
Abstract
To report the association of autoimmune polyglandular syndrome type 1 (APS1) with cone dystrophy in a large Saudi family. This is a Retrospective chart review and prospective genetic testing and ophthalmic examination of a large multiplex consanguineous family. Genetic testing was performed on 14 family members, seven of whom had detailed ophthalmic examinations. Medical history, ocular history and evaluation, visual field testing, full-field electroretinogram (ERG), and Whole Exome Sequencing (WES) results were analyzed. Three family members were homozygous for c.205_208dupCAGG;p.(Asp70Alafs*148) in AIRE and homozygous for c.481-1G>A in PDE6C. One additional family member was homozygous for only the AIRE variant and another additional family member was homozygous for only the PDE6C variant. All patients with homozygosity for the PDE6C variant had cone dystrophy, and all patients with homozygosity for the AIRE variant had APS1. In addition, two of the family members who were homozygous for the PDE6C and AIRE variants had reduced rod function on ERG. We report the co-inheritance for APS1 and PDE6C-related cone dystrophy, an unusual example of two seemingly independent recessive conditions coinciding within a family. Dual molecular diagnosis must be taken into account by ophthalmologists facing unusual constellations of findings, especially in consanguineous families.
Similar content being viewed by others

Introduction
Autoimmune Polyglandular Syndrome type1 (APS1; OMIM 240300) is a subtype of Autoimmune Polyendocrine Syndromes (APS) which are autoimmune disorders involving several endocrine organs. APS1 is characterized by a triad of Addison’s disease, hypoparathyroidism and mucocutaneous candidiasis among several other endocrine and nonendocrine manifestations1. It is associated with biallelic (recessive) or monoallelic (dominant) pathogenic variants in the human autoimmune regulatory (AIRE) gene2, which encodes a 545-amino-acid proline-rich (APECED) protein with a molecular weight of 58 kD. APECED protein is mainly localized to the cell nucleus, where it plays a role in regulating genetic transcription in normal cells3. Pathogenic variants in AIRE gene lead to changes in intracellular localization of APECED protein, altering its role in transcription4.
Ocular manifestations of APS1 include autoimmune keratopathy, dry eyes, and pigmentary retinopathy5. The mechanisms for the development of ocular surface disease in patients with APS1 are not completely understood. APECED is thought to regulate the expression of non-thymic proteins within the thymus to facilitate the elimination of self-reactive T cells6. APECED deficiency leads to infiltration of CD4+ and CD8+ T cells on the ocular surface and meibomian glands of Aire-mutant mice6. An increased expression of proinflammatory cytokines by ocular surface cells was also seen7. Therefore, immune-mediated mechanisms may play a role in the loss of ocular surface barrier function, decreased goblet cell density and increased epithelial stratification7. These factors, in turn, lead to severe blepharitis and keratoconjunctivitis in patients with APS1.
In addition, a target eye antigen [interphotoreceptor retinoid-binding protein (IRBP)] was found to be a dominant eye autoantigen in Aire-mutant mice8. This may underlie a possibly autoimmune retinopathy in patients with APS1.
APS1 has not been previously associated with cone dystrophy. Here we report for the first time the association of APS1 with PDE6C-related cone dystrophy, in a large inbred Saudi family. This is a rare co-occurrence of two homozygously inherited recessive conditions. We discuss the clinical implications of this phenomenon in the practice of ophthalmology especially in highly consanguineous populations.
Methods
This study was conducted at King Khaled Eye Specialist Hospital (KKESH), according to the tenets of the Declaration of Helsinki, and approved by the Institutional Review Board at KKESH (Project 22088-R). Consent for genetic testing and participation in this study was obtained from all participants (KFSHRC RAC#2070 023). Rare (allele frequency ≤ 0.001) homozygous and compound heterozygous loss of function variants or variants with damaging in silico predictions that lie within the coding or splicing regions of protein-coding genes were considered. After identification of the index case through discovering pathogenic variants in AIRE and PDE6C genes, all available family members were recruited for further genetic testing. Due to the lack of ophthalmic facilities which allow for proper ophthalmic evaluation of the family members in their home city, prospective ophthalmic examination was performed on 7 available family members to document the ophthalmic findings. Collected data included age, gender, past medical and family history including parental consanguinity; best-corrected visual acuity (VA), refractometry, slit-lamp biomicroscopy, and fundus examination; full-field electroretinography (ERG) according to the institution’s protocol previously reported9; multimodal imaging including macular spectral-domain optical coherence tomography (SD-OCT, Spectralis OCT, Heidelberg Engineering, Inc., Heidelberg, Germany), color fundus photos (Topcon TRC-50DX, Topcon Medical Systems, Inc., NJ, US), ultra-widefield pseudocolor fundus photos and fundus autofluorescence (FAF) (Optos PLC, Dunfermline, UK). Genetic analysis was performed using whole exome sequencing in the index case and targeted variant analysis by Sanger sequencing in the other family members. Both vaiants in AIRE and PDE6C genes which have led to the diagnosis of APS1 and cone dystrophy, respectively in this paper were described previously by some of the authors10.
Ethical approval
Ethical approval was obtained from the Institutional Review Board (IRB) at King Khaled Eye Specialist Hospital (KKESH) with approval ID: Project 22088-R. Written informed consents to participate in this study and for their clinical details to be known were obtained from all patients and legal guardians of participants below 16 years. This study was conducted in compliance with the guidelines of the Declaration of Helsinki.
Informed consent
Written informed consents for publication of this study, the details and accompanying images were obtained from all involved patients.
Results
Detailed ophthalmic and systemic history was obtained for 31 family members (Fig. 1). After identification of the proband, 13 family members were brought for genetic testing using targeted variant analysis (Table 1). Ophthalmic examinations were performed on seven family members (Table 2). Figure 2 shows the ffERG which could be obtained from five family members.

A partial pedigree of the Saudi family showing members with APS1 and/or cone dystrophy.

Electroretinogram of 5 family members showing flat photopic and markedly reduced scotopic responses in patient III:11 who is homozygous for the AIRE and PDE6C variants (the proband). Patient III:14 who is homozygous for the AIRE variant and heterozygous for the PDE6C variant showed normal scotopic and photopic responses (the proband’s sister). Photopic responses showed Profoundly reduced b wave with delayed implicit time with flat photopic ERG in patient III:15 who is homozygous for the AIRE and PDE6C variants. Patient IV:5 who is heterozygous for the AIRE variant and homozygous for the PDE6C variant who had low amplitude and delayed implicit time photopic responses while scotopic ffERG was normal. Patient IV:6 who is homozygous for the AIRE and PDE6C variants had low amplitude and delayed implicit time photopic responses while scotopic ffERG was normal.
Cone dystrophy was documented in three patients, including one presymptomatic patient, who were homozygous for a pathogenic PDE6C variant. Three family members were homozygous for both NM_000383.4:c.205_208dupCAGG; p.(Asp70Alafs*148) (Human Gene Mutation Database; HGMD CI991953) in AIRE and homozygous for NM_006204.3:c.481-1G>A (HGMD CS2015060) in PDE6C, while another two family members were homozygous for either variant. The c.205_208dupCAGG: p.(Asp70Alafs*148) variant in AIRE gene causes a frameshift, which alters the proteins amino acid sequence beginning at position 70 and leads to a premature termination codon 148 amino acids downstream therefore completely abolishing the downstream SAND domain and the two Zinc Finger domains. It is predicted to cause a truncated or absent AIRE protein, while the c.481-1G>A variant in PDE6C leads to a canonical splicing site (-1) predicted to be deleterious (loss of acceptor site) by in silico tools .Homozygosity for the PDE6C variant was associated with cone dystrophy and homozygosity for the AIRE variant was associated with APS1. Both variants were described previously10. In addition, two family members who were homozygous for the PDE6C and AIRE variants showed reduced rods function on ERG.
Patient III:11 (the proband)
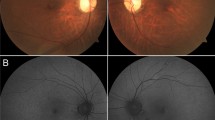
A 25-year-old male who is a known case of APS1 presented to the ophthalmology clinic at KKESH with gradual decrease in vision, photophobia, and burning sensation. Systematic review revealed Addison disease, hypothyroidism and hypoparathyroidism. Medication history included oral steroids, thyroid hormone replacement, calcium, and vitamin D. Best-corrected visual acuity (BCVA) was 20/200 in the right eye and 20/300 in the left eye. Corneal examination showed autoimmune keratitis with moderate corneal opacities. Fundus examination showed Bull's eye maculopathy (Fig. 3A,B). Fundus Autofluorescence showed a broad ring of macular hyperautofluorescence and a dull hypoautofluorescent central reflex (Fig. 3C,D). SD-OCT showed decreased subfoveal thickness and profound loss of the inner segment ellipsoid (ISe) in the foveal area (Fig. 3E,F). Goldmann visual field showed loss of peripheral visual field in the right eye and a central scotoma in the left eye. FfERG showed flat photopic and markedly reduced scotopic responses (Fig. 2; Patient III:11). Genetic testing with whole exome sequencing showed two homozygous pathogenic loss of function variants in known disease-related genes: a c.205_208dupCAGG;p.(Asp70Alafs*148) frameshift indel in exon 2 of AIRE and a c.481-1G > A canonical splicing variant upstream of exon 2 of PDE6C. The patient was treated with topical cyclosporine A 1% as described previously11.

Detailed ophthalmic examination and multimodal imaging in a 25-year-old male patient who is homozygous for the AIRE and PDE6C variants and manifested both the autoimmune polyglandular syndrome type 1 (APS1) and cone dystrophy (the proband; patient III:11). (A,B) Are color fundus photos showing Bull’s eye maculopathy. (C,D) Are Fundus Autofluorescence photos showing broad ring of macular hyperautofluorescence and a dull hypoautofluorescent central reflex. (E,F) Are Spectral Domain Optical Coherence Tomography (SD-OCT) showing decreased subfoveal thickness and profound loss of the inner segment ellipsoid (ISe) in the foveal area.
Genetic and ophthalmic examinations of further family members
Patient III:14 (proband’s sister)
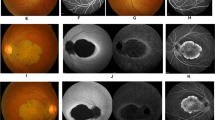
An 18-year-old female who is following with endocrinology as a case of an APS1. Systematic review revealed Addison disease, and hypoparathyroidism for which she was taking cortisol, calcium, and vitamin D. Her BCVA was 20/400 in the right eye and 20/300 in the left eye. Corneal examination showed autoimmune keratitis with moderate corneal opacities. Dilated fundus examination was normal in both eyes (Fig. 4A,B). Fundus autofluorescence showed normal autofluorescence in both eyes (Fig. 4C,D). SD-OCT was normal in both eyes (Fig. 4E,F). FfERG was normal in both eyes (Fig. 2; patient III:14). Genetic testing showed homozygosity and heterozygosity for the familial AIRE and PDEC6C variants, respectively. The patient was treated with topical cyclosporine A 1% as described previously11.

Detailed ophthalmic examination and multimodal imaging in an 18-year-old female patient who is homozygous for the AIRE variant and heterozygous for the PDE6C variant who manifested APS1 (the proband’s sister, patient III:14). (A,B) Are color fundus photos showing normal examination in both eyes. (C,D) Are Fundus autofluorescence showing normal autofluorescence in both eyes. (E,F) Are SD-OCT showing normal retinal layers in both eyes.
Patient IV:5 (proband’s nephew’s child)
A 5 year-old boy whose parents denied any systemic or visual problems was examined and showed normal anterior segment, while fundus examination showed attenuated foveal reflexes in both eyes (Fig. 5A,B). Fundus autofluorescence revealed Marked foveal hypoautofluorescence in both eyes (Fig. 5C,D) and SD-OCT revealed foveal thinning and Mild attenuation of ellipsoid zone at fovea both eyes (Fig. 5E,F). Photopic ffERG showed low amplitude and delayed implicit time while scotopic ffERG was normal (Fig. 2; patient IV:5). Genetic testing showed heterozygosity and homozygosity for the familial AIRE and PDE6C variants, respectively.

Detailed ophthalmic examination and multimodal imaging in a 5-year-old male who is heterozygous for the AIRE and homozygous for the PDE6C variant and manifested cone dystrophy (patient IV:5). (A,B) Are color fundus photos showing attenuated foveal reflexes in both eyes. (C,D) Are Fundus autofluorescence showing Marked foveal hypoautofluorescence in both eyes. (E,F) Are SD-OCT showing foveal thinning and Mild attenuation of ellipsoid zone at fovea both eyes.
Discussion
In this paper, we describe the ocular findings of a Saudi family with pathogenic variants in AIRE and PDE6C. The PDE6C gene belongs to the Phosphodiesterase 6 (PDE6) family. PDE6 is one of 21 enzymes which regulate the intracellular concentration of cyclic nucleotides12,13. PDE6 plays an important role in converting light to electrical signals within the neural retina14. When the PDE6 enzyme is activated, intracellular cGMP is hydrolyzed, leading to propagation of the visual cycle15. While rods have a PDE6 catalytic core composed of a heterodimer of PDE6A and PDE6B subunits which are inhibited by PDE6G, cones have a catalytic homodimer of PDE6C, which is inhibited by PDE6H subunits16. This explains why pathogenic variants in PDE6A (OMIM #180071), PDE6B (OMIM #180072) and PDE6G (OMIM #180073) cause predominantly rod-involving retinal dystrophies, whereas pathogenic variants in PDE6C and PDE6H (OMIM #600827 and #601190) lead to retinal dystrophies which predominantly affect cones17,18,19.
Cone dystrophy was documented in three patients, including one presymptomatic, who were homozygous for the pathogenic PDE6C variant. One patient had flat ERG with poor visibility of the fundus due to totally opaque corneas in a patient who was homozygous for the pathogenic variants in both AIRE and PDE6C. The corneal opacification in this patient could potentially contribute to the flat ERG appearance in this patient. Although peripheral retinitis pigmentosa-like picture was described in APS1, none of our patients had that manifestation20,21,22. Bourgault et al.22 found retinitis pigmentosa changes in a series of 5 patients with APS1 with elevated antiretinal antibodies. Additionally, some reports found elevated antiretinal antibodies, raising the possibility of autoimmune retinopathy23,24. Histopathological examination of retinal pigmentations in APS1 found focal areas of pigmentation in the inner retina along the retinal blood vessels surrounded by areas of an atrophy of the outer retina25. Although these changes were similar to the histopathological findings in retinitis pigmentosa, the exact mechanism which leads to pigment deposition is not known25,26.
Among the five cases described by Bourgault et al., three showed macular thinning on examination which were documented by decreased subfoveal thickness and disruption of external limiting membrane and inner segment ellipsoid in SD-OCT22. In addition, four out of five patients had reduced or nonrecordable cone function. Limited screening for Leber Congenital Amaurosis (LCA) genes in one of the five patients was negative22. Furthermore, macular dystrophic changes were found in one Japanese patient with APS127. All these findings indicate that APS1 patients can manifest cone dystrophy. Cone-rod dystrophies could also be found in other of systemic conditions including endocrinopathies and metabolic diseases, such as the Alstrom syndrome, ciliopathies, fucosidosis, neuronal ceroid lipofuscinosis, infantile phytanic acid storage disease, and methylmalonic aciduria with homocystinuria28.
Two patients (patients III:11 and III:15) who are homozygous for c.205_208dupCAGG;p.(Asp70Alafs*148) variant in exon 2 of AIRE and homozygous for the c.481-1G>A variant in exon 2 of PDE6C, showed rod dysfunction on ERG in addition to the impaired cone responses. The reason for this is not clear. One possibility is that this is caused by an autoimmune retinopathy associated with APS1, similar to what has been suggested previously in the literature20,21,22. The diagnosis of autoimmune retinopathy is confirmed by measurements of the levels of antiretinal antibodies which were not performed in this study. Furthermore, although WES was performed, we cannot completely rule out further intronic variants in other or novel retinopathy genes which could cause this finding. Another less likely possibility is the association with the homozygous PDE6C variant17. It is unknown whether the c.205_208dupCAGG;p.(Asp70Alafs*148) variant in AIRE has affected the rods function and aggravated the retinal phenotype which is affected by the c.481-1G>A variant in PDE6C.
Notably, The clinical findings in patient III:11 (the proband) including fundus photos, FAF and SD-OCT did not show signs of auto immune retinopathy including narrowing of the retinal vasculature, retinal pigmentary changes, optic nerve pallor, diffuse posterior pole hyperautofluorescence which spares the fovea and peripheral outer retinal loss (Fig. 3)29. Furthermore, the proband’s sister (patient III:14) who is homozygous for the AIRE variant and heterozygous for the PDE6C variant has got completely normal retinal exam despite having the APS1 disease which further reduces the possibility of auto immune retinopathy in this study.
The increasing use of untargeted genome sequencing assays such as whole exome and whole genome sequencing has greatly increased the visibility of “multilocus” diseases in which the patient’s phenotype is attributed to pathogenic variants in two more genes causing the simultaneous occurrence of more than one monogenic disease in the same patient30. Although all combinations of inheritance modes (autosomal, X-linked, recessive and dominant) have been observed, the co-occurrence of two or more independent recessive conditions is particularly prevalent in consanguineous families as shown by us and others10,31,32,33. This should not be surprising since consanguineous couples share a significant proportion of their genomes (e.g. 1/8 in the case of first cousins) so the likelihood of the shared carrier status of more than one pathogenic recessive variant is greatly increased. Indeed, we have recently shown that the classic counseling of consanguineous couples of the 25% recurrence risk of a familial variant overlooks the small but significant recurrence risk for other autosomal recessive variants even when family history is lacking10. Therefore, consanguinity may prompt the search of more than one disease-related gene in families in whom the disease phenotype is atypical. This is especially challenging in the absence of family relatives in whom the different components of the phenotype segregate separately.
Limitations of this study include the limited number of family members who were available for ophthalmic examination. In addition, evaluation of this family did not include anti-retinal antibodies testing to explore the possibility of an autoimmune retinopathy which could underlie the rod dysfunction which was observed in 2 of the patients.
In summary we present for the first time the co-occurrence of PDE6C-related cone dystrophy with APS1. We suggest that ophthalmologists who encounter unusual presentations of diseases with well-established phenotypic spectrum to consider the possibility of a multilocus disease especially in the setting of consanguinity.
Data availability
The datasets generated and/or analyzed during the current study are not publicly available due to limitations of ethical approval involving the patient data and anonymity but are available from the corresponding author on reasonable request.
References
Ahonen, P., Myllarniemi, S., Sipila, I. & Perheentupa, J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N. Engl. J. Med. 322, 1829–1836 (1990).
Nagamine, K. et al. Positional cloning of the APECED gene. Nat. Genet. 17, 393–398 (1997).
Björses, P. et al. Localization of the APECED protein in distinct nuclear structures. Hum. Mol. Genet. 8, 259–266 (1999).
Björses, P. et al. Mutations in the AIRE gene: Effects on subcellular location and transactivation function of the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy protein. Am. J. Hum. Genet. 66(2), 378–392 (2000).
Couturier, A. & Brézin, A. P. Ocular manifestations of autoimmune polyendocrinopathy syndrome type 1. Curr. Opin. Ophthalmol. 27(6), 505–513. https://doi.org/10.1097/ICU.0000000000000306 (2016).
Yeh, S. et al. Spontaneous T cell mediated keratoconjunctivitis in Aire-deficient mice. Br. J. Ophthalmol. 93(9), 1260–1264. https://doi.org/10.1136/bjo.2008.153700 (2009).
Li, S. et al. Small proline-rich protein 1B (SPRR1B) is a biomarker for squamous metaplasia in dry eye disease. Investig. Ophthalmol. Vis. Sci. 49(1), 34–41. https://doi.org/10.1167/iovs.07-0685 (2008).
DeVoss, J. et al. Spontaneous autoimmunity prevented by thymic expression of a single self-antigen. J. Exp. Med. 203(12), 2727–2735. https://doi.org/10.1084/jem.20061864(2006).Erratum.In:J.Exp.Med.204(1),203 (2007).
Magliyah, M. S., AlSulaiman, S. M., Schatz, P. & Nowilaty, S. R. Evolution of macular hole in enhanced S-cone syndrome. Doc. Ophthalmol. 142(2), 239–245. https://doi.org/10.1007/s10633-020-09787-8 (2021).
AlAbdi, L. et al. Residual risk for additional recessive diseases in consanguineous couples. Genet. Med. 23(12), 2448–2454. https://doi.org/10.1038/s41436-021-01289-5 (2021).
AlAbbasi, O., Magliyah, M. S. & Ahad, M. Long term keratits treatment with topical cyclosporin a in autoimmune polyglandular syndrome type 1. Am. J. Ophthalmol. Case Rep. 21, 101009. https://doi.org/10.1016/j.ajoc.2020.101009 (2021).
Bender, A. T. & Beavo, J. A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 58, 488–520 (2006).
Lagman, D., Franzén, I. E., Eggert, J., Larhammar, D. & Abalo, X. M. Evolution and expression of the phosphodiesterase 6 genes unveils vertebrate novelty to control photosensitivity. BMC Evol. Biol. 16, 124. https://doi.org/10.1186/s12862-016-0695-z (2016).
Arshavsky, V. Y., Lamb, T. D. & Pugh, E. N. G proteins and phototransduction. Annu. Rev. Physiol. 64, 153–187 (2002).
Cote, R. H. Characteristics of photoreceptor PDE (PDE6): Similarities and differences to PDE5. Int. J. Impot. Res. 16, S28–S33 (2004).
Gopalakrishna, K. N., Boyd, K. & Artemyev, N. O. Mechanisms of mutant PDE6 proteins underlying retinal diseases. Cell Signal. 37, 74–80 (2017).
Daich Varela, M. et al. PDE6C: Novel mutations, atypical phenotype, and differences among children and adults. Investig. Ophthalmol. Vis. Sci. 61(12), 1. https://doi.org/10.1167/iovs.61.12.1 (2020).
Thiadens, A. A. et al. Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders. Am. J. Hum. Genet. 85, 240–247. https://doi.org/10.1016/j.ajhg.2009.06.016 (2009).
Kohl, S. et al. A nonsense mutation in PDE6H causes autosomal-recessive incomplete achromatopsia. Am. J. Hum. Genet. 91, 527–532. https://doi.org/10.1016/j.ajhg.2012.07.006 (2012).
Gass, J. D. The syndrome of keratoconjunctivitis, superficial moniliasis, idiopathic hypoparathyroidism, and Addison’s disease. Am. J. Ophthalmol. 54, 660–674 (1962).
Orlova, E. M. et al. Autoimmune polyglandular syndrome type 1 in Russian patients: Clinical variants and autoimmune regulator mutations. Horm. Res. Paediatr. 73, 449–457 (2010).
Bourgault, S. et al. Retinal degeneration in autoimmune polyglandular syndrome type 1: A case series. Br. J. Ophthalmol. 99(11), 1536–1542 (2015).
Breunig, A. et al. Autoimmune retinopathy in a patient with autoimmune polyendocrine syndrome type I. Ocul. Immunol. Inflamm. 21(2), 153–157 (2013).
Wood, L. W., Jampol, L. M. & Daily, M. J. Retinal and optic nerve manifestations of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Arch. Ophthalmol. 109, 1065 (1991).
Culp, C. J. et al. Clinical, histological and genetic findings in a donor with a clinical history of type 1 autoimmune polyendocrinopathy syndrome. Am. J. Ophthalmol. Case Rep. 25, 101266. https://doi.org/10.1016/j.ajoc.2022.101266 (2022).
Milam, A. H., Li, Z. Y. & Fariss, R. N. Histopathology of the human retina in retinitis pigmentosa. Prog. Retin. Eye Res. 17(2), 175–205 (1998).
Haruta, M., Tsuji, T. & Yoshida, S. Ultra-widefield OCT in retinopathy of autoimmune polyendocrine syndrome type 1. Ophthalmol. Retina 5(1), 9. https://doi.org/10.1016/j.oret.2020.06.007 (2021).
Traboulsi, E. I. Genetic Diseases of the Eye 2nd edn. (Oxford University Press, 2012).
Canamary, A. M. Jr., Takahashi, W. Y. & Sallum, J. M. F. Autoimmune retinopathy: A review. Int. J. Retina Vitreous 4, 1. https://doi.org/10.1186/s40942-017-0104-9 (2018).
Posey, J. E. et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J Med. 376(1), 21–31. https://doi.org/10.1056/NEJMoa1516767 (2017).
Mitani, T. et al. High prevalence of multilocus pathogenic variation in neurodevelopmental disorders in the Turkish population. Am. J. Hum. Genet. 108(10), 1981–2005. https://doi.org/10.1016/j.ajhg.2021.08.009 (2021).
Monies, D. et al. Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. Am. J. Hum. Genet. 104(6), 1182–1201. https://doi.org/10.1016/j.ajhg.2019.04.011 (2019). Erratum in: Am. J. Hum. Genet. 105(4), 879 (2019).
Monies, D. et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum. Genet. 136(8), 921–939. https://doi.org/10.1007/s00439-017-1821-8 (2017).
Acknowledgements
The authors thank the study family for their enthusiastic participation. We thank Firdous Abdulwahab for her help as research coordinators and Hessa Al-Saif for her technical help. The authors extend their appreciation to the King Salman Center For Disability Research for funding this work through Research Group no RG-2022-011.
Author information
Authors and Affiliations
Contributions
Literature search and figures: A.B., M.M. and L.A. Data collection and interpretation: A.B., M.M., O.A., and L.A. Manuscript drafting: A.B., M.M., F.A., P.S., H.B. and M.M. and approval. Manuscript revision: All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Badawi, A., Magliyah, M., Alabbasi, O. et al. Cone dystrophy associated with autoimmune polyglandular syndrome type 1. Sci Rep 13, 11223 (2023). https://doi.org/10.1038/s41598-023-38419-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-38419-9
- Springer Nature Limited