
Abstract
Free Fecal Liquid (FFL), also termed Fecal Water Syndrome (FWS), is an ailment in horses characterized by variable solid and liquid (water) phases at defecation. The liquid phase can be excreted before, during, or after the solid defecation phase. While the underlying causes of FFL are unknown, hindgut dysbiosis is suggested to be associated with FFL. Three European studies investigated dysbiosis in horses with FFL using 16S rRNA sequencing and reported results that conflicted between each other. In the present study, we also used 16S rRNA sequencing to study the fecal microbial composition in 14 Canadian horses with FFL, and 11 healthy stable mate controls. We found no significant difference in fecal microbial composition between FFL and healthy horses, which further supports that dysbiosis is not associated with FFL.
Similar content being viewed by others


Introduction
Free Fecal Liquid (FFL), also known as fecal water syndrome (FWS), is a condition in domestic horses in which defecation occurs in both a liquid and solid phase1,2,3. The liquid phase can occur either before, during, or after defecation4. Horses with FFL sometimes display signs of discomfort during defecation2 and persistently have soiled hindquarters1, as well as a high risk of developing dermatitis, bloated abdomen, and weight loss in severe cases2,3. While the exact causes of FFL are unknown, several factors including social stress1,2 endoparasitic infection, poor dentition, and diet have been suggested as at least being contributing factors5. Diet6, environmental conditions6,7 and stress4,6,8 also have a significant impact on the composition of the microbial population (microbiota) within the intestines. An imbalance in equine gut microbiota composition (dysbiosis) is already well documented to affect the normal health and function of the gastrointestinal tract and is associated with diseases like colic9, colitis10, laminitis11,12, and diarrhea in foals13,14. As such, dysbiosis has been suspected to play a significant role in FFL.
Recent studies have compared the microbial composition of horses with FFL to healthy control subjects and found conflicting results. Two studies in particular, Lausten et al.1 and Schoster et al.3, concluded no statistical difference in microbial composition between healthy horses and those with FFL. However, another study found statistically significant differences in the population contribution of genera Alloprevotella and Bacillus between FFL horses and stable-matched controls, while the overall population diversity of the microbiota remained similar between the groups5. Notably, all previous studies regarding the relationship between FFL and the microbiome have been carried out in Europe. Therefore, the objective of this study was to expand the current understanding of FFL and extend sampling to North America, by examining fecal microbiome profiles of both healthy horses and those exhibiting FFL in the Fraser Valley region of British Columbia, Canada. Given the reported associations of dysbiosis with other gastrointestinal tract diseases, we hypothesized that dysbiosis may also be associated with FFL in Canadian horses. However, our results support the conclusion that there is no significant difference between the microbiome of feces from FFL animals and controls.
Methods
Animals and sample collection
A case-controlled study was performed by sampling horses suffering with FFL and healthy stable matched controls. Fecal samples were collected and provided by an Agwest Veterinary Group Ltd. veterinarian in Fall 2020 during routine care of horses distributed across British Columbia’s Fraser Valley. The veterinarian collected approximately 40 mL of feces from each horse using the rectal grab technique. During collection, the health status, age, breed, sex, feed, and farm location were also noted. Following collection, the fecal samples were immediately placed on ice and kept for a maximum of eight hours before storing at -80°C until DNA extraction.
DNA Extraction and 16S rRNA sequencing
DNA was extracted from fecal samples using the QIAamp PowerFecal DNA Kit (Qiagen) according to the manufacturer’s instructions. DNA quality was assessed by NanoDrop™ One Spectrophotometer (Thermo Scientific™) and DNA quantity was determined using the Qubit 4 Fluorometer (Invitrogen).
DNA was amplified using PCR targeting multiple hypervariable regions of the 16S rRNA bacterial gene using the V2-4–8 and V3-6,7–9 primer sets included with the Ion 16S™ Metagenomics Kit (Life Technologies). Amplicons were purified by Agencourt® AMPure® XP Kit (Beckman Coulter) and subsequently pooled by sample. Library preparation was conducted using the Ion Plus Fragment Library Kit (Thermo Fisher Scientific) according to the manufacturer’s recommendations for barcoded libraries using the Ion Xpress™ Barcode Adapters 1–96 Kit (Thermo Fisher Scientific). Libraries were quantified using the Ion Universal Library Quantification Kit (Life Technologies), normalized to 10pM, and then pooled. Template generation occurred within the Ion Chef instrument (Ion Torrent™) using the pooled libraries and an Ion 520™ Chip (Ion Torrent™). The loaded Ion 520™ Chip was sequenced using the Ion GeneStudio™ S5 System (Ion Torrent™). All extraction, library preparation, and sequencing steps were performed at the Applied Genomics Centre at Kwantlen Polytechnic University in Surrey, BC.
Statistical analysis
Sequence data was analysed using the QIIME2 pipeline, as outlined in the QIIME2 documentation (https://docs.qiime2.org/2023.5/)16. Demultiplexed fastq files collected from the GenoStudio™ S5 System were imported into QIIME2 using the Single End Fastq Manifest Phred 33V2 format. Sequences were filtered by quality scores and ambiguous base calls17, then dereplicated using VSEARCH18 to produce a feature table and feature representative sequences. Contigs were aligned to SILVA 16S rRNA (138 release) sequence reference database19,20 and classified as operational taxonomic units (OTUs) using the closed reference feature clustering approach18. Chimeric representative sequences were detected using UCHIME18 and filtered out. The OTUs were classified against the SILVA 16S rRNA (138 release) sequence reference database19,20 using the VSEARCH-based consensus taxonomy classifier pipeline18 within the QIIME2 feature-classifier plugin21. Representative sequences were used to construct a rooted phylogenetic tree using the MAFFT-FastTree pipeline22,23. The OTU feature table, taxonomic feature data, and rooted phylogenetic tree QIIME2 artifacts were exported.
The exported OTU feature table, taxonomic feature data, rooted phylogenetic tree, and the sample metadata file were imported into R24,25 loaded with the phyloseq26, vegan27, and tidyverse28 packages. The files were used as components to construct a phyloseq object, which was subsequently filtered based on taxonomic classification status26. The phyloseq object was used to display the relative abundance of each taxonomic classification with a mean above 1% for comparison28. Normality of the data was tested using the Shapiro-Wilks test29. Abundances were compared between FFL status, sex, and geographical location using the Kruskal–Wallis test30 and the Wilcoxon rank sum test31, using the Benjamini–Hochberg procedure using α = 0.0532 to adjust for false discovery rates (FDR). The phyloseq object was then rarefied for downstream diversity metrics testing26. Alpha diversities were quantified using the Shannon index26,33, Simpson index26,34, and Chao richness26,35 and were compared by FFL status, sex, and geographical location using the Kruskal–Wallis test30 and Wilcoxon rank sum hypothesis testing31. The Benjamini–Hochberg procedure using α = 0.0532 of adjustment was used to control FDR. Beta diversity, the dissimilarity in community composition, was quantified for FFL status and geographical locations using the Bray–Curtis index27,36 and Jaccard distance27. Quantified beta diversity metrics were compared using one-way permutational multivariate analysis of variance (PERMANOVA)37 and analysis of similarities (ANOSIM)38, each using 999 permutations. The dissimilarity between FFL status, sex, and geographical location was visualized by principal coordinate analysis (PCoA) plots27,28.
Ethics declaration
The samples were collected by an Agwest Veterinary Group Ltd. veterinarian in accordance with the Canadian Association for Laboratory Animal Medicine (CALAM) Standards of Veterinary Care, which adheres to the ARRIVE guidelines15.
Results
Animal demographic data
A total of 25 horses were examined, 14 horses with FFL and 11 stable-mate controls. Among the affected horses, five were male. Among the control group, seven were male. The horses sampled were from one of six geographical locations within British Columbia’s Fraser Valley: Richmond (7), Surrey (2), Langley (6), Maple Ridge (1), Abbotsford (7), and Matsqui Flats (2). The horses included in the study had a mean age of 14 and a half years and a range of 2 to 29 years. Detailed information on horse metadata is presented in Supplementary Table S1.
Sequencing output
Sequencing analysis of the 16S rRNA gene yielded a total of 208,907 sequence reads that passed all quality control filters. All 25 samples returned more than 1,000 reads and were retained for further analyses and had a median sequencing depth of 7,963 reads. Specifically, the control horses had a total of 97,754 sequencing reads (median; 8,114 reads), while the FFL horses had 111,153 sequencing reads (median: 7,837 reads).
Microbial population composition
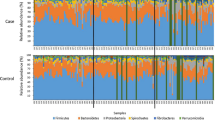
The microbial population for all 25 horses in the study was determined. We identified 7 phyla, 8 classes, 12 orders, 17 families, and 17 genera which exhibited a relative abundance greater than 1%. The seven most abundant phyla are Bacillota (median: 37.155 ± 12.465%), Bacteroidota (median: 37.838 ± 10.429%), Alphaproteobacteriota (median: 3.986 ± 6.874%), Verrucomicrobiota (median: 5.657 ± 5.025%), Spirochaetota (median: 3.968 ± 4.316%), Fibrobacterota (median: 1.442 ± 1.857%), and Patescibacteria (median: 0.940 ± 0.691%). The most abundant phyla, classes, and orders between FFL status and geographic location are presented in Fig. 1. The most abundant families and genera are displayed in Supplementary Fig. 1. Relative abundances above 1% at all classification levels for each horse in the study are presented in Supplementary Fig. 2. Additionally, the most abundant taxa at all classification levels between sex are depicted in Supplementary Fig. 3.

Bar charts illustrating the mean relative abundance of phyla (a and b), classes (c and d), and orders (e and f) between FFL status (a, c, and e) and geographical location (b, d, and f). All named taxa displayed have a mean relative abundance above 1%.
Alpha diversity
Alpha diversity indices between horses with FFL and the control group were compared using the Wilcoxon rank sum test and found no statistically significant differences in Shannon index (p = 0.767), Simpson index (p = 0.999), and Chao richness (p = 0.809). Furthermore, the alpha diversity indices between geographical location were compared using the Kruskal–Wallis test and found no statistically significant differences in Shannon index (p = 0.571), Simpson index (p = 0.059), and Chao richness (p = 0.201). Boxplots of all diversity indices for both FFL status and geographical location are outlined in Fig. 2.

Boxplots overlayed with all data points of alpha diversity index measure estimates using Shannon Evenness (a), Simpson Diversity (b), and Chao Richness (c) between FFL status and geographical locations groupings. Alpha diversity indices between groups did not show statistical significance (p > 0.05).
Beta diversity
Based on the Bray–Curtis and Jaccard PCoA plots (Fig. 3), no distinct clusters were visually evident between FFL horses and healthy controls. This was confirmed through ANOSIM and PERMANOVA analyses, which indicate no significant difference in community composition and structure (Table 1, Bray–Curtis ANOSIM: p = 0.829, Jaccard ANOSIM: p = 0.638, Bray–Curtis PERMANOVA: p = 0.904, Jaccard PERMANOVA: p = 0.934). Additionally, we compared overall differences between sex and also found no statistical differences between groups (Table 1, Bray–Curtis ANOSIM: p = 0.556, Jaccard ANOSIM: p = 0.238, Bray–Curtis PERMANOVA: p = 0.411, Jaccard PERMANOVA: p = 0.207). The PCoA plots outlining the differences between sex using the Bray–Curtis and Jaccard indices is depicted in Supplementary Fig. 4. However, significant difference was observed when comparing overall geographical location (Table 1, Bray–Curtis ANOSIM: p = 0.002, Jaccard ANOSIM: p = 0.001, Bray–Curtis PERMANOVA: p = 0.001, Jaccard PERMANOVA: p = 0.001). Furthermore, relative differences between the Richmond and Abbotsford sample communities are evident in the PCoA plots (Fig. 3). This was confirmed statistically by Bray–Curtis pairwise ANOSIM (p = 0.015, R = 0.43), Jaccard pairwise ANOSIM (p = 0.015, R = 0.39), Bray–Curtis pairwise PERMANOVA (p = 0.015, R2 = 0.167, F = 2.422) and Jaccard pairwise PERMANOVA (p = 0.015, R2 = 0.103, F = 1.376) analysis. Other geographical location combinations did not indicate pairwise statistical significance for either ANOSIM or PERMANOVA analyses (p > 0.05).

Principal coordinate analyses (PCoA) of the microbial composition and diversity between FFL status and geographical locations using the Bray–Curtis index (a) and the Jaccard index (b). Circular icons indicate healthy control horses and triangular icons indicate those with FFL. Each colour represents a different geographical location.
FFL Status microbial composition comparison
The overall relative abundance at all taxonomic ranks were compared in FFL and healthy horses using the Kruskal–Wallis test. No statistically significant differences at any of the taxonomic levels (phylum: p = 0.877, class: p = 0.839, order: p = 0.464, family: p = 0.140, and genus: p = 0.106) between FFL horses and healthy controls indicated in Fig. 4a (red). Given prior findings suggested that the genera Alloprevotella and Bacillus have differences in FFL status, we further investigated their abundances. The Wilcoxon rank sum test was used to compare the genera and found no differences. Specifically, the median relative abundance of Alloprevotella accounted for 0.474 ± 1.130% in control horses and 0.737 ± 2.465% in FFL horses (p = 0.202, Fig. 4b), whereas Bacillus accounted for 0 ± 0.027% in control horses and 0.023 ± 0.019% in FFL horses (p = 0.327, Fig. 4c). The taxonomic lineages up to phylum of both genera were compared using the Wilcoxon rank sum test and found to have no statistical differences (p > 0.05).

Line plot of taxonomic classification levels (phylum to genus) and -log10(p) of the p-values from the Kruskal–Wallis tests for FFL status (red), sex (yellow), and geographical location (blue) (a). The dashed line represents the p-value threshold at 0.05. Boxplots comparing FFL Status at the genera Alloprevotella (b) and Bacillus (c). Boxplot comparing Sex at the genus Roseburia (d). Boxplots of relative abundances for Richmond (R), Langley (L), and Abbotsford (A) for the six taxa Bacteroidales I, Enterobacterales (f) Lachnospirales (g), MVP-15 (h), Saccharimonadales (i), and WCHB1-41 (j). The boxplots outline the p-values of all statistically significant (p < 0.05) differences as determined by the Wilcoxon rank sum test.
Sex microbial composition comparison
The overall relative abundance at all taxonomic levels were compared between male and female horses using the Kruskal–Wallis test. All levels, except for genus (p = 0.003) were non-significant (phylum: p = 0.817, class: p = 0.992, order: p = 0.266, and family: p = 0.051) as depicted in Fig. 4a (yellow). The Wilcoxon rank sum test was used to compare the relative abundance for each taxon. A statistically significant difference in only the genus Roseburia (p = 0.030) was found between male (0.04 ± 0.02%) and female (0.08 ± 0.03%) horses (Fig. 4d).
Geographical location microbial composition comparison
The overall differences in the relative abundance between geographical locations was assessed using the Kruskal–Wallis test. We found no statistical differences at the phylum (p = 0.832) and class (p = 0.234) taxonomic levels, however, there were statistical differences at the order (p = 0.001), family (p = 3.31 × 10−6), and genus (p = 1.05 × 10−8) taxonomic levels, as illustrated in Fig. 4a (blue). Further investigation on the individual taxa was done within the order taxonomic level using the Wilcoxon rank sum test. We found statistically significant differences in the relative abundance between Richmond, Langley, and Abbotsford at the orders Bacteroidales (Richmond vs Abbotsford: p = 0.017), Enterobacterales (Richmond vs Abbotsford: p = 0.011), Lachnospirales (Richmond vs Abbotsford: p = 0.026), MVP-15 (Richmond vs Langley: p = 0.038), Saccharimonadales (Richmond vs Langley: p = 0.035, Richmond vs Abbotsford: p = 0.001), and WCHB1-41 (Richmond vs Langley: p = 0.035). Boxplots outlining the relative abundance at each of the three locations for each taxon are depicted in Fig. 4e-j. Other comparisons between these three locations for the six taxa were not statistically significant (p > 0.05) as outlined in Table 2.
Discussion
To test the hypothesis that dysbiosis is associated with FFL, recent studies have compared the fecal microbial composition of horses with FFL against healthy animals. However, the results of these studies are conflicting. While Lausten et al.1 and Schoster et al.3 conclude no statistical significance in microbial composition between healthy horses and those with FFL, Lindroth et al.5 found statistical differences in the genera Alloprevotella and Bacillus. These three studies examining the association between FFL and the microbiome have exclusively been conducted in Europe. To our knowledge, our current study is the first to investigate the correlation between dysbiosis and FFL in Canadian horses. Our results support the conclusion that there is no significant difference between the microbiome of feces from FFL animals and controls. These findings support that dysbiosis is likely not associated with FFL. Our results show that fecal samples from FFL and control animals have similar microbial community composition and structure when visualized using PCoA. Statistical analysis using ANOSIM and PERMANOVA substantiate these results. Furthermore, no individual taxa were identified as having any significant differences in relative abundance using Wilcoxon rank sum hypothesis testing. These results strongly suggest no difference between the microbial composition of the FFL and healthy horses in this Canadian study. Our findings are consistent with Laustsen et al.1 who conducted a similar study while monitoring symptom severity after fecal transplants and Schoster et al.3 who compared FFL and healthy horses in both the autumn and spring seasons.
The current study did however identify differences in microbial composition when comparing sex and some of the geographical locations. Differences between male and female horses was only visible when comparing individual taxa at the genus level and not in alpha and beta diversity metrics. Specifically, the genus Roseburia was found to be more abundant in female horses. Genus Roseburia is known to be a butyrate-producing, gram-positive anaerobic bacteria39. Specifically, the species R. intestinalis has been labelled as a beneficial gut organism and is considered to have therapeutic role in various human diseases39. Differences in microbial communities between sex of horses have been previously reported, however, indicating different taxa40. Differences between geographical location were also evident from the ANOSIM and PERMANOVA analyses identifying differences between the Richmond and Abbotsford locations. The PCoA plots indicated that a relatively low percentage of variation among groups could be accounted for by geographic location, despite the apparent clustering of Richmond and Abbotsford groups. This was further confirmed when comparing taxa at each taxonomic rank, where we see differences in microbial composition at the order classification level and below. Specifically, the orders Bacteroidales, Enterobacterales, Lachnospirales, and Saccharimonadales had different relative abundances between Richmond and Abbotsford horses. We also visualize differences in the orders MVP-15, Saccharimonadales, and WCHB1-41 between Richmond and Langley. Richmond and Abbotsford are the farthest from each other within the study group, being 66 km apart and Richmond and Langley are the second farthest distance at 41 km apart from each other. Microbial community differences have also been reported in different geographical locations, however, at a much greater magnitude than the distances between the Richmond, Langley, and Abbotsford locations41.
Limitations of this study could provide sources of error that prevent the detection of small, yet significant, differences in the microbiota of horses with FFL. For instance, variance introduced from collecting samples from different geographical locations and the relatively small sample size. The microbial population differences found between the locations could be masking minor changes between horses with FFL and healthy controls. However, in comparison to other studies between gut microbiome and FFL in horses1,3,5, the locations that the samples analyzed in the present study were taken from a smaller land area. While the sample size is also relatively small, it is difficult to obtain more samples due to prevalence of the condition. FFL severity between cases may have also obscured the detection of differences as samples collected from horses with both acute and chronic FFL symptoms were grouped together. It should also be noted that Lindroth et al.5 found differences using a much larger sample size in comparison to this study and others that these results are consistent with1,3. Additionally, diet is known to be a factor that contributes to variations in microbial composition42. As horses were from different farms, their diets varied.
FFL could be a condition that is associated with multiple factors3. Some reports point towards stress4 or disturbances associated by gastric disorders43 or feeding practices42, in addition to microbial composition3,5. To assess these factors, it may be valuable to do a longitudinal study monitoring diet, stress, and health, collecting fecal matter over a longer period to observe changes in microbial composition over the course of the experiment. Microbial composition and diversity may not be the central factor that affects FFL, but rather simply a contributor towards the metabolomic environment that both the horse and microbiota generate within the hindgut. Recently, in human microbiome research, there has been a shift in focus from microbial composition and diversity to functional aspects of the microbiota44. In concordance with this, fecal transplants from clinically healthy donors containing microbes and metabolites were shown to reduce the severity of FFL symptoms in horses1. Transcriptomic analyses of both the horse and its microbiota might illuminate possible tissue-microbe interactions that lead to FFL. Recent studies have determined transcriptomic profiles in equine gastrointestinal tissues; however, no studies have involved transcriptomics of the equine microbiota45,46.
The results of this study indicated no differences in the microbial composition and diversity between healthy and FFL horses, consistent with other findings1,3. However, differences in the composition and diversity of samples between sex and the geographic locations Richmond and Abbotsford as well as Richmond and Langley were found. It is important to find the root cause of FFL, so that effective treatments can also be determined. Further research is needed to determine whether dysbiosis or other factors lead to FFL, and exactly what role the gut microbiota and its metabolites play.
Data availability
The sequencing data that supports the findings of this study are readily accessible on NCBI’s Sequence Read Archive repository at https://www.ncbi.nlm.nih.gov/sra/PRJNA1034666, BioProject accession number PRJNA1034666.
References
Laustsen, L. et al. Free faecal water: Analysis of horse faecal microbiota and the impact of faecal microbial transplantation on symptom severity. Animals 11, 2776 (2021).
Lindroth, K. M. et al. Differential defecation of solid and liquid phases in horses—A descriptive survey. Animals. 10(1), 76 (2020).
Schoster, A., Weese, J. S., Gerber, V. & Nicole Graubner, C. Dysbiosis is not present in horses with fecal water syndrome when compared to controls in spring and autumn. J. Vet. Intern. Med. 34, 1614–1621 (2020).
Kienzle, E. et al. Field study on risk factors for free fecal water in pleasure horses. J. Equine Vet. Sci. 44, 32–36 (2016).
Lindroth, K. M., Dicksved, J., Pelve, E., Båverud, V. & Müller, C. E. Faecal bacterial composition in horses with and without free faecal liquid: a case control study. Sci. Rep. 11, 4745 (2021).
Garber, A., Hastie, P. & Murray, J.-A. Factors influencing equine gut microbiota: Current knowledge. J. Equine Vet. Sci. 88, 102943 (2020).
Chaucheyras-Durand, F., Sacy, A., Karges, K. & Apper, E. Gastro-intestinal microbiota in equines and its role in health and disease: The black box opens. Microorganisms 10, 2517 (2022).
Faubladier, C., Chaucheyras-Durand, F., da Veiga, L. & Julliand, V. Effect of transportation on fecal bacterial communities and fermentative activities in horses: Impact of Saccharomyces cerevisiae CNCM I-1077 supplementation1. J. Animal Sci. 91, 1736–1744 (2013).
Weese, J. S. et al. Changes in the faecal microbiota of mares precede the development of post partum colic. Equine Vet. J. 47, 641–649 (2015).
Costa, M. C. et al. Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3–V5 region of the 16S rRNA gene. PLOS ONE 7, e41484 (2012).
Milinovich, G. J. et al. Fluorescence in situ hybridization analysis of hindgut bacteria associated with the development of equine laminitis. Environ. Microbiol. 9, 2090–2100 (2007).
Steelman, S. M., Chowdhary, B. P., Dowd, S., Suchodolski, J. & Janečka, J. E. Pyrosequencing of 16S rRNA genes in fecal samples reveals high diversity of hindgut microflora in horses and potential links to chronic laminitis. BMC Vet. Res. 8, 231 (2012).
Kuhl, J. et al. Changes in faecal bacteria and metabolic parameters in foals during the first six weeks of life. Vet. Microbiol. 151, 321–328 (2011).
Schoster, A., Staempfli, H. R., Guardabassi, L. G., Jalali, M. & Weese, J. S. Comparison of the fecal bacterial microbiota of healthy and diarrheic foals at two and four weeks of life. BMC Vet. Res. 13, 1–10 (2017).
du Sert, N. P. et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLOS Biol. 18, 3000410 (2020).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Bokulich, N. A. et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59 (2013).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 4, e2584 (2016).
Yilmaz, P. et al. The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucl. Acids Res. 42, D643–D648 (2014).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucl. Acids Res. 41, D590–D596 (2013).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90 (2018).
Katoh, K. & Standley, D. M. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLOS ONE 5, e9490 (2010).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/ (2023).
Posit team. Rstudio: Integrated development environment for R. Posit Software, PBC, Boston, MA. URL http://www.posit.co/ (2023).
McMurdie, P. J. & Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLOS ONE 8, e61217 (2013).
Oksanen, J. et al. Vegan: Community Ecology Package. R package Version 2.6–4. https://CRAN.R-project.org/package=vegan (2022).
Wickham, H. et al. Welcome to the Tidyverse. J. Open Source Softw. 4, 1686 (2019).
Shapiro, S. S. & Wilk, M. B. An analysis of variance test for normality (complete samples)†. Biometrika 52, 591–611 (1965).
Kruskal, W. H. & Wallis, W. A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 47, 583–621 (1952).
Wilcoxon, F. Individual comparisons by ranking methods. Biom. Bull. 1, 80–83 (1945).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Methodol. 57, 289–300 (1995).
Shannon, C. E. & Weaver, W. The Mathematical Theory of Communication (The University of Illinois Press, 1949).
Simpson, E. H. Measurement of diversity. Nature 163, 688–688 (1949).
Chao, A. Estimating the population size for capture-recapture data with unequal catchability. Biometrics 43, 783–791 (1987).
Bray, J. R. & Curtis, J. T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 27, 326–349 (1957).
McArdle, B. H. & Anderson, M. J. Fitting multivariate models to community data: A comment on distance-based redundancy analysis. Ecology 82, 290–297 (2001).
Clarke, K. R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18, 117–143 (1993).
Nie, K. et al. Roseburia intestinalis: A beneficial gut organism from the discoveries in genus and species. Front Cell Infect. Microbiol. 11, 757718 (2021).
Hu, D. et al. Effect of gender bias on equine fecal microbiota. J. Equine Vet. Sci. 97, 103355 (2021).
Ang, L. et al. Gut microbiome characteristics in feral and domesticated horses from different geographic locations. Commun. Biol. 5, 172 (2022).
Fernandes, K. A. et al. Faecal microbiota of forage-fed horses in New Zealand and the population dynamics of microbial communities following dietary change. PLOS ONE 9, e112846 (2014).
Ertelt, A. & Gehlen, H. Free fecal water in the horse—An unsolved problem. Pferdeheilkunde 31, 261–268 (2015).
Jandhyala, S. M. et al. Role of the normal gut microbiota. World J. Gastroenterol. 21, 8787–8803 (2015).
Coleman, M. C. et al. Non-invasive evaluation of the equine gastrointestinal mucosal transcriptome. PLOS ONE 15, e0229797 (2020).
Mach, N. et al. Mining the equine gut metagenome: poorly-characterized taxa associated with cardiovascular fitness in endurance athletes. Commun. Biol. 5, 1–15 (2022).
Funding
Natural Sciences and Engineering Research Council of Canada
Author information
Authors and Affiliations
Contributions
P.J.A. and C.C.K. planned the study and obtained financial support. L.E.J. managed the collection of fecal samples and P.J.A., and G.C.M. collected horse metadata. G.C.M. performed the DNA extractions from fecal samples. L.L.B. and R.J.W. conducted the 16S rRNA sequencing workflow. R.J.W. carried out the bioinformatics and biostatistics analyses. R.J.W. and P.J.A. interpreted the data. R.J.W. prepared all figures, tables, and drafted the main text. R.J.W., P.J.A., and L.L.B. drafted the manuscript and contributed to the discussion. All authors reviewed the manuscript before article submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wester, R.J., Baillie, L.L., McCarthy, G.C. et al. Dysbiosis not observed in Canadian horses with free fecal liquid (FFL) using 16S rRNA sequencing. Sci Rep 14, 12903 (2024). https://doi.org/10.1038/s41598-024-63868-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-63868-1
- Springer Nature Limited