
Abstract
Earliella scabrosa is a pantropical species of Polyporales (Basidiomycota) and well-studied concerning its morphology and taxonomy. However, its pantropical intraspecific genetic diversity and population differentiation is unknown. We initiated this study to better understand the genetic variation within E. scabrosa and to test if cryptic species are present. Sequences of three DNA regions, the nuclear ribosomal internal transcribed spacer (ITS), the large subunit ribosomal DNA (LSU), and the translation elongation factor (EF1α) were analysed for 66 samples from 15 geographical locations. We found a high level of genetic diversity (haplotype diversity, Hd = 0.88) and low nucleotide diversity (π = 0.006) across the known geographical range of E. scabrosa based on ITS sequences. The analysis of molecular variance (AMOVA) indicates that the genetic variability is mainly found among geographical populations. The results of Mantel tests confirmed that the genetic distance among populations of E. scabrosa is positively correlated with the geographical distance, which indicates that geographical isolation is an important factor for the observed genetic differentiation. Based on phylogenetic analyses of combined dataset ITS-LSU-EF1α, the low intraspecific divergences (0–0.3%), and the Automated Barcode Gap Discovery (ABGD) analysis, E. scabrosa can be considered as a single species with five different geographical populations. Each population might be in the process of allopatric divergence and in the long-term they may evolve and become distinct species.
Similar content being viewed by others


Introduction
Data on genetic variation within a population is useful to discuss species concepts, cryptic species, breeding patterns, degree of relatedness, differentiation, and gene pool disruptions1,2,3,4. Low levels of genetic diversity may reduce the ability of populations to cope with environmental changes and other threats, leading species to become endangered or even extinct5,6,7. High genetic diversity allows species to adapt to environmental changes8. It is therefore essential to understand the genetic diversity and population structure of a species. This is important in particular for species with a wide geographical distribution as these may include multiple genetic lineages or cryptic species9,10.
Earliella Murrill (Basidiomycota, Polyporales) includes a single species Earliella scabrosa (Pers.) Gilb. & Ryvarden. The species was described as Polyporus scabrosus Pers. on the basis of a specimen collected in the Marianas islands (exact position not known)—about 1500 km east of the Philippine islands (south of Japan) in the Pacific Ocean. Earliella scabrosa is a saprotrophic species that colonises dead wood. It is also reported to be an opportunistic pathogen of plants and humans. As a human pathogen, it can infect and cause endophthalmitis and cutaneous fungal septic emboli11,12. Earliella scabrosa is also recognized as a medicinal species with anticancer, antifungal and antimicrobial activities as well as wound healing capabilities13,14,15. The species produces enzymes for the biotransformation of dyes in solid state fermentation and for the removal of bromocresol green, thus purifying water16,17.
Morphologically, E. scabrosa can be easily recognized due to its effuse-reflexed basidiomata with reddish cuticle and irregular, elongated or sinuous pores18. It is a common fungal species distributed all over the tropical regions of both hemispheres (Fig. 1A). Although the shape and colour of the basidiomata vary considerably (Fig. 1b), E. scabrosa is considered as a single species. Information on intraspecific diversity and genetic variability within and among populations is not available.

(A) Distribution map of Earliella scabrosa. Occurrences of all collected specimens during our mycological surveys as well as other occurrences available on the GBIF (Global Biodiversity Information Facility) website and those of the sequences obtained from the NCBI website were projected onto the map using the QGIS v3.28.0. (B) Morphological variability of basidiomata of Earliella scabrosa. Scale bar 2 cm.
Studies on many fungi previously thought to belong to a single species like Cryptococcus neoformans (San Felice) Vuill.19,20, Paxillus involutus (Batsch) Fr.21, Serpula himantioides (Fr.) P. Karst.22, and Trametes elegans (Spreng.) Fr.23,24 have been identified as species complexes that each contain two or more cryptic species. The above-mentioned consideration could also account for E. scabrosa with a wide distribution (pantropical), but, until now, we have no information on the genetic diversity within this species.
The most widely used approaches to assess genetic diversity from DNA sequences are nucleotide diversity and haplotype diversity25,26,27. Several highly variable DNA molecular markers are used for intra- and inter-specific genetic characterisation in plants, animals, and fungi28,29,30,31. In fungi, the internal transcribed spacer (ITS) region of nuclear ribosomal DNA has been most intensively sequenced and used32,33,34,35. It has been widely used for species identification, phylogenetic analyses, and the investigation of genetic variability36,37,38,39,40. In general, the fungal ITS marker provides higher interspecific resolution, with some degree of intraspecific variability34,36.
The goal of this study is to examine the extent of genetic divergence within E. scabrosa throughout most its geographical range and discuss these results in light of the potential existence of cryptic species within this taxon. At present, there is no DNA sequence for African specimens of E. scabrosa in any public database. Therefore, we focused our attention on African specimens in order to answer the following questions: (i) Is there genetic variation among individuals of E. scabrosa from the same geographical area? (ii) Is there genetic differentiation between populations in different geographic areas? (iii) If so, is the level of genetic differentiation positively correlated with geographic distances? (iv) Do the different populations of E. scabrosa form a single species or does this species contain cryptic (hidden) species?
Materials and methods
Specimen assembly
A total of 20 specimens of E. scabrosa were collected in Benin, Congo, and Guinea. Specimens were photographed in their natural environment before recording using a Sony camera, model DSC-HX400V. The geographic coordinates of occurrence of each specimen were recorded (Fig. 1). Small pieces of fresh basidiomata were placed in plastic bags half-filled with silica gel for gentle drying. The rest of the basidiomata were air- or oven-dried at 45–50 °C. Specimens are deposited at the mycological herbarium of the University of Parakou (UNIPAR) in Benin. For specimen data, see Supplementary S1.
DNA extraction, amplification, sequencing, and genetic distances analysis
DNA extraction, amplification, and sequencing
Genomic DNA from dried specimen was extracted using the microwave method41 or Analytik Jena kit when the first technic did not yield good results. The extracted genomic DNA was amplified targeting two nuclear ribosomal DNA regions, the internal transcribed spacer region of rDNA (ITS) with the primer pair ITS-1F/ITS442,43 and the D1–D4 domain of large subunit (LSU; 28S rDNA) with the primers LR0R/LR544. Additionally, the protein-coding gene translation elongation factor 1-alpha (EF1α) was amplified using the primers EF1-983/EF1-221845. The polymerase chain reaction (PCR) procedure for ITS was as follows: initial denaturation at 95 °C for 3 min, followed by 35 cycles at 95 °C for 30 s, 52 °C for 30 s and 68 °C for 1 min, and a final extension at 68 °C for 3 min. The PCR procedure to amplify the LSU rDNA sequence differed from the one for the ITS rDNA only by the annealing temperature (55 °C instead of 52 °C) and an increased cycle extension time (90 s per cycle). To amplify the protein-coding gene EF1α, the touchdown PCR protocol following Justo and Hibbett46 was used. The PCR products were further cleaned with QIAquick PCR Purification Kit according to the manufacturer’s instructions (QIAGEN GmbH, Hilden, Germany) and then sequenced at the company Eurofins Genomics Germany GmbH (https://www.eurofinsgenomics.eu/). All sequences used in this study are listed in Table 1 and the newly generated sequences deposited in GenBank.
Checking the affiliation of the name Earliella scabrosa applied to sequences in GenBank
On July 16th, 2022, all sequences of the ITS region labelled as E. scabrosa in GenBank (n = 76) were downloaded. The 76 ITS sequences of E. scabrosa from GenBank were aligned with 20 ITS sequences of E. scabrosa newly generated in this study. Sequences of the type species of the genera Daedalea and Trametes are added as outgroup. A maximum likelihood analysis was performed using IQ-tree 1.6.12 (http://www.iqtree.org/). Sequences of specimens from Mariana islands (type locality of E. scabrosa) are not available. However, all sequences (n = 66) of E. scabrosa that cluster together to form a single clade with sequences of specimens from other Pacific Ocean islands like French Polynesia are retained as E. scabrosa and were used for further analysis. Twenty-three sequences that were labelled E. scabrosa in GenBank did not form part of this clade. They are considered misidentifications and were not used for the present analysis (Supplementary S2).
Genetic diversity and relationships between geographic populations of Earliella scabrosa
A total of 66 ITS sequences belonging to E. scabrosa and annotated with data on geographic origin were used for the genetic analyses. These sequences were aligned and the resulting alignment was manually adjusted using Aliview. After this, the molecular diversity indices, such as the number of haplotypes (Nh), haplotype diversity (Hd), and nucleotide diversity (π) were estimated using pegas package47. To detect genetic differentiation within and among populations of E. scabrosa, an analysis of molecular variance (AMOVA) was performed using the alignment of 66 ITS sequences from 15 countries. The correlation between genetic and geographic distances was evaluated using a Mantel test48 and a linear regression. The phylogenetic relationships among the detected haplotypes are presented in a haplotype network. Statistical analyses have been implemented in the Integrated Development Environment RStudio (RStudio Team, 2021) for R software v4.1.2 (R Core Team, 2021).
Species delimitation within Earliella scabrosa
The same alignment with 66 sequences used in previous analyses is used here for species delimitation using the Automated Barcode Gap Discovery (ABGD) analysis49. The ABGD is a model-based method that delimit partitions of taxa, which can be recognized as species entities49. It sorts the sequences into hypothetical species based on the barcode gap. The ABGD analysis was performed using the Jukes-Cantor (JC69) distance with the relative gap width set to 1.0 and all other parameters were kept in default mode.
Phylogenetic relationship analyses
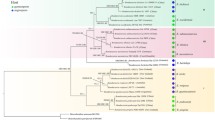
For phylogenetic analyses, sequences of ITS, LSU, and EF1α were used. Sequences of Daedalea quercina (L.) Pers. and Trametes suaveolens (L.) Fr. were used as outgroup. The sequences were aligned separately for each marker using the online mode of MAFFT version 750. The resulting multiple sequence alignments were checked in Geneious 5.6.7 (Kearse et al.51, https://www.geneious.com), where the ends rich in gaps were manually trimmed. Further, the multiple sequence alignments were viewed and some bases were manually corrected using AliView52. The combination of ITS, LSU and EF1α alignments was used for phylogenetic relationship analyses. The best-fit evolutionary model was estimated for each region using ModelFinder implemented in IQ-tree 1.6.12 http://www.iqtree.org/53. Based on estimated evolutionary models, the phylogenetic tree inference of Maximum likelihood (ML) and Bayesian Inference (BI) were performed to verify the phylogenetic relationship between all sequences of E. scabrosa. The Maximum likelihood analysis was performed with Ultrafast Bootstrap (UFBoot)54 and the branch support was evaluated with 5000 replicates using the IQ-tree 1.6.12. The BI was executed using MrBayes v. 3.2.7 in command line mode (https://github.com/NBISweden/MrBayes) for five million generations until the standard deviation of split frequencies reached 0.01. Chain convergence was determined using Tracer v. 1.7.1 (http://tree.bio.ed.ac.uk/software/tracer/) and the first 25% (5000) of the trees were discarded as burn-in. The remaining trees were used to build the consensus tree using the Phylogenetic Tree Summarization (SumTrees) program within DendroPy v. 4.3.0., https://github.com/jeetsukumaran/DendroPy55. The topology of species resulting from ML is used, and to add the posterior probabilities (PP) of BI on the ML tree, the Phylogenetic Tree Summarization (SumTrees) program within DendroPy v. 4.3.0., was used. Then, the UFBoot values were added to the ML best tree that already having the posterior probabilities using IQ-tree56. The resulting tree is presented below in the Fig. 5 and the support values of UFBoot/PP are indicated on each node.
Results
Newly generated sequences
A total of 28 new sequences were generated, namely 20 ITS sequences, four LSU sequences, and four EF1α sequences. Sequences have been generated for the first time for specimens from Africa.
Genetic diversity analysis
Haplotype diversity and distribution
The analysis of 66 ITS sequences of E. scabrosa with information on geographic origin revealed 24 haplotypes. The probability that two randomly selected alleles are different (haplotype diversity) is 0.880. The nucleotide diversity, which is the mean difference in nucleotide per base position when comparing DNA sequences pairwisely, is π = 0.0058. The highest number of haplotypes (Nh = 6) is recorded for China, French Polynesia and Taiwan while the highest haplotype diversity (haplotypes diversity = 1) is discovered for Sri Lanka (Fig. 2).

Haplotype diversity in countries with high scores.
The haplotype distribution in all populations is shown in Fig. 3 and suggests a limited gene flow between populations between geographic areas. Of the 24 haplotypes in total, only four haplotypes namely h1, h2, h4, and h20 occur in several populations in different areas. Haplotype 20 (h20) is only distributed in three African countries, namely Benin, Congo Republic, and Guinea. Almost all haplotypes are linked to h3 which is from French Polynesia (Fig. 3). Our haplotype analyses revealed a high number of unique haplotypes mostly found in Taiwan and French Polynesia. The most common haplotypes are h20, h1, h6 and h4 which correspond to respectively 29%, 15%, 11%, and 9% of the individual DNA sequences included in the analysis.

Haplotype network based on ITS sequences of Earliella scabrosa, including information on the geographical origin of the sequences by different colours. The circles represent different haplotypes. The sizes of the circles as well as the sizes of the pies reflect the frequency of detection of each haplotype. The lines connecting haplotypes represent their genetic distance and the short perpendicular lines indicate the number of nucleotide differences.
Population structure and genetic differentiation
The results from the analysis of molecular variance (AMOVA) showed significant genetic differentiation between the sequences of E. scabrosa from the source countries (ΦST = 0.82, p = 0, Table 2). The analysis suggests that nearly 82% of the genetic variation is explained by between country differences while only 18% of the genetic variation is explained by within country differences (Table 2). The result of the Mantel test shows a significant correlation between geographical distance and genetic distance. This is confirmed by the linear regression fit which indicates a positive correlation of the genetic distance and the geographic distance (Fig. 4).

Influence of geographic distance on genetic distance in Earliella scabrosa populations.
Species delimitation
The ABGD partitioned the ITS dataset into three initial partitions. The first two partitions with an intraspecific divergence of 0–0.2% divided sequences of E. scabrosa into 18 groups with each group representing a hypothetical species. All sequences from African specimens clustered together and formed a single hypothetical species (Supplementary S3). However, with an intraspecific divergence of only 0.3%, all sequences of E. scabrosa should be considered as one single species (Supplementary S4).
Phylogenetic analysis
The alignment of ITS, LSU, and EF1α sequence data of specimens of E. scabrosa includes 68 sequences with 2195 characters, 336 distinct patterns, 77 parsimony-informative sites, 301 singleton sites, and 1817 constant sites. The topology of species resulting from ML and BI analyses is congruent. Different groups that reflect populations of E. scabrosa with moderate to high support values and evident geographical patterns regarding the origin of collection are highlighted (Fig. 5).

This multilocus ML/BI tree was constructed from the combined alignment of ITS, LSU, and EF1α sequences showing phylogenetic relationships between the 66 collections of Earliella scabrosa. The groups highlighted bear the names of the origins of most of the sequences in the respective group.
Discussion
We used the combination of haplotype networks and phylogenetic trees to analyse the genetic diversity and population differentiation within and among samples of Earliella scabrosa from its pantropical range of distribution. The phylogenetic tree revealed distinct lineages among the populations of E. scabrosa. Meanwhile, the haplotype network provided additional quantitative information on how haplotypes are shared between populations of E. scabrosa.
Haplotype diversity, population structure and genetic differentiation
In this study, we found a high level of genetic diversity (haplotype diversity, Hd = 0.88) across the known geographical range of E. scabrosa based on ITS sequences. Similar levels of genetic variability are commonly observed in wild populations with broad ecological niches, and/or wide geographical distribution, such as Funneliformis mosseae (T.H. Nicolson & Gerd.) C. Walker & A. Schüßler63 and Schizophyllum commune Fr.38,64. However, the level of genetic diversity detected in our study could be the direct consequence of a single gene used or the nuclear ribosomal region used as other studies on genetic diversity have revealed a difference between the ribosomal and the ISSR markers7,65. Here, we were not able to combine both regions or several gene loci because corresponding sequence data are not available in GenBank. Future studies to sequence and combine more genes from different populations of E. scabrosa will increase our understanding on the genetic diversity of this species.
The 24 haplotypes detected in our analyses are revealed for the first time, and among them, two haplotypes (h20 and h21) are detected from African specimens. Our haplotype analyses reveal a dominance of unique haplotypes (20 out of 24 in total) which are mostly found in Taiwan and French Polynesia. Because our sample sizes are relatively small for several sites, it is possible that more ITS haplotypes might be found if more extensive sampling were conducted and that wider distributions of some of these unique haplotypes might be revealed.
Within the studied populations of E. scabrosa, haplotypes are hardly shared. Of the 24 haplotypes detected, a few of them (h1, h2, h4, h20) were found distributed in multiple geographical areas (Fig. 3). This result is consistent with a certain degree of gene flow between geographical populations of this species in nature. While evidence for certain degree of gene flow has been found in E. scabrosa, our population genetic analyses also suggest that, overall, gene flow was somewhat limited. Previous studies on the gene flow of fungal have shown that gene flow can be short distance or long distance and both of them are generally mediated by wind, human or animal activities63,66. In this study, h20 is only shared between African specimens from Congo Republic, Guinea and Benin. The distances between Benin and Guinea on the one hand and Benin and Congo Republic on the other are about 1500 km and 2000 km. Similar spore dispersals (up to 2000 km) were reported in several fungi67,68,69. Thus, we can assume that wind-driven dispersal of basidiospores is probably responsible for the observed gene flow in the African samples. Unlike h20, which is distributed only among samples from the same continent, h1 is shared between America and China, h2 between Sri Lanka and Venezuela, and h4 between Asia and French Polynesia. The geographic distance between these abovementioned populations was much larger than the short dispersal distance of fungal spores. However, even if long-distance gene flow was relatively much rarer70, this idea cannot be ruled out as several fungi are known to be influenced by human activity, including plant pathogens71,72 and fungi that are found on substrates used by humans like E. scabrosa22. For example, in natural populations of the button mushroom Agaricus bisporus (J.E. Lange) Imbach, the sharing of certain genotypes was found for strains from different regions, countries and even continents73,74. Because those genotypes were identical or very similar to cultivated strains, it was suggested that human-aided dispersal was responsible for such long-distance gene flow in A. bisporus. A similar process could account for the wide distribution of h1, h2, and h4 in our samples.
Phylogenetics and taxonomy
We detected some level of intraspecific variation within the population of E. scabrosa even at the fine geographical scale (country level). For example, with an intraspecific divergence of 0–0.2%, populations of E. scabrosa in China, Venezuela, Taiwan, and French Polynesia might be split into several hypothetical species. However, considering an intraspecific divergence of 0.3%, all sequences of E. scabrosa sequences are grouped into a single species. This means ITS region may include considerable intraspecific variation, which can lead to oversplitting of species during DNA barcoding analyses75. The intraspecific divergence found in this study corresponds well with data available in literature on intraspecific ITS variability of 0–3% in the fungal kingdom36,76. Seena et al.77, when proposing the internal transcribed spacer gene region as a barcode for identifying aquatic hyphomycete species, found also a 0.3% variation within Articulospora tetracladia Ingold. The difference in numbers of hypothetical species according to the intraspecific divergence suggests that these hypothetical species should not be used as phylogenetic species, but an extra clustering step is needed to approach species-level resolution. Phylogenetic analyses grouped E. scabrosa sequences in several groups of which five displaying well evidenced geographical patterns. The phylogenetic patterns within E. scabrosa can be attributed to geographic hypotheses. Our five well distinct groups showed a preference or tendency for geographic regions. The African group is geographically restricted to specimens from African countries. The Polynesian group is mostly represented by Polynesian samples with an additional collection from Taiwan. Samples forming the Asia group are widely distributed across Asia countries with one collection from French Polynesia. The America group includes American samples as well as multiple samples from China and Sri Lanka. The heterogeneity within some geographical groups confirmed the sharing haplotypes between these populations. Only specimens from the African group are available and examination of these has not revealed any anatomical differences, although distinct morphological characters (presence or absence of reddish cuticles on the basidiomata, the effused basidiomata in some and resupinate in others) have been observed within the specimens of this group. As with the ABGD, the 20 sequences from the African specimens formed a well-defined group and no other sequences from other areas fell into this group. We therefore suspect that geographically separated populations of E. scabrosa are genetically divergent, but difficult to separate based on morphology. The results from the Mantel tests and linear regression supports the hypothesis that the genetic relationship among populations is closely associated with geographic distance (Table 2, Fig. 4). AMOVA tests also confirmed that geographic separation contributed significantly to the observed sequence variation and approximately 82% of observed differentiations were partitioned among populations. Similar types of geographic patterns of DNA sequence variation have been observed in many other fungal groups. For example, distinct alleles were observed in geographically separated populations of Russula brevipes Peck78,79. Each group identified here and especially the African one could become a distinct phylogenetic species knowing that long-term geographical isolation could favour the process of allopatric divergence of different populations80.
Conclusion
This study revealed five distinct groups within E. scabrosa and a correlation between genetic and geographic distances. The intraspecific divergences (0–0.3%), however, are relatively low, so cryptic taxa apparently are not present. The different groups may eventually evolve into phylogenetically distinct species in the long term, as geographical isolation could favour the process of allopatric divergence. More extensive sampling and analyses combining several makers may reveal additional distinct lineages as well as novel distribution patterns within this species in its geographical areas of distribution.
Data availability
Newly generated sequences are available in GenBank and the accession numbers are given in Table1. Alignment, phylogenetic tree, and accession numbers of newly generated sequences will be public after the paper is published. Collected specimens are available at the mycological herbaria of the University of Parakou (UNIPAR) in Benin.
References
Sah, S., Barat, A., Pande, V. & Sati, J. Population structure of Indian Hill Trout (Barilius bendelisis) inferred from variation in mitochondrial DNA sequences tissue specific transcriptome of mahseer view project malarial drug targets view project. Adv. Biol. Res. 5, 93–98 (2011).
Jin, C., Huixia, K. & Shubin, D. Population genetic structure and gene flow of rare and endangered Tetraena mongolica Maxim. revealed by reduced representation sequencing. BMC Plant Biol. 20, 1–13 (2020).
Hopley, T. & Byrne, M. Gene flow and genetic variation explain signatures of selection across a climate gradient in two riparian species. Genes (Basel) 10, 579 (2019).
Lacy, R. C., Journal, S. & May, N. American society of mammalogists importance of genetic variation to the viability of mammalian populations importance of genetic variation to the viability of. Society 78, 320–335 (2010).
Maki, M. & Horie, S. Random amplified polymorphic DNA (RAPD) markers reveal less genetic variation in the endangered plant Cerastium fischerianum var. molle than in the widespread conspecific C. fischerianum var. fischerianum (Caryophyllaceae). Mol. Ecol. 8, 145–150 (1999).
Spielman, D., Brook, B. W. & Frankham, R. Most species are not driven to extinction before genetic factors impact them. Proc. Natl. Acad. Sci. USA 101, 15261–15264 (2004).
Qin, A. et al. Low genetic diversity and population differentiation in Thuja sutchuenensis Franch., an extremely endangered rediscovered conifer species in southwestern China. Glob. Ecol. Conserv. 25, e01430 (2021).
Baranova, M. A. et al. Extraordinary genetic diversity in a wood decay mushroom. Mol. Biol. Evol. 32, 2775–2783 (2015).
Jargeat, P. et al. Phylogenetic species delimitation in ectomycorrhizal fungi and implications for barcoding: The case of the Tricholoma scalpturatum complex (Basidiomycota). Mol. Ecol. 19, 5216–5230 (2010).
Douhan, G. W., Fuller, E., McKee, B. & Pond, E. Genetic diversity analysis of avocado (Persea americana Miller) rootstocks selected under greenhouse conditions for tolerance to phytophthora root rot caused by Phytophthora cinnamomi. Euphytica 182, 209–217 (2011).
Lim, D. S. W., Tan, P. L., Jureen, R. & Tan, K. B. Cutaneous Emboli of invasive basidiomycosis in a child with aplastic anemia. Am. J. Dermatopathol. 39, 204–207 (2017).
He, H., Chen, X., Liu, H., Wu, J. & Zhong, X. Earliella scabrosa-associated postoperative Endophthalmitis after Phacoemulsification with intraocular lens implantation: A case report. BMC Ophthalmol. 18, 18–21 (2018).
Zmitrovich, I. V., Ezhov, O. N., Ranadive, K. R. & Wasser, S. P. Profiles of little-known medicinal polypores: Earliella scabrosa (agaricomycetes). Int. J. Med. Mushrooms 19, 1023–1027 (2017).
Peng, Y. T. & Don, M. M. Antifungal activity of in-vitro grown Earliella scabrosa, a Malaysian fungus on selected wood-degrading fungi of rubberwood. J. Phys. Sci. 24, 21–33 (2013).
Kithiyon, M., Pannerselvam, B., Balasaubramaniyam Madhukumar, S. S., Sridharan, J. & Alagumuthu, T. S. Efficacy of mycosynthesised AgNPs from Earliella scabrosa as an in vitro antibacterial and wound healing agent. IET Nanobiotechnol. 13, 339–344 (2019).
Guerra, G. et al. Production of laccase and manganese peroxidase by white-rot fungi from sugarcane bagasse in solid bed: Use for dyes decolourisation. Sugar Tech. 10, 260–264 (2008).
Sintakindi, A. & Ankamwar, B. Biosorption of Bromocresol green from aqueous solution by Earliella scabrosa fungal biomass in removal of environmental pollutants. Int. J. Environ. Sci. Technol. https://doi.org/10.1007/s13762-022-04218-4 (2022).
Gilbertson, R. L. & Ryvarden, L. North American polypores. Vol. I. Abortiporus-Lindtneria. Vol. I (Fungiflora A/S, 1986).
Xu, J., Vilgalys, R. & Mitchell, T. G. Multiple gene genealogies reveal recent dispersion and hybridization in the human pathogenic fungus Cryptococcus neoformans. Mol. Ecol. 9, 1471–1481 (2000).
Bovers, M., Hagen, F., Kuramae, E. E. & Boekhout, T. Six monophyletic lineages identified within Cryptococcus neoformans and Cryptococcus gattii by multi-locus sequence typing. Fungal Genet. Biol. 45, 400–421 (2008).
Hedh, J., Samson, P., Erland, S. & Tunlid, A. Multiple gene genealogies and species recognition in the ectomycorrhizal fungus Paxillus involutus. Mycol. Res. 112, 965–975 (2008).
Kauserud, H. et al. Asian origin and rapid global spread of the destructive dry rot fungus Serpula lacrymans. Mol. Ecol. 16, 3350–3360 (2007).
Olou, B. A., Krah, F., Piepenbring, M., Yorou, N. S. & Langer, E. Diversity of Trametes (Polyporales, Basidiomycota) in tropical Benin and description of new species Trametes parvispora. MycoKeys 65, 25–47 (2020).
Carlson, A., Justo, A. & Hibbett, D. S. Species delimitation in Trametes : A comparison of ITS, RPB1, RPB2 and TEF1 gene phylogenies. Mycologia 106, 735–745 (2014).
Goodall-Copestake, W. P., Tarling, G. A. & Murphy, E. J. On the comparison of population-level estimates of haplotype and nucleotide diversity: A case study using the gene cox1 in animals. Heredity (Edinb). 109, 50–56 (2012).
Fan, P. et al. An approach for estimating haplotype diversity from sequences with unequal lengths. Methods Ecol. Evol. 12, 1658–1667 (2021).
Miraldo, A. et al. An anthropocene map of genetic diversity. Science (80-) 353, 1532–1535 (2016).
Sari, A., Duran, M., Sen, A. & Bardakci, F. Investigation of chironomidae (Diptera) relationships using mitochondrial COI gene. Biochem. Syst. Ecol. 59, 229–238 (2015).
Fang, D. A. et al. Genetic diversity and population differentiation of naked carp (Gymnocypris przewalskii) revealed by cytochrome oxidase subunit I and D-loop. Front. Ecol. Evol. 10, 1–11 (2022).
Amiteye, S. Basic concepts and methodologies of DNAa marker systems in plant molecular breeding. Heliyon 7, e08093 (2021).
Egydio Brandão, A. P. M., Yamaguchi, L. F., Tepe, E. J., Salatino, A. & Kato, M. J. Evaluation of DNA markers for molecular identification of three piper species from Brazilian Atlantic rainforest. PLoS One 15, 1–17 (2020).
Badotti, F. et al. Effectiveness of ITS and sub-regions as DNA barcode markers for the identification of Basidiomycota (Fungi). BMC Microbiol. 17, 42 (2017).
Raja, H. A., Miller, A. N., Pearce, C. J. & Oberlies, N. H. Fungal identification using molecular tools: A primer for the natural products research community. J. Nat. Prod. 80, 756–770 (2017).
Schoch, C. L. et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 109, 6241–6246 (2012).
Hoang, M. T. V. et al. Dual DNA barcoding for the molecular identification of the agents of invasive fungal infections. Front. Microbiol. 10, 1–13 (2019).
Nilsson, R. H., Kristiansson, E., Ryberg, M., Hallenberg, N. & Larsson, K. H. Intraspecific ITS variability in the Kingdom Fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol. Bioinform. 2008, 193–201 (2008).
Garnica, S. et al. Determining threshold values for barcoding fungi: Lessons from Cortinarius (Basidiomycota), a highly diverse and widespread ectomycorrhizal genus. FEMS Microbiol. Ecol. 92, 1–16 (2016).
Choi, Y., Kim Nguyen, H. T., Lee, T. S., Kim, J. K. & Choi, J. Genetic diversity and dye-decolorizing spectrum of Schizophyllum commune population. J. Microbiol. Biotechnol. 30, 1525–1535 (2020).
de Rollemberg, N. C. et al. Identification of fungi in flaxseed (L. usitatissimum L.) using the ITS1 and ITS2 intergenic regions. Microbiol. Res. (Pavia) 13, 315–322 (2022).
D’Andreano, S., Cuscó, A. & Francino, O. Rapid and real-Time identification of fungi up to species level with long amplicon nanopore sequencing from clinical samples. Biol. Methods Protoc. 6, 1–6 (2021).
Dörnte, B. & Kües, U. Fast microwave-based DNA extraction from vegetative Mycelium and fruiting body tissues of agaricomycetes for PCR amplification. Curr. Trends Biotechnol. Pharm. 7, 825–836 (2013).
White, T. J., Bruns, S., Lee, S. & J, T. Amplification and direct sequencing of fungal ribo- somal RNA genes for phylogenetics. In: PCR Protocols: A Guide to Methods and Applications. 315–322 (Acad. Press., New York, 1990) doi:citeulike-article-id:671166.
Gardes, M. & Bruns, T. D. ITS primers with enhanced specificity for basidiomycetes—Application to the identification of mycorrhizae and rusts. Mol. Ecol. 2, 113–118 (1993).
Vilgalys, R. & Hester, M. Rapid genetic identification and mapping of enzymatically amplified ribosomal DNA from several Cryptococcus species. J. Bacteriol. 172, 4238–4246 (1990).
Rehner, S. A. & Buckley, E. A Beauveria phylogeny inferred from nuclear ITS and EF1-sequences: Evidence for cryptic diversification and links to Cordyceps teleomorphs. Mycologia 97, 84–98 (2008).
Justo, A. & Hibbett, D. S. Phylogenetic classification of Trametes (Basidiomycota, Polyporales) based on a five-marker dataset. Taxon 60, 1567–1583 (2011).
Paradis, E. Pegas: An R package for population genetics with an integrated-modular approach. Bioinformatics 26, 419–420 (2010).
Mantel, N. The detection of disease clustering and a generalized approach. Cancer Res. 27, 209–220 (1967).
Puillandre, N., Lambert, A., Brouillet, S. & Achaz, G. ABGD, automatic barcode gap discovery for primary species delimitation. Mol. Ecol. 21, 1864–1877 (2012).
Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. https://doi.org/10.1093/bib/bbx108 (2017).
Kearse, M. et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278 (2014).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Hoang, D. T., Chernomor, O., Von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Sukumaran, J. & Holder, M. T. DendroPy: A Python library for phylogenetic computing. Bioinformatics 26, 1569–1571 (2010).
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235 (2016).
Justo, A. et al. A revised family-level classification of the Polyporales (Basidiomycota). Fungal Biol. 121, 798–824 (2017).
Sayers, E. W. et al. GenBank. Nucleic Acids Res. 48, D84–D86 (2020).
Osmundson, T. W., Bergemann, S. E., Rasmussen, R. & Garbelotto, M. M. Using point data to assess biogeographical signal, endemicity and factors associated with macrofungal diversity in the data-poor Pacific oceanic island bioregion. J. Biogeogr. 49, 891–903 (2022).
Hage, H. et al. Gene family expansions and transcriptome signatures uncover fungal adaptations to wood decay. Environ. Microbiol. 23, 5716–5732 (2021).
Pang, K. L. et al. Insights into fungal diversity of a shallowwater hydrothermal vent field at Kueishan Island, Taiwan by culture-based and metabarcoding analyses. PLoS One 14, e0226616 (2019).
de Pádua, A. P. S. L. et al. Fungal endophyte diversity in the leaves of the medicinal plant Myracrodruon urundeuva in a Brazilian dry tropical forest and their capacity to produce L-asparaginase. Acta Bot. Brasilica 33, 39–49 (2019).
Rosendahl, S., McGee, P. & Morton, J. B. Lack of global population genetic differentiation in the arbuscular mycorrhizal fungus Glomus mosseae suggests a recent range expansion which may have coincided with the spread of agriculture. Mol. Ecol. 18, 4316–4329 (2009).
James, T. Y., Moncalvo, J.-M., Li, S. & Vilgalys, R. Polymorphism at the ribosomal DNA spacers and its relation to breeding structure of the widespread mushroom schizophyllum commune. Genetics 157, 149–161 (2001).
Liu, J., Shi, S., Chang, E., Yang, W. & Jiang, Z. Genetic diversity of the critically endangered Thuja sutchuenensis revealed by ISSR markers and the implications for conservation. Int. J. Mol. Sci. 14, 14860–14871 (2013).
Bertolino, S., Vizzini, A., Wauters, L. A. & Tosi, G. Consumption of hypogeous and epigeous fungi by the red squirrel (Sciurus vulgaris) in subalpine conifer forests. For. Ecol. Manag. 202, 227–233 (2004).
Saville, B. J., Yoell, H. & Anderson, J. B. Genetic exchange and recombination in populations of the root-infecting fungus Armillaria gallica. Mol. Ecol. 5, 485–497 (1996).
Xu, J., Guo, H. & Yang, Z. L. Single nucleotide polymorphisms in the ectomycorrhizal mushroom Tricholoma matsutake. Microbiology 153, 2002–2012 (2007).
Xu, J., Sha, T., Li, Y. C., Zhao, Z. W. & Yang, Z. L. Recombination and genetic differentiation among natural populations of the ectomycorrhizal mushroom Tricholoma matsutake from southwestern China. Mol. Ecol. 17, 1238–1247 (2008).
Zhao, M. et al. Genetic variation and population structure of the mushroom Pleurotus ferulae in China inferred from nuclear DNA analysis. J. Integr. Agric. 15, 2237–2246 (2016).
Couch, B. C. et al. Origins of host-specific populations of the blast pathogen Magnaporthe oryzae in crop domestication with subsequent expansion of pandemic clones on rice and weeds of rice. Genetics 170, 613–630 (2005).
Kohn, L. M. Mechanisms of fungal speciation. Annu. Rev. Phytopathol. 43, 279–308 (2005).
Xu, J., Kerrigan, R. W., Callac, P., Horgen, P. A. & Anderson, J. B. Genetic structure of natural populations of Agaricus bisporus, the commercial button mushroom. J. Hered. 88, 482–488 (1997).
Xu, J. et al. Mitochondrial DNA variation in natural populations of the mushroom Agaricus bisporus. Mol. Ecol. 7, 19–33 (1998).
Estensmo, E. L. F. et al. The influence of intraspecific sequence variation during DNA metabarcoding: A case study of eleven fungal species. Mol. Ecol. Resour. 21, 1141–1148 (2021).
Smith, M. E., Douhan, G. W. & Rizzo, D. M. Intra-specific and intra-sporocarp ITS variation of ectomycorrhizal fungi as assessed by rDNA sequencing of sporocarps and pooled ectomycorrhizal roots from a Quercus woodland. Mycorrhiza 18, 15–22 (2007).
Seena, S., Pascoal, C., Marvanová, L. & Cássio, F. DNA barcoding of fungi: A case study using ITS sequences for identifying aquatic hyphomycete species. Fungal Divers. 44, 77–87 (2010).
Bergemann, S. E., Douhan, G. W., Garbelotto, M. & Miller, S. L. No evidence of population structure across three isolated subpopulations of Russula brevipes in an oak/pine woodland. New Phytol. 170, 177–184 (2006).
Bergemann, S. E. & Miller, S. L. Size, distribution, and persistence of genets in local populations of the late-stage ectomycorrhizal basidiomycete, Russula Brevipes. New Phytol. 156, 313–320 (2002).
Pildain, M. B. et al. Molecular phylogeny of Armillaria from the Patagonian Andes. Mycol. Prog. 8, 181–194 (2009).
Acknowledgements
The authors are grateful to Christian Printzen for his advice on how to handle the data of this study. This research was funded by the German Federal Ministry of Education and Research, BMBF (Grant No. 01D20015). Boris Armel Olou also received support from the German Academic Exchange Service, DAAD (Grant No. 91651629), and the MBZ Species Conservation Fund (Grant No. 2225290065).
Author information
Authors and Affiliations
Contributions
B.A.O. had the initial idea of the study. B.A.O., M.P., and N.S.Y. designed the study and B.O.A. implemented it. B.O.A. conducted field work, molecular lab works, and phylogenetic analysis. B.A.O. and A.R.H. planned statistical analysis and analyzed the data. B.A.O. wrote the first draft of the manuscript and M.P. and N.S.Y. commented and revised the manuscript. B.A.O. wrote then the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Olou, B.A., Hègbè, A.D.M.T., Piepenbring, M. et al. Genetic diversity and population differentiation in Earliella scabrosa, a pantropical species of Polyporales. Sci Rep 13, 23020 (2023). https://doi.org/10.1038/s41598-023-50398-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-50398-5
- Springer Nature Limited