covered our sample concentration range. Briefly, samples were diluted (serum 1:4, brain tissue 1:2), in sample diluent, and incubated with detection antibodies coupled to magnetic beads, washed using a Bio-Plex Pro wash station and incubated in streptavidin–phycoerythrin before wells were quantified using a Xponent software package (Luminex, Austin, TX). Provided standards generated a five-parameter standard curve for all 48 cytokines and unknown concentrations were calculated with Bio-Plex Manager software 6.1. The intra-assay %CV for the serum plate was 2.68–4.97 (average: 3.51) and for the brain tissue plate 3.65–6.42 (average: 4.86). The inter-assay %CV for the two plates was 4.48–13.57 (average: 7.92).
Statistical analysis
All statistical analyses were performed using SPSS statistical software (IBM, Chicago, IL, United States). A multivariate analysis (general linear model), covarying for age and sex, was used to determine differences in the cytokine levels in FTD (N = 10) and control (N = 11) with posthoc statistical significance set at P < 0.05. Principal component factor analyses (PCA) were performed to determine if significantly altered cytokines, were clustering in the same group of variance for serum and brain cytokines. To be considered significant, cytokines required a loading score of > 0.7 and to be responsible for > 10% of variance. PCA was first performed on serum cytokines to determine if cytokines that were altered in the serum of bvFTD cohort were clustering together. This is followed by analysis on brain cytokines to examine if similar components of variance were observed in both brain and serum.
Results
Altered cytokine levels in FTD serum
Forty-eight cytokines were measured in bvFTD (N = 10) serum and controls (N = 10) using a multiplex assay. Two samples, year 1 and year 2 (i.e. 12-months apart), from each individual were assessed. Firstly, we assessed the cytokines independent of time and found that IL-2Rα, IP10, macrophage inflammatory protein 1-alpha (MIP-1α) and stem cells growth factor-beta (SCGF-BB) were significantly increased in bvFTD compared to controls (Fig. 1). Of the 48 cytokines, IL-10, IL-12 (p40), IL-5, IL-15, IL-16, monocyte chemotactic protein-3 (MCP-3) and vascular endothelial growth factor (VEGF) were not detected by the multiplex assay. Secondly, we assessed the cytokines longitudinally and found that five chemokines (GRO-α/CXCL1, monocyte chemotactic protein-1 (MCP-1), macrophage inflammation protein 1-beta (MIP-1β), RANTES and SDF-1α) (Fig. 2A), one interleukin (IL-18) (Fig. 2B), one interferon (interferon alpha-2 (IFNα-2)) (Fig. 3A) and one growth factor (platelet derived growth factor BB (PDGF-BB)) (Fig. 3B) were significantly altered in year 2 compared to year 1 in bvFTD. None of the cytokines were altered in year 2 compared to year 1 in controls (Figs. 2 and 3).

Serum cytokines levels in bvFTD and control cases. Serum samples collected from year 1 and year 2 of FTD (20 samples) and control cases (20 samples) were quantified for cytokine levels and the results were grouped according to disease state. White bars show control and light grey are bvFTD. Data represent mean and SE as error bars, *P < 0.05, **P < 0.01, ***P < 0.001.

Serum chemokines and interleukins levels in year 1 and year 2 of bvFTD and control cases. Serum samples collected from year 1 and year 2 of FTD (20 samples) and control cases (20 samples) were quantified and the levels of from each year were grouped for control and bvFTD cases. The results for (A) chemokines and (B) Interleukins were shown here. White bars show results from year 1 and light grey are results from year 2. Data represent mean and SE as error bars, *P < 0.05, **P < 0.01, ***P < 0.001.

Serum interferons, growth factors and colony stimulating factors levels in year 1 and year 2 of bvFTD and control cases. Serum samples collected from year 1 and year 2 of FTD (20 samples) and control cases (20 samples) were quantified and the levels of from each year were grouped for control and bvFTD cases. The results for (A) interferons and (B) growth factors and colony stimulating factors were shown here. White bars show results from year 1 and light grey are results from year 2. Data represent mean and SE as error bars, *P < 0.05, **P < 0.01, ***P < 0.001.
Cytokine analysis of FTD brain tissue
We were also interested in changes in the cytokines in bvFTD brain and assessed the same cytokines, using the same multiplex assay, in the superior frontal cortex, a disease-affected region, of FTD (N = 10) and controls (N = 11). Of the 48 cytokines, HGF and IL-18 were significantly elevated in bvFTD compared to controls (Fig. 4) with IL-5 being undetectable. We then categorized bvFTD into two groups based on neuropathological severity, i.e. early stage 1 bvFTD (N = 5) and later stage 2/3 bvFTD (N = 5), and divided the cytokines into functional groups, i.e. chemokines, interleukins, interferons, growth factors and colony stimulating factors. In terms of chemokines and interleukins, GRO-α (Fig. 5A) and IL-16 (Fig. 5B) were significantly elevated in late bvFTD compared to controls, whereas IL-18 was more significantly increased in early bvFTD relative to late bvFTD (Fig. 5B). In terms of interferons, there were no changes in either early or late bvFTD compared to controls (Fig. 6A). In terms of growth factors, HGF was increased in early bvFTD and further increased in late bvFTD compared to controls (Fig. 6B). In summary, GRO-α and IL-18 are elevated in both serum and brain in bvFTD compared to controls.

Brain cytokines levels in bvFTD and control cases. Cytokine levels in the frontal cortex of FTD (10 samples) and control cases (10 samples) were quantified and the results were grouped according to disease state. White bars show control and light grey are bvFTD. Data represent mean and SE as error bars, *P < 0.05, **P < 0.01, ***P < 0.001.

Chemokines and interleukins levels in early and late bvFTD brains compared to control cases. Cytokine levels in the frontal cortex of FTD (10 samples) and control cases (10 samples) were quantified and the bvFTD cases were further separated into early cases (5 cases) and late cases (5 cases). The results for (A) chemokines and (B) Interleukins were shown here. White bars show results from year 1 and light grey are results from year 2. Data represent mean and SE as error bars, *P < 0.05, **P < 0.01, ***P < 0.001.

Interferons, growth factors and colony stimulating factors levels in early and late bvFTD brains compared to control cases. Cytokine levels in the frontal cortex of FTD (10 samples) and control cases (10 samples) were quantified and the bvFTD cases were further separated into early cases (5 cases) and late cases (5 cases). The results for (A) interferons and (B) growth factors and colony stimulating factors were shown here. White bars show results from year 1 and light grey are results from year 2. Data represent mean and SE as error bars, *P < 0.05, **P < 0.01, ***P < 0.001.
Analysis of variance in brain and serum cytokines
To identify clustering groups of cytokines with similar functional characteristics in bvFTD serum and brain (> 10% variance from controls), principal factor component analysis (PCA) was performed on the serum (Fig. 7A) and brain (Fig. 7B) datasets. The serum PCA revealed two components of variance. The larger component (loading score > 0.7, accounting for 39% of variance) comprised of beta-nerve growth factor (β-NGF), granulocyte colony stimulating factor (G-CSF), IFN-α2, IL-17A, IL-1α, IL-2, IL-3, IL-4, IL-7, IL-8, IL-9, MIP-1β and tumor necrosis factor-alpha (TNF-α) (Table 3). The other component consisted of only one cytokine, RANTES.

Principal components Analysis (PCA) on significantly altered cytokines in (A) serum and (B) brain. PCA were performed to determine whether cytokines with significantly altered levels in FTD samples were clustering in the same group of variance. To be considered significant, cytokines required a loading score of > 0.7 and to be responsible for > 10% of variance. PCA on (A) serum and (B) brain datasets both reveal two components of variance. Cytokines in component 1 are represented by blue circles while cytokines in component 2 are represented by red circles (component 2).
To determine whether similar components of variance occurred in serum and brain, PCA analysis was then performed on all brain samples, from which two components of variance emerged (Table 4). The analysis showed that the greatest component of variance in FTD brain consisted of β -NGF, eotaxin, G-CSF, granulocyte–macrophage colony stimulating factor (GM-CSF), IFN-α2, IFN-γ, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-15, IL-16, IL-17A, IL-1α, IL-2Rα, IL-3, IL-4, IL-6, IL-7, IL-8, IL-9, LIF, MCP-3, monocyte induced by gamma interferon (MIG/ CXCL9), MIP-1α, MIP-1β, PDGF-BB, stem cell factor (SCF), stromal cell derived factor-1 alpha (SDF1α), TNF-α, TNF-β and VEGF (loading score > 0.7, accounting for 51% of variance) (Table 4). The second largest component of variance consisted of fibroblast growth factor (FGF basic), GRO-α, hepatocyte growth factor (HGF), IL-16, IL-18, M-CSF, macrophage migration inhibitory factor (MIF) and SCGF-β (loading score > 0.7, accounting for 15% of variance). Interestingly, the second largest component of variance comprised of all four cytokines that were altered in the brain—GRO-α, HGF, IL-16 and IL-18. In addition, all the cytokines from the first components of variance in the serum samples were also found in the largest component of variance in the brain.
Discussion
Neuroinflammation is known to play a major role in the neuropathology of neurodegenerative diseases including bvFTD24. Since cytokines are integral to neuroinflammation, an improved understanding of cytokines in bvFTD could provide valuable insights into the pathogenesis of bvFTD and other neurodegenerative diseases. While changes in brain/CSF cytokines levels in bvFTD have been well documented, reports on serum cytokine concentrations in bvFTD are scarce, and the number of cytokines measured in these studies small. The present study is the first to directly compare the serum and brain levels of a comprehensive range of cytokines in bvFTD. The side-by-side assessment of serum and brain cytokine concentrations allowed the identification of common cytokines that are altered in both the blood and brain. The relative ease of serum collection over that of CSF makes these cytokines good candidates for neuroinflammation biomarkers. In addition, the grouping of functionally similar cytokines enabled the identification of novel neuroinflammation pathways that could contribute to the pathogenesis of bvFTD.
In agreement with previous studies, this study also showed cytokine level changes in serum and brain of bvFTD patients compared to those of healthy controls. In the serum, the concentrations of four cytokines, IL-2Rα, IP-10, MIP-1α and SCGF-BB, were elevated in bvFTD compared to controls, while GRO-α, IFN-α2, IL-18, MCP-1, MIP-1β and PDGF-BB have shown time-dependent increase in serum concentration with disease progression. Of note, GRO-α and IL-18 levels were also increased in bvFTD brain, in addition to HGF and IL-16. The fact that both bvFTD brain and serum have elevated levels of GRO-α and IL-18 suggests that these two cytokines are involved in pathways crucial in the pathogenesis of bvFTD. Principle component analysis on brain cytokines levels have placed GRO-α and IL-18 in the group 2 of brain cytokines (Table 4), suggesting that these two cytokines are functionally related in bvFTD brain. IL-18 and GRO-α (also known as CXCL1) are both pro-inflammatory cytokines. IL-18 is involved in the activation of mast cells and CD8 + T cells, production of IFN-γ and Th2 cytokines and inducing innate-type allergic inflammation70 while GRO-α binds to its receptor CXCR2 to promote neutrophil recruitment and activation at the site of infection. In addition, both cytokines are involved in NLRP3 inflammasome pathways. While IL-18 release is mediated by the NLRP3 inflammasome activation, which recruits caspases-1 to cleave IL-18 pro-peptide to active IL-1871,72, GRO-α has been shown to promote the activation of NLRP3 inflammasome73. Although dysregulation of these two cytokines were unknown in bvFTD, an increase in IL-1874,75 and GRO-α76 have been reported in other neurodegenerative diseases77,78.
Apart from IL-18 and GRO-α, the second components of variance for brain cytokines (Group 2) also contained HGF, M-CSF, MIF, FGF basic, IL-16 and SCGF-β. Of note, GRO-α, HGF, IL-16 and IL-18 levels were all altered (Fig. 3), which is suggestive of their significant physiological roles in bvFTD brain. Apart from SCGF-β—a recently discovered protein for which very little is known–the other cytokines in the clusters are involved in the NLRP3 inflammasome pathway. FGF has been shown to upregulate the NLRP3 inflammasome79, while MIF is required for NLRP3 activation80. Interestingly, HGF is known to inhibit the NFκB pathway81 leading to non-expression of RANTES, MCP-1, IL-1β, TNF-α, IL-1 and IL-681. Significantly, the NFκB pathway is known to activate the NLRP3 inflammasome82. On the other hand, M-CSF have been shown to activate NFκB83. Thus, the cytokines clustered in group 2 of brain cytokines are involved in the NLRP3 inflammasome pathway, either directly or through the NFκB pathway, thus underscoring the importance of NLRP3 inflammasome in the etiology of bvFTD.
The significance of the NFκB pathway in the pathogenesis of FTD was further confirmed by results emerged from the principal component analyses, in which cytokines from the first component of variance for brain cytokines, group 1 (Table 4), are all related to the NFκB signaling pathway, either as NFκB regulators and/or downstream effectors of the NFκB pathway. IL-2R α, IFNγ, TNFα, IL-2, IL-17A, IL-4, IL-1α, TNF-β, IFN-α2, IL-13, IL-12 (p70), SDF-1α, G-CSF, PDGF-BB, β-NGF, IL-15, IL-1β are all activators of NFκB84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102, while IL-9, IL-10 and IL-13 are reported to suppress NFκB103,104,105. In turn, some of these cytokines are activated by NFκB: TNF-α, IL-3, IL-8, MIP-1β, eotaxin, IL-6 and GM-CSF, MIP-1α106,107,108,109,110,111. In addition, LIF, SCF, IL-7 MCP-3, MIG, VEGF and IL-12 (p40) are regulated by the NFκB pathway112,113,114,115,116,117. Moreover, several of these cytokines also have a direct link to the NLRP3 inflammasome: the secretion of IL-1α and β are mediated by the NLRP3 inflammasome54, while IL-4 is reported to inhibit inflammasome assembly118 and SDF-1α inhibits inflammasome activation119,120. Notably, cytokines from the entire first components of variance for serum (Group 1), β-NGF, TNFα, G-CSF, IL-1α, IL-2, IL-4, IL-8, IL-3, MIP-1β, IL-9, IL-17A, and IL-7, are all present in the first component of variance for brain cytokines, again confirming the prominence of the NFκB pathway in the etiology of bvFTD. The second component for serum cytokines only consisted of one cytokine, RANTES, and it is also regulated by NFκB121. Of note, some of the above cytokines are also responsible for regulation of other cytokines. For example, IL-15 induces IL-8 production100, SDF1-α upregulates IL-6122 and GM-CSF signaling increases IL-1 production123, implying secondary regulatory mechanisms within these cytokines. Thus, the current study showed that significantly altered cytokines in bvFTD are all part of an intricate network that revolves around the NLRP3 inflammasome, either directly or via the NFκB pathway.
Activation of the NLRP3 inflammasome has been associated with neurodegenerative diseases124,125,126, including FTD55. Indeed, amyloid-β and α-synuclein were reported to induce NLRP3 inflammasome activation in Alzheimer’s58,59,127 and Parkinson’s60 disease, respectively. In addition, mutant SOD1 and TDP-43 proteins have also been reported to activate NLRP3 inflammasome128 while aggregated tau56 and TDP-4357,129 are also known to activate NLRP3 inflammasome. Interestingly, a recent study implicated NLRP3 inflammasome activation in driving tau pathology55. Indeed, inflammasome inhibitors have been shown to inhibit α-synuclein pathology130 and reduce amyloid-β accumulation131 in mouse models. Unsurprisingly, there is increasing interest in using NLRP3 inflammasome inhibitors as a therapeutic target for neurodegenerative diseases132,133,134. Pilot studies using inhibitors of NLRP3 in mouse models of neurodegenerative diseases have proved this approach effective130,131,135,136,137,138. In an FTD mouse model, the inflammasome inhibitor MCC950 improves inflammation and endoplasmic reticular stress signaling, in addition to partially normalizing the levels of phosphorylated tau139.
Taken together, the current study has revealed evidence of cytokine dysregulation in bvFTD serum and brain. In particular, the levels of IL-18 and GRO-α appear to be changed in both serum and brain of bvFTD patients, making these two cytokines possible inflammation biomarkers for bvFTD. Interestingly, these two cytokines are both involved in the NLRP3 inflammasome pathway, which has been associated with other neurodegenerative diseases. Furthermore, principal component analysis performed on serum and brain cytokines have revealed that all significantly altered cytokines are associated with the NLRP3 inflammasome and/or the NFκB pathway, which is a known activator for the NLRP3 inflammasome. Thus, our data show that NLRP3 inflammasome signaling occurs early in the pathogenesis of bvFTD. Given the recent interest in using NLRP3 inflammasome inhibitors as therapeutics against neurodegenerative diseases, and the promising outcomes of these molecules in mouse models, a better understanding on the role of cytokines in NLRP3 inflammasome activation could provide valuable insights into the pathogenesis of bvFTD and its diagnosis and treatment.
Conclusions
In conclusion, our results showed that cytokine dysregulation is evident in bvFTD brain and serum. Importantly, GRO-α and IL-18 appear to be increased in both serum and brain in bvFTD, making them possible candidates as neuroinflammation biomarkers for bvFTD. The cytokines that are altered in bvFTD serum and/or brain are all related to NLRP3 inflammasome activation or NFκB pathway, which regulates NLRP3. These results therefore suggest that the NLRP3 inflammasome could be important in bvFTD pathogenesis.
Data availability
All relevant data are available from the corresponding author upon reasonable request. Other patient data cannot be made publicly available because the ethical approval and the informed consent from the patients included in this study did not cover placing the data into publicly open repositories.
References
Younes, K. & Miller, B. L. Frontotemporal dementia: Neuropathology, genetics, neuroimaging, and treatments. Psychiatr. Clin. N. Am. 43, 331–344 (2020).
Seeley, W. W. Behavioral variant frontotemporal dementia. Continuum (Minneap. Minn.) 25, 76–100 (2019).
Bright, F., Chan, G., van Hummel, A., Ittner, L. M. & Ke, Y. D. TDP-43 and inflammation: Implications for amyotrophic lateral sclerosis and frontotemporal dementia. Int. J. Mol. Sci. 22, 7781 (2021).
Hartnell, I. J., Blum, D., Nicoll, J. A. R., Dorothee, G. & Boche, D. Glial cells and adaptive immunity in frontotemporal dementia with tau pathology. Brain 144, 724–745 (2021).
Woodburn, S. C., Bollinger, J. L. & Wohleb, E. S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflamm. 18, 258 (2021).
de Araújo Boleti, A. P. et al. Neuroinflammation: An overview of neurodegenerative and metabolic diseases and of biotechnological studies. Neurochem. Int. 136, 104714 (2020).
Subhramanyam, C. S., Wang, C., Hu, Q. & Dheen, S. T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 94, 112–120 (2019).
Guzman-Martinez, L. et al. Neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 10, 1008 (2019).
Haukedal, H. & Freude, K. Implications of microglia in amyotrophic lateral sclerosis and frontotemporal dementia. J. Mol. Biol. 431, 1818–1829 (2019).
Halliwell, B. Oxidative stress and neurodegeneration: Where are we now?. J. Neurochem. 97, 1634–1658 (2006).
Butler, C. A. et al. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 158, 621–639 (2021).
Nixon, R. A. The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997 (2013).
Lin, M. T. & Beal, M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795 (2006).
Lant, S. B. et al. Patterns of microglial cell activation in frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol. 40, 686–696 (2014).
Bellucci, A., Bugiani, O., Ghetti, B. & Spillantini, M. G. Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. Neurodegener. Dis. 8, 221–229 (2011).
Bevan-Jones, W. R. et al. Neuroinflammation and protein aggregation co-localize across the frontotemporal dementia spectrum. Brain 143, 1010–1026 (2020).
Woollacott, I. O. C. et al. Cerebrospinal fluid YKL-40 and chitotriosidase levels in frontotemporal dementia vary by clinical, genetic and pathological subtype. Dement. Geriatr. Cogn. Disord. 49, 56–76 (2020).
Venneti, S., Wang, G., Nguyen, J. & Wiley, C. A. The positron emission tomography ligand DAA1106 binds with high affinity to activated microglia in human neurological disorders. J. Neuropathol. Exp. Neurol. 67, 1001–1010 (2008).
Cagnin, A., Rossor, M., Sampson, E. L., Mackinnon, T. & Banati, R. B. In vivo detection of microglial activation in frontotemporal dementia. Ann. Neurol. 56, 894–897 (2004).
Kreisl, W. C. et al. PET imaging of neuroinflammation in neurological disorders. Lancet Neurol. 19, 940–950 (2020).
Pascual, B. et al. Neuroinflammation is highest in areas of disease progression in semantic dementia. Brain 144, 1565–1575 (2021).
Malpetti, M. et al. In vivo PET imaging of neuroinflammation in familial frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 92, 319–322 (2021).
Pottier, C., Ravenscroft, T. A., Sanchez-Contreras, M. & Rademakers, R. Genetics of FTLD: Overview and what else we can expect from genetic studies. J. Neurochem. 138(Suppl 1), 32–53 (2016).
Bright, F. et al. Neuroinflammation in frontotemporal dementia. Nat. Rev. Neurol. 15, 540–555 (2019).
Ahmed, Z., Mackenzie, I. R., Hutton, M. L. & Dickson, D. W. Progranulin in frontotemporal lobar degeneration and neuroinflammation. J. Neuroinflamm. 4, 7 (2007).
Lall, D. & Baloh, R. H. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Invest. 127, 3250–3258 (2017).
Hashimoto, K., Jahan, N., Miller, Z. A. & Huang, E. J. Neuroimmune dysfunction in frontotemporal dementia: Insights from progranulin and C9orf72 deficiency. Curr. Opin. Neurobiol. 76, 102599 (2022).
Bevan-Jones, W. R. et al. In vivo evidence for pre-symptomatic neuroinflammation in a MAPT mutation carrier. Ann. Clin. Transl. Neurol. 6, 373–378 (2019).
Milanesi, E. et al. Molecular signature of disease onset in granulin mutation carriers: A gene expression analysis study. Neurobiol. Aging 34, 1837–1845 (2013).
Leyns, C. E. G. et al. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA 114, 11524–11529 (2017).
Liu, W. et al. Trem2 promotes anti-inflammatory responses in microglia and is suppressed under pro-inflammatory conditions. Hum. Mol. Genet. 29, 3224–3248 (2020).
Swift, I. J. et al. Fluid biomarkers in frontotemporal dementia: Past, present and future. J. Neurol. Neurosurg. Psychiatry 92, 204–215 (2021).
Holdsworth, S. R. & Gan, P. Y. Cytokines: Names and numbers you should care about. Clin. J. Am. Soc. Nephrol. 10, 2243–2254 (2015).
Altan-Bonnet, G. & Mukherjee, R. Cytokine-mediated communication: A quantitative appraisal of immune complexity. Nat. Rev. Immunol. 19, 205–217 (2019).
Becher, B., Spath, S. & Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 17, 49–59 (2017).
Allan, S. M. & Rothwell, N. J. Cytokines and acute neurodegeneration. Nat. Rev. Neurosci. 2, 734–744 (2001).
Chen, X., Hu, Y., Cao, Z., Liu, Q. & Cheng, Y. Cerebrospinal fluid inflammatory cytokine aberrations in Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis: A systematic review and meta-analysis. Front. Immunol. 9, 2122 (2018).
Swardfager, W. et al. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 68, 930–941 (2010).
Griffin, W. S. et al. Microglial interleukin-1 alpha expression in human head injury: Correlations with neuronal and neuritic beta-amyloid precursor protein expression. Neurosci. Lett. 176, 133–136 (1994).
Hull, M., Berger, M., Volk, B. & Bauer, J. Occurrence of interleukin-6 in cortical plaques of Alzheimer’s disease patients may precede transformation of diffuse into neuritic plaques. Ann. N. Y. Acad. Sci. 777, 205–212 (1996).
Dickson, D. W. The pathogenesis of senile plaques. J. Neuropathol. Exp. Neurol. 56, 321–339 (1997).
Ono, S., Hu, J., Shimizu, N., Imai, T. & Nakagawa, H. Increased interleukin-6 of skin and serum in amyotrophic lateral sclerosis. J. Neurol. Sci. 187, 27–34 (2001).
Sekizawa, T. et al. Cerebrospinal fluid interleukin 6 in amyotrophic lateral sclerosis: Immunological parameter and comparison with inflammatory and non-inflammatory central nervous system diseases. J. Neurol. Sci. 154, 194–199 (1998).
Wijeyekoon, R. S. et al. Cerebrospinal fluid cytokines and neurodegeneration-associated proteins in Parkinson’s disease. Mov. Disord. 35, 1062–1066 (2020).
Hirsch, E. C. & Hunot, S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection?. Lancet Neurol. 8, 382–397 (2009).
Rentzos, M. et al. IL-15 is elevated in cerebrospinal fluid of patients with Alzheimer’s disease and frontotemporal dementia. J. Geriatr. Psychiatry Neurol. 19, 114–117 (2006).
Tennakoon, A. et al. Normal aging, motor neurone disease, and Alzheimer’s disease are characterized by cortical changes in inflammatory cytokines. J. Neurosci. Res. 100, 653–669 (2022).
Bossu, P. et al. Loss of function mutations in the progranulin gene are related to pro-inflammatory cytokine dysregulation in frontotemporal lobar degeneration patients. J. Neuroinflamm. 8, 65 (2011).
Galimberti, D. et al. Inflammatory molecules in frontotemporal dementia: Cerebrospinal fluid signature of progranulin mutation carriers. Brain Behav. Immun. 49, 182–187 (2015).
Galimberti, D. et al. Intrathecal levels of IL-6, IL-11 and LIF in Alzheimer’s disease and frontotemporal lobar degeneration. J. Neurol. 255, 539–544 (2008).
Rentzos, M. et al. Interleukin-12 is reduced in cerebrospinal fluid of patients with Alzheimer’s disease and frontotemporal dementia. J. Neurol. Sci. 249, 110–114 (2006).
Gibbons, L. et al. Plasma levels of progranulin and interleukin-6 in frontotemporal lobar degeneration. Neurobiol. Aging 36(1603), e1601-1604 (2015).
Sjogren, M., Folkesson, S., Blennow, K. & Tarkowski, E. Increased intrathecal inflammatory activity in frontotemporal dementia: Pathophysiological implications. J. Neurol. Neurosurg. Psychiatry 75, 1107–1111 (2004).
Swanson, K. V., Deng, M. & Ting, J. P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489 (2019).
Ising, C. et al. NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673 (2019).
Stancu, I. C. et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 137, 599–617 (2019).
Zhuang, J. et al. TDP-43 upregulation mediated by the NLRP3 inflammasome induces cognitive impairment in 2 2’,4,4’-tetrabromodiphenyl ether (BDE-47)-treated mice. Brain Behav. Immun. 65, 99–110 (2017).
Liu, Y. et al. Beta-amyloid activates NLRP3 inflammasome via TLR4 in mouse microglia. Neurosci. Lett. 736, 135279 (2020).
Halle, A. et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–865 (2008).
Codolo, G. et al. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 8, e55375 (2013).
Ahmed, R. M. et al. Systemic metabolism in frontotemporal dementia. Neurology 83, 1812–1818 (2014).
Rascovsky, K. et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134, 2456–2477 (2011).
Cairns, N. J. et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 114, 5–22 (2007).
Mackenzie, I. R. et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update. Acta Neuropathol. 119, 1–4 (2010).
Montine, T. J. et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 123, 1–11 (2012).
Hyman, B. T. et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 8, 1–13 (2012).
Tan, R. H. et al. Classification of FTLD-TDP cases into pathological subtypes using antibodies against phosphorylated and non-phosphorylated TDP43. Acta Neuropathol. Commun. 1, 33 (2013).
Bhatia, S., Kim, W. S., Shepherd, C. E. & Halliday, G. M. Apolipoprotein D upregulation in Alzheimer’s disease but not frontotemporal dementia. J. Mol. Neurosci. 67, 125–132 (2019).
Broe, M. et al. Staging disease severity in pathologically confirmed cases of frontotemporal dementia. Neurology 60, 1005–1011 (2003).
Yasuda, K., Nakanishi, K. & Tsutsui, H. Interleukin-18 in health and disease. Int. J. Mol. Sci. 20, 649 (2019).
Palomo, J., Dietrich, D., Martin, P., Palmer, G. & Gabay, C. The interleukin (IL)-1 cytokine family—Balance between agonists and antagonists in inflammatory diseases. Cytokine 76, 25–37 (2015).
Voet, S., Srinivasan, S., Lamkanfi, M. & van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 11, 10248 (2019).
Boro, M. & Balaji, K. N. CXCL1 and CXCL2 regulate NLRP3 inflammasome activation via G-protein-coupled receptor CXCR2. J. Immunol. 199, 1660–1671 (2017).
Bossu, P. et al. Interleukin-18, from neuroinflammation to Alzheimer’s disease. Curr. Pharm. Des. 16, 4213–4224 (2010).
Sutinen, E. M., Pirttila, T., Anderson, G., Salminen, A. & Ojala, J. O. Pro-inflammatory interleukin-18 increases Alzheimer’s disease-associated amyloid-beta production in human neuron-like cells. J. Neuroinflamm. 9, 199 (2012).
Craig-Schapiro, R. et al. Multiplexed immunoassay panel identifies novel CSF biomarkers for Alzheimer’s disease diagnosis and prognosis. PLoS ONE 6, e18850 (2011).
Kadhim, H., Deltenre, P., Martin, J. J. & Sebire, G. In-situ expression of Interleukin-18 and associated mediators in the human brain of sALS patients: Hypothesis for a role for immune-inflammatory mechanisms. Med. Hypotheses 86, 14–17 (2016).
Italiani, P. et al. Evaluating the levels of interleukin-1 family cytokines in sporadic amyotrophic lateral sclerosis. J. Neuroinflamm. 11, 94 (2014).
ZhuGe, D. L., Javaid, H. M. A., Sahar, N. E., Zhao, Y. Z. & Huh, J. Y. Fibroblast growth factor 2 exacerbates inflammation in adipocytes through NLRP3 inflammasome activation. Arch. Pharm. Res. 43, 1311–1324 (2020).
Lang, T. et al. Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat. Commun. 9, 2223 (2018).
Giannopoulou, M. et al. Hepatocyte growth factor exerts its anti-inflammatory action by disrupting nuclear factor-kappaB signaling. Am. J. Pathol. 173, 30–41 (2008).
Boaru, S. G. et al. NLRP3 inflammasome expression is driven by NF-kappaB in cultured hepatocytes. Biochem. Biophys. Res. Commun. 458, 700–706 (2015).
Wang, Y. et al. M-CSF induces monocyte survival by activating NF-kappaB p65 phosphorylation at Ser276 via protein kinase C. PLoS ONE 6, e28081 (2011).
Chan, K. K. et al. Interleukin-2 induces NF-kappaB activation through BCL10 and affects its subcellular localization in natural killer lymphoma cells. J. Pathol. 221, 164–174 (2010).
Lin, Y., Jamison, S. & Lin, W. Interferon-gamma activates nuclear factor-kappa B in oligodendrocytes through a process mediated by the unfolded protein response. PLoS ONE 7, e36408 (2012).
Arima, N., Kuziel, W. A., Grdina, T. A. & Greene, W. C. IL-2-induced signal transduction involves the activation of nuclear NF-kappa B expression. J. Immunol. 149, 83–91 (1992).
Cheshire, J. L. & Baldwin, A. S. Jr. Synergistic activation of NF-kappaB by tumor necrosis factor alpha and gamma interferon via enhanced I kappaB alpha degradation and de novo I kappaBbeta degradation. Mol. Cell Biol. 17, 6746–6754 (1997).
Moe, K. T. et al. Tumor necrosis factor-alpha-induced nuclear factor-kappaB activation in human cardiomyocytes is mediated by NADPH oxidase. J. Physiol. Biochem. 70, 769–779 (2014).
Yang, Z. Y. & Yuan, C. X. IL-17A promotes the neuroinflammation and cognitive function in sevoflurane anesthetized aged rats via activation of NF-kappaB signaling pathway. BMC Anesthesiol. 18, 147 (2018).
Zamorano, J., Mora, A. L., Boothby, M. & Keegan, A. D. NF-kappa B activation plays an important role in the IL-4-induced protection from apoptosis. Int. Immunol. 13, 1479–1487 (2001).
Thieu, V. T. et al. IL-4-stimulated NF-kappaB activity is required for Stat6 DNA binding. J. Leukoc. Biol. 82, 370–379 (2007).
Hartupee, J., Li, X. & Hamilton, T. Interleukin 1alpha-induced NFkappaB activation and chemokine mRNA stabilization diverge at IRAK1. J. Biol. Chem. 283, 15689–15693 (2008).
Freudlsperger, C. et al. TGF-beta and NF-kappaB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene 32, 1549–1559 (2013).
Grohmann, U. et al. IL-12 acts directly on DC to promote nuclear localization of NF-kappaB and primes DC for IL-12 production. Immunity 9, 315–323 (1998).
Yang, C. H. et al. IFNalpha/beta promotes cell survival by activating NF-kappa B. Proc. Natl. Acad. Sci. USA 97, 13631–13636 (2000).
Han, Y., He, T., Huang, D. R., Pardo, C. A. & Ransohoff, R. M. TNF-alpha mediates SDF-1 alpha-induced NF-kappa B activation and cytotoxic effects in primary astrocytes. J. Clin. Invest. 108, 425–435 (2001).
Ebner, K., Bandion, A., Binder, B. R., de Martin, R. & Schmid, J. A. GMCSF activates NF-kappaB via direct interaction of the GMCSF receptor with IkappaB kinase beta. Blood 102, 192–199 (2003).
Olashaw, N. E., Kowalik, T. F., Huang, E. S. & Pledger, W. J. Induction of NF-kappa B-like activity by platelet-derived growth factor in mouse fibroblasts. Mol. Biol. Cell 3, 1131–1139 (1992).
Maggirwar, S. B., Sarmiere, P. D., Dewhurst, S. & Freeman, R. S. Nerve growth factor-dependent activation of NF-kappaB contributes to survival of sympathetic neurons. J. Neurosci. 18, 10356–10365 (1998).
McDonald, P. P., Russo, M. P., Ferrini, S. & Cassatella, M. A. Interleukin-15 (IL-15) induces NF-kappaB activation and IL-8 production in human neutrophils. Blood 92, 4828–4835 (1998).
Stone, K. P., Kastin, A. J. & Pan, W. NFkB is an unexpected major mediator of interleukin-15 signaling in cerebral endothelia. Cell Physiol. Biochem. 28, 115–124 (2011).
Scholz, C. C. et al. Regulation of IL-1beta-induced NF-kappaB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc. Natl. Acad. Sci. USA 110, 18490–18495 (2013).
Lentsch, A. B., Shanley, T. P., Sarma, V. & Ward, P. A. In vivo suppression of NF-kappa B and preservation of I kappa B alpha by interleukin-10 and interleukin-13. J. Clin. Invest. 100, 2443–2448 (1997).
Richard, M., Louahed, J., Demoulin, J. B. & Renauld, J. C. Interleukin-9 regulates NF-kappaB activity through BCL3 gene induction. Blood 93, 4318–4327 (1999).
Driessler, F., Venstrom, K., Sabat, R., Asadullah, K. & Schottelius, A. J. Molecular mechanisms of interleukin-10-mediated inhibition of NF-kappaB activity: A role for p50. Clin. Exp. Immunol. 135, 64–73 (2004).
Wurster, A. L., Precht, P. & Pazin, M. J. NF-kappaB and BRG1 bind a distal regulatory element in the IL-3/GM-CSF locus. Mol. Immunol. 48, 2178–2188 (2011).
Elliott, C. L., Allport, V. C., Loudon, J. A., Wu, G. D. & Bennett, P. R. Nuclear factor-kappa B is essential for up-regulation of interleukin-8 expression in human amnion and cervical epithelial cells. Mol. Hum. Reprod. 7, 787–790 (2001).
Matsukura, S. et al. Activation of eotaxin gene transcription by NF-kappa B and STAT6 in human airway epithelial cells. J. Immunol. 163, 6876–6883 (1999).
Zhu, X. et al. Upregulation of CCL3/MIP-1alpha regulated by MAPKs and NF-kappaB mediates microglial inflammatory response in LPS-induced brain injury. Acta Neurobiol. Exp. (Wars) 76, 304–317 (2016).
Collart, M. A., Baeuerle, P. & Vassalli, P. Regulation of tumor necrosis factor alpha transcription in macrophages: Involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol. Cell Biol. 10, 1498–1506 (1990).
Widmer, U., Manogue, K. R., Cerami, A. & Sherry, B. Genomic cloning and promoter analysis of macrophage inflammatory protein (MIP)-2, MIP-1 alpha, and MIP-1 beta, members of the chemokine superfamily of proinflammatory cytokines. J. Immunol. 150, 4996–5012 (1993).
Yeh, J. X., Schultz, K. L. W., Calvert, V., Petricoin, E. F. & Griffin, D. E. The NF-kappaB/leukemia inhibitory factor/STAT3 signaling pathway in antibody-mediated suppression of Sindbis virus replication in neurons. Proc. Natl. Acad. Sci. USA 117, 29035–29045 (2020).
Reber, L., Vermeulen, L., Haegeman, G. & Frossard, N. Ser276 phosphorylation of NF-kB p65 by MSK1 controls SCF expression in inflammation. PLoS ONE 4, e4393 (2009).
Miller, M. L. et al. Basal NF-kappaB controls IL-7 responsiveness of quiescent naive T cells. Proc. Natl. Acad. Sci. USA 111, 7397–7402 (2014).
Thompson, W. L. & Van Eldik, L. J. Inflammatory cytokines stimulate the chemokines CCL2/MCP-1 and CCL7/MCP-3 through NFkB and MAPK dependent pathways in rat astrocytes [corrected]. Brain Res. 1287, 47–57 (2009).
Horton, M. R., Boodoo, S. & Powell, J. D. NF-kappa B activation mediates the cross-talk between extracellular matrix and interferon-gamma (IFN-gamma) leading to enhanced monokine induced by IFN-gamma (MIG) expression in macrophages. J. Biol. Chem. 277, 43757–43762 (2002).
Murphy, T. L., Cleveland, M. G., Kulesza, P., Magram, J. & Murphy, K. M. Regulation of interleukin 12 p40 expression through an NF-kappa B half-site. Mol. Cell Biol. 15, 5258–5267 (1995).
Huanosta-Murillo, E. et al. NLRP3 regulates IL-4 expression in TOX(+) CD4(+) T cells of cutaneous T cell lymphoma to potentially promote disease progression. Front. Immunol. 12, 668369 (2021).
Roosen, K. et al. CXCL12 inhibits inflammasome activation in LPS-stimulated BV2 cells. Brain Res. 1763, 147446 (2021).
Wang, S. et al. Exogenous stromal cell-derived factor-1 (SDF-1) suppresses the NLRP3 inflammasome and inhibits pyroptosis in synoviocytes from osteoarthritic joints via activation of the AMPK signaling pathway. Inflammopharmacology 29, 695–704 (2021).
Hirano, F. et al. Nuclear factor-kappaB regulates RANTES chemokine expression in response to tumor necrosis factor-alpha in fibroblast-like synoviocytes. Mod. Rheumatol. 12, 37–43 (2002).
Lu, D. Y. et al. SDF-1alpha up-regulates interleukin-6 through CXCR4, PI3K/Akt, ERK, and NF-kappaB-dependent pathway in microglia. Eur. J. Pharmacol. 613, 146–154 (2009).
Khameneh, H. J., Isa, S. A., Min, L., Nih, F. W. & Ruedl, C. GM-CSF signalling boosts dramatically IL-1 production. PLoS ONE 6, e23025 (2011).
Saresella, M. et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 11, 23 (2016).
Panicker, N. et al. Neuronal NLRP3 is a parkin substrate that drives neurodegeneration in Parkinson’s disease. Neuron 110, 2422-2437e2429 (2022).
Gugliandolo, A., Giacoppo, S., Bramanti, P. & Mazzon, E. NLRP3 inflammasome activation in a transgenic amyotrophic lateral sclerosis model. Inflammation 41, 93–103 (2018).
Jung, E. S. et al. Amyloid-beta activates NLRP3 inflammasomes by affecting microglial immunometabolism through the Syk-AMPK pathway. Aging Cell 21, e13623 (2022).
Deora, V. et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 68, 407–421 (2020).
Zhao, W. et al. TDP-43 activates microglia through NF-kappaB and NLRP3 inflammasome. Exp. Neurol. 273, 24–35 (2015).
Gordon, R. et al. Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 10, 4066 (2018).
Dempsey, C. et al. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav. Immun. 61, 306–316 (2017).
Zhang, Y., Dong, Z. & Song, W. NLRP3 inflammasome as a novel therapeutic target for Alzheimer’s disease. Signal Transduct Target Ther. 5, 37 (2020).
Singh, S. S. et al. NF-kappaB-mediated neuroinflammation in Parkinson’s disease and potential therapeutic effect of polyphenols. Neurotox. Res. 37, 491–507 (2020).
Haque, M. E. et al. Targeting the microglial NLRP3 inflammasome and its role in Parkinson’s disease. Mov. Disord. 35, 20–33 (2020).
Haseeb, M. et al. Novel small-molecule inhibitor of NLRP3 inflammasome reverses cognitive impairment in an Alzheimer’s disease model. ACS Chem. Neurosci. 13, 818–833 (2022).
Lonnemann, N. et al. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 117, 32145–32154 (2020).
Ren, Y., Wang, Q., Yang, Z., Feng, L. & Zhang, Y. MCC950 ameliorates the dementia symptom at the early age of line M83 mouse and reduces hippocampal alpha-synuclein accumulation. Biochem. Biophys. Res. Commun. 611, 23–30 (2022).
Huang, S. et al. A selective NLRP3 inflammasome inhibitor attenuates behavioral deficits and neuroinflammation in a mouse model of Parkinson’s disease. J. Neuroimmunol. 354, 577543 (2021).
Hull, C. et al. NLRP3 inflammasome inhibition with MCC950 improves insulin sensitivity and inflammation in a mouse model of frontotemporal dementia. Neuropharmacology 180, 108305 (2020).
Acknowledgements
We are grateful to the ForeFront research participants in the Frontier research clinic and the associated brain donor programs, and Nicole Mueller for coordinating these donor programs. We also thank Katherine Phan and Ying He for blood and brain tissue processing and technical assistance. Brain tissues were received from the Sydney Brain Bank at Neuroscience Research Australia (supported by Neuroscience Research Australia) and the New South Wales Brain Tissue Resource Centre at the University of Sydney (supported by National Institutes of Health grant funding indicated below). Research reported in this publication was supported by the National Institute on Alcohol Abuse and Alcoholism of the National Institutes of Health under Award Number R28AA012725. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding
This work was supported by funding to ForeFront, a collaborative research group dedicated to the study of frontotemporal dementia and motor neuron disease, from the National Health and Medical Research Council of Australia (NHMRC) program grants (#1037746 & 1132524). G.M.H. is a NHMRC Senior Leadership Fellow (#1176607). O.P. is supported by a NHMRC Leadership Fellowship (GNT2008020).
Author information
Authors and Affiliations
Contributions
J.S.K. carried out the cytokine assays, analyzed the data, and wrote the manuscript. H.C.L. analyzed the data, prepared the figures and wrote the manuscript. O.P. and J.R.H. recruited the patients, performed the clinical examinations and determined clinical diagnosis. Y.F. provided expert advice and reviewed the manuscript. G.M.H. directed the ForeFront research program, recruited the patients for the blood and brain tissue studies, provided final neuropathological diagnoses through working at the Sydney Brain Bank, provided expert advice and revised the manuscript. W.S.K. conceived, designed and supervised the project, analyzed the data, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lok, H.C., Katzeff, J.S., Hodges, J.R. et al. Elevated GRO-α and IL-18 in serum and brain implicate the NLRP3 inflammasome in frontotemporal dementia. Sci Rep 13, 8942 (2023). https://doi.org/10.1038/s41598-023-35945-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-35945-4
- Springer Nature Limited
This article is cited by
-
Immunological aspects of central neurodegeneration
Cell Discovery (2024)