
Abstract
Pancreatic cancer (PC) is one of the most common malignant tumors of the digestive tract and has a very high mortality rate worldwide. Different PC patients may respond differently to therapy and develop therapeutic resistance due to the complexity and variety of the tumor microenvironment. The Eph/ephrin signaling pathway is extensively involved in tumor-related biological functions. However, the key function of the Eph/ephrin signaling pathway in PC has not been fully elucidated. We first explored a pan-cancer overview of Eph/ephrin signaling pathway genes (EPGs). Then we grouped the PC patients into 3 subgroups based on EPG expression levels. Significantly different prognoses and tumor immune microenvironments between different subtypes further validate Eph/ephrin's important role in the pathophysiology of PC. Additionally, we estimated the IC50 values for several commonly used molecularly targeted drugs used to treat PC in the three clusters, which could help patients receive a more personalized treatment plan. Following a progressive screening of optimal genes, we established a prognostic signature and validated it in internal and external test sets. The receiver operating characteristic (ROC) curves of our model exhibited great predictive performance. Meanwhile, we further validated the results through qRT-PCR and immunohistochemistry. Overall, this research provides fresh clues on the prognosis and therapy of PC as well as the theoretical groundwork for future Eph/ephrin signaling pathway research.
Similar content being viewed by others


Introduction
Pancreatic cancer (PC) is a common malignant tumor of the digestive tract characterized by aggressiveness and poor prognosis1. Numerous studies have largely established that the mechanisms driving its onset and progression are primarily genetic in nature. Despite continued advances in early detection and therapy, patients have a 5-year survival rate of approximately 12%2. The high mortality rate of PC is mainly due to the lack of biomarkers for early detection and the poor efficacy of chemotherapy. Chemotherapy and surgical excision are the main treatments for PC. Nevertheless, most individuals have metastases by the time of diagnosis. Recently, advanced PC has been treated clinically with molecularly targeted drug therapy. And probably due to tumor heterogeneity, no single molecularly targeted therapy has achieved consistent success3. Because of this, it is very important to find new biomarkers to predict the outcome of PC patients and give them personalized treatment.
Eph/ephrin signaling pathway is essential to tumor biology and is the focus of studies on cancer targeted therapy. Eph receptor is the most common subfamily of receptor tyrosine kinases, which triggers signal propagation through interactions with ephrin ligands that are also present on the cell surface4,5. Synaptic plasticity, neuron regeneration, cancer progression, immune system, lymphangiogenesis, angiogenesis, and some key physiologic functions of the pancreas are all influenced by Eph receptors and ephrins in adults6,7. Abnormal expression of Eph and ephrin has been found in several types of cancer in recent years, like neuroblastoma8, breast carcinomas9,10,11, lung cancer12, gastric cancer13,14, prostate cancer15, ovarian cancer16, colon cancer17, PC7,18, and melanoma19. Eph receptors are becoming recognized as master modulators capable of enhancing or suppressing the activity of the cancer signaling network, and Eph family members have been recognized as potential anticancer targets. Meanwhile, Eph and ephrin expression have the potential to be a strong predictor of prognosis and treatment sensitivity. For instance, elevated EPHA2 expression could promote susceptibility to dasatinib and resistance to trastuzumab20,21,22. Therefore, the development of PC risk stratification tools based on the Eph/ephrin signaling pathway genes (EPGs) is promising.
We first analyzed the genomic variations and expression profiles of the EPGs across different cancer types to explore the characterization of the Eph/ephrin signaling pathway in tumors. To explore EPG-related molecular subtypes in PC patients, we categorized PC samples into different subtypes according to the expression level of EPG and investigated their association with prognosis, immune microenvironment, and drug sensitivity. To further identify the genes that characterize the different subtypes, we performed differential expression analysis among the PC subtypes. Differentially expressed genes (DEGs) were then filtered by tumor and normal tissue difference analysis and prognostic analysis. Finally, a novel signature including 6 genes was constructed using LASSO-Cox regression and expression status of model genes were then tested by qRT-PCR and immunohistochemistry. Overall, this research provides fresh clues on the prognosis and therapy of PC as well as the theoretical groundwork for future Eph/ephrin signaling pathway research.
Methods
Data collection and processing
The TCGA, GEO, ICGC, ArrayExpress, and GTEx databases were utilized to collect gene expression data and clinical annotations for the datasets. Individuals that lacked survival information were not included in the analysis. Batch effects were corrected by the “SVA (version 3.42.0)” package23. We combined the GSE5749524, GSE2873525,26, GSE6245227, E-MTAB-613428, and TCGA-PAAD (https://portal.gdc.cancer.gov/) sets, which included 635 PC individuals, and randomly divided into a training set (319 samples) and an internal validation set (316 samples) for future research. Meanwhile, we combined the ICGC-CA and ICGC-AU for an external test, with 295 samples serving as the external validation set29. The Molecular Signatures Database was used to obtain the EPGs30, 37 EPGs were found in 930 PC individuals were included in this analysis. R (version 4.13) and “Bioconductor (version 3.14)” package were used for this analysis31.
Pan-cancer analysis
The variations of EPGs in different cancers are not well summarized. Copy number variant (CNV), single nucleotide variation (SNV), and DNA methylation data taken from the TCGA were analyzed and visualized for a pan-cancer analysis of variants of EPGs. Additionally, differential mRNA expression (FoldChange > 1, FDR < 0.05) and univariate Cox regression analysis were applied to study the expression status and prognostic values of EPGs in multiple human tumors using “limma” and “survival” packages32.
Cluster analysis according to Eph/ephrin scores
Eph/ephrin pathway enrichment scores were calculated by ssGSEA. We calculated high or low expression thresholds for each gene in PC samples to determine their expression states. Subsequently, the obtained expression state matrix was used for clustering and visualization. Both rows and columns were clustered using the Ward.D method, and the samples were categorized into three subtypes with reference to previous literature33,34,35. The “kruskal.test” function was then conducted to test the differences in Eph/ephrin scores among the three clusters. We utilized the “ggpubr” package to show the score levels of different clusters. According to the Eph/Ephrin scores of the three clusters, we named them as Eph/ephrin-inactive cluster (C3), Eph/ephrin-normal cluster (C1), and Eph/ephrin-active cluster (C2). Finally, KM survival curves were generated using the “survminer (version 0.4.9)” package to compare the prognostic differences among the three clusters by log rank test.
Drug sensitivity analysis
“OncoPredict (version 0.2)” is the package that predicts in vivo medication responses and biomarkers in cancer patients using data from cell line screening. By combining the GDSC database and the “oncoPredict (version 0.2)” package, we assessed the variations in common chemotherapeutic and targeted drug sensitivity among various subtypes36.
Associations of the Eph/ephrin scores with the immune cell infiltration (ICI)
The set of 29 immune-related gene sets were evaluated using ssGSEA. The R programs “ggplot2” and “dplyr” were used to create a heatmap showing the link between EPGs and ICI, with statistical analysis using Spearman's coefficient. In addition to EPGs, the relation between the Eph/ephrin signaling pathway scores and immune cells was determined based on the results of ssGSEA. Finally, scatter plots were constructed by “ggscatterstats” function of the “ggstatsplot (version 0.11.1)” package to reveal the correlation between the most relevant classical immune cell populations and Eph/ephrin signaling pathway scores.
Discrepancy in ICI and immune checkpoint genes (ICGs) expression across different subtypes
Using transcriptome data, the “ESTIMATE” package was utilized to assess the difference in immune characteristics among three PC clusters37. The infiltration abundance of each immune cell type was studied in depth using various immunocorrelation algorithms (including TIMER、QUANTISEQ、MCPCOUNTER、XCELL、EPIC, CIBERSORT). The “kruskal.test” in R was utilized to compare the differences in ICI abundance and expression levels of common ICGs between the different subtypes, showing only statistically significant findings (p < 0.05).
Construction of the prognostic model
We used the “limma” package to find DEGs among three PC subtypes (FDR < 0.05). Then 160 DEGs that were also differentially expressed between tumor and normal tissues for subsequent analysis (|log2(FC)|> 1, FDR < 0.05) were included in the follow-up analysis. Later, these genes underwent univariate Cox regression analysis with a filtering criterion of p < 0.05. The LASSO regression analysis was conducted to address overfitting by “glmnet” package38. Given that Cox regression does not require assumptions about the shape of the baseline risk function, we further create signatures through multivariate Cox regression analysis and calculate risk scores by “predict” R function. To assess the predictive power of the model and to ensure that the high- and low-risk groups were numerically balanced, individuals were categorized into high- and low-risk groups based on the median risk score of the training cohort.
The following analyses were performed on all cohorts for the internal and external tests of our model: (1) to visualize sample classification by t-distributed stochastic neighbor embedding (t-SNE) and principal component analysis (PCA) analysis; (2) to create a heatmap of gene expression using R's “pheatmap”; (3) KM approach was utilized to perform a survival analysis; (4) receiver operating characteristic (ROC) curves were created for evaluating the predictive performance of our signature.
The discrepancy of drug sensitivity, ICI, and ICG expression between different risk subgroups
Each PC patient's drug sensitivity was predicted using the R “OncoPredict (version 0.2)” package. Only agents that achieved statistical significance across all training and test groups were regarded as authentic and credible targeted agents. In addition, we compared the differences in ICI and ICGs in high- and low-risk groups by similar methods as above.
Isolation of total RNA with qRT-PCR and immunohistochemistry
The American Type Culture Collection supplied the HPDE6-C7 normal human pancreatic ductal cell line. Bxpc-3 and CFPAC-1 are two human PC cell lines that were supplied by our research laboratory. The HPDE6-C7 and BxPC-3 lines' cells were cultured in DMEM supplemented with 10% FBS. IMDM supplemented with 10% FBS was used to culture CFPAC-1 cells. TRIzol (Accurate Biotechnology) was used to extract total RNA from human PC cell lines. Reverse transcription was then performed on the RNA to produce cDNA using the Evo M-MLV RT Kit. The SYBR Green Premix Pro Taq HS qPCR Kit was used to quantify the expression levels of 6 genes. The RNA level expression was analyzed and quantified using the ΔΔCt method, and differences in the expression of target genes in normal and pancreatic cancer cell lines were compared by t-test. The primer sequences employed were as follows, which were obtained from Gene Pharma (Suzhou) Co., Ltd.: for human KRT17, 5′-AGGTGAAGATCCGTGACTGG-3′ (Forward), 5′-TCTTGTTCTGCAGCTCCTCA-3′ (Reverse); for human DSG2, 5′-ACGTAGAAGTTACGCGCATAAA-3′ (Forward), 5′-GGGTCACAATTCCTTCGTTAGTT-3′ (Reverse); for human LOXL2, 5′-CCATCCTCTCAACCTACCGC-3′ (Forward), 5′-GTCTTGCCCTCCTTCACCTC-3′ (Reverse); for human DCBLD2, 5′-AGCAAGGTGATGGATGTGGA-3′ (Forward), 5′-TGCGAACTCTCTCTCCCATC-3′ (Reverse); for human DKK1, 5′-ATAGCACCTTGGATGGGTATTCC-3′ (Forward), 5′-CTGATGACCGGAGACAAACAG-3′ (Reverse); for human LGR5, 5′-CACTTATGACCTGCCTCCCA-3′ (Forward), 5′-ACAGAGGAAAGATGGCAGCT-3′ (Reverse); for human GAPDH, 5′-GGTCTCCTCTGACTTCAACA-3′ (Forward), 5′-GTGAGGGTCTCTCTCTTCCT-3′ (Reverse).
Tissue samples from 10 PC patients who underwent surgery from January 2023 to September 2023 were collected from our hospital. The formalin-fixed paraffin-embedded PC tissues and paired paracancerous tissues of ten patients were subjected to immunohistochemical analysis. Primary antibodies were incubated with the tissue slices for a whole night at 4 °C, including DCBLD2 (PTG, 13168), DKK1 (PTG, 21112), DSG2 (PTG, 21880), KRT17 (Servicebio, GB12363), and LOXL2 (Servicebio, GB11849). Subsequently, the slices were treated for fifty minutes at room temperature with the matching secondary antibody. Lastly, representative pictures were collected using an Olympus scanning microscope, and the mean optical density (MOD) values of the cancer and controls were compared by t-tests. All methods were performed in accordance with the relevant guidelines and regulations.
Consent to participate and ethics approval
The Ethics Committee of Dalian Medical University's First Affiliated Hospital examined and authorized the investigations involving human subjects (ID: PJ-KS-KY-2023-482). Written informed consent was given by the participants to take part in this research.
Results
Pan-cancer landscape of the EPGs
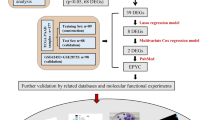
The current research is illustrated in the flow chart (Fig. 1). The function of the Eph/ephrin pathway in tumor development has not been clarified, and EPGs' whole-cancer characteristics are not well-summarized. Therefore, thorough research of EPGs in a variety of tumors from the perspectives of CNV, SNV, mRNA expression features, prognostic performance, and DNA methylation is necessary. WASL, MMP9, EPHB3, EPHB4, LIMK1, EPHA1, ACTB, and other genes exhibit an extensive expansion of CNV in multiple cancers. EPHA2, TIAMI, AP2A2, and EPHA7 exhibited extensive defects (Fig. 2A). Moreover, most EPGs in UCEC have a greater degree of SNV (Fig. 2B). We also discovered that all EPGs were expressed differently in at least one cancer type (Fig. 2C). Then we developed a survival landscape of EPGs according to TCGA's connection between patient survival and gene expression. It can be found that most of the EPGs act as protective genes in LUSC and KIRC and as risky genes in PC, ACC, LIHC, KICH, and other types of tumors (Fig. 2D). Considering that aberrant DNA methylation in promoters is also closely related to tumorigenesis39, we analyzed DNA methylation of EPGs in different tumors. We observed that EPGs showed mostly hypomethylation in 20 cancer types (Fig. 2E).

The present study's workflow.

Panoramic view of the Eph/ephrin signaling pathway genes (EPGs) in pan-cancer. (A) The frequency of copy number variant (CNV) for each gene in each cancer type is represented as a histogram. (B) The heatmap showed the single nucleotide variation (SNV) of EPGs. (C) The heatmap depicts the fold change and FDR of EPGs in different tumors. (D) The survival landscape for EPGs was visualized using a heatmap. Risky (HR > 1 and p < 0.05) and protective (HR < 1 and p < 0.05). (E) The heatmap shows the differential methylation of EPGs in cancers.
Classification of PC into three subtypes based on Eph/ephrin scores
To further analyze the correlation between EPGs and PC, 930 PC individuals were split into three groups according to the EPGs' expression levels (Fig. 3A). The violin plot displayed the enrichment scores for 3 groups (C2 > C1 > C3, p < 2.2e − 16) (Fig. 3B). In addition, KM analysis on three clusters showed significantly varied PC survival rates (Fig. 3C). Cluster 3 had a much better prognosis than clusters 1 and 2, which is identical to the situation represented in Fig. 2D. Most EPGs are risky in PC, and the patients with low Eph/ephrin scores had longer survival.

Cluster analysis according to the Eph/ephrin scores. (A) The heatmap depicts three clusters of gene data. (B) The enrichment score of three clusters is shown by a violin plot. (C) Survival curves showed differences in prognosis among the three clusters.
Differences in drug sensitivity in different subtypes of PC patients
Considering that chemotherapy and molecularly targeted therapies are now commonly used to treat PC, we assessed the drug sensitivity of these three molecular subtypes with the help of the “OncoPredict” package. The results demonstrated the following differences in drug sensitivity among Eph/ephrin clusters: 5-Fluorouracil: C2 > C3 (p = 1.9e − 06); cisplatin: C1 > C2 (p = 4.2e − 08); gemcitabine: C1 > C2 (p = 0.0069); irinotecan: C3 > C2 (p = 1e − 09); oxaliplatin: C3 > C2 (p = 2.1e − 07); paclitaxel: C2 > C3 (p = 7.1e − 05); camptothecin: C3 > C2 (p = 1.3e − 05); dasatinib: C1 > C3 (p = 8.5e − 14); sorafenib: C3 > C2 (p = 9.6e − 11) (Fig. 4).

The plot shows the estimated IC50 for common chemotherapeutic drugs.
Associations of the Eph/ephrin scores with the immune cells
The prognosis of cancer is associated with immune cells, which are key components of the tumor microenvironment (TME) and effective anticancer treatment targets40. The Eph/ephrin pathway is also important in controlling the tumor immune microenvironment41. To learn more about how the Eph/ephrin pathway and immunity are related, we conducted a relevance analysis between the EPGs and ICI in PC patients (Fig. 5A). Most EPGs were found to be associated with ICI. Among them, genes with significant positive correlation included TIAM1, SDC2, MMP9, ARPC5, ACTR3, LIMK1, and those with significant negative correlation included EFNA2, EFNA3, EFNA5, EPHA1, EPHB4. Next, the relationship between Eph/ephrin score and ICI was analyzed (Fig. 5B). It was found to have a positive relationship with macrophage infiltration, parainflammation, and APC co-stimulation, but a negative relationship with Tfh cells (Fig. 5C–F).

The relationships between EPGs and immune cell infiltration (ICI). (A) The heatmap depicts the relationship between the EPGs and ICI. (B) The relationship between the Eph/ephrin score and different immune cells. (C–F) The relationship between the Eph/ephrin score and four immune-infiltration-related substances including macrophage, parainflammation, APC co-stimulation, and Tfh.
Discrepancy in ICI and ICGs expression across three subtypes of PC patients
We then compared differences in the immune microenvironment among three subtypes. The findings demonstrated that C3 had lower levels of immuneScore, stromalScore, and estimateScore than C1 and C2 (Kruskal–Wallis test, p < 2.2e − 16) (Fig. 6A). Meanwhile, we found that macrophages, cancer associated fibroblast, and myeloid dendritic cells show a lower expression in the C3 subgroup, while C1 exhibited higher levels (Fig. 6B). In addition, we also found the ICGs expression was significantly increased in the C1 (Fig. 6C).

Immune status in three molecular subtypes. (A) Comparison of immuneScore, stromalScore, and estimateScore among the three subtypes. (B) Discrepancy analysis of tumor ICI in different groups. (C) Comparison of immune checkpoint gene (ICG) expression among the three subtypes. (*p < 0.05; **p < 0.01; ***p < 0.001).
Establishment of the prognostic model consisting of six genes
We first analyzed the DEGs of the three PC subtypes, and a total of 8449 DEGs were obtained. Then we obtained 160 genes from them that were differentially expressed not only between the three PC subtypes but also between PC and normal individuals in the three datasets (Fig. S1A). Subsequently, the DEGs were subjected to univariate Cox regression analysis to obtain 80 genes with prognostic value for subsequent analysis. Then we employed LASSO regression to create a prediction signature to predict the prognosis of PC individuals. Eight genes were selected from 80 genes by the LASSO regression (Fig. S1B,C). Finally, six genes were preserved for conducting multivariate Cox regression analysis (Fig. S1D).
All PC individuals were further split into low- and high-risk subgroups according the median risk score. Samples with higher risk score had a higher chance of dying (Fig. 7A,B). Then, T-SNE and PCA results illustrated that low- and high-risk individuals could be effectively distinguished (Fig. 7C,D). The heatmap showed that the remaining five genes except LGR5 were more highly expressed in the high-risk group (Fig. 7E). The high-risk group had shorter overall survival (Fig. 7F). Meanwhile, the AUC of ROC curves for 1-year, 2-year, and 3-year survival were 0.713, 0.718, and 0.7 in the train cohort (Fig. 7G).

Evaluation of the risk score in the training cohort. (A) Based on the median risk score, the training cohort was separated into different groups. (B) Distributions of risk scores and the survival status. (C, D) t-distributed stochastic neighbor embedding (t-SNE) and principal component analysis (PCA) analysis showed that samples in high and low risk groups could be clearly distinguished. (E) The heatmap was generated to illustrate the expression levels of six model-related genes. (F) Survival curves illustrating significant differences in prognosis between high and low risk patients. (G) The receiver operating characteristic (ROC) curves illustrate the good predictive value of our model.
Internal and external verification of our model
To assess the signature's reliability and validity in the internal and external validation sets, risk scores were created and individuals were split into high- and low-risk subgroups based on the median risk scores of the training cohort (Figs. S2A and S3A). The distributions of survival status and risk scores were similar to the training set (Figs. S2B and S3B). Through t-SNE and PCA, it has been shown that two categories can be differentiated (Figs. S2C,D and S3C,D). Heatmaps revealed the presence of highly expressed genes (KRT17, DSG2, LOXL2, DCBLD2, and DKK1) and genes with low expression (LGR5) in the high-risk subgroup in both internal and external verification sets (Figs. S2E and S3E). In addition, high-risk individuals have a poorer prognosis in test sets (Figs. S2F and S3F). The ROC curves indicate that our signature's diagnostic value is satisfied (Figs. S2G and S3G).
In addition, Sankey diagram showed the relationship between molecular subtypes and risk subgroups (Fig. S4A). It can be found that the number of high- and low-risk individuals in C1 and C2 is similar, while C3 is mainly composed of low-risk patients. To further explore the prognostic value of the prognostic model in different EPH-related subtypes, we compared the prognostic differences between samples from high and low risk subgroups in different subtypes. Due to the small sample of C3 subtype and the majority of low-risk patients, only C1 and C2 were analyzed, and it could be found that the prognosis of high-risk patients was equally worse than that of the low-risk group in C1 and C2 (Fig. S4B,C).
Differences in drug sensitivity, ICI, and ICG expression between individuals in high- and low-risk subgroups
Similarly, we compared whether there were differences in the drug sensitivity of individuals in the high- and low-risk subgroups to the nine drugs mentioned above. The results revealed that low-risk individuals might benefit from camptothecin, cisplatin, gemcitabine, irinotecan, oxaliplatin, and sorafenib; however, high-risk populations might benefit from dasatinib in all sets (Fig. S5).
Several immune-related algorithms were used to investigate immune responses, which were displayed in heatmaps (Fig. 8A–C). After combining all of the variations in training and test cohorts between the two groups, we discovered that several anti-tumor immune cells had lower proportions in the high-risk subgroup, including B cells and CD4+ T cells. In addition, M1 macrophage expression was upregulated in the high-risk group. Because ICGs are so important in immunotherapy, we then studied the differences in ICG expression between the two subgroups. CD70, TNFSF4, CD80, CD274 and PDCD1LG2 were upregulated in the high-risk subgroup, while JAK2 and CD160 were reversed (Fig. 8D–F).

ICI and ICGs differences between high and low risk subgroups. (A–C) The ICI distribution landscape in training, internal and external validation cohorts. (D–F) ICGs differential expression between high and low risk subgroups in three cohorts. (*p < 0.05; **p < 0.01; ***p < 0.001).
Verification of six signature genes by qRT-PCR and immunohistochemistry
We found that six genes in our model were significantly upregulated in PC, according to the outcomes of the differential expression analysis (Fig. S6). We then used qRT-PCR to validate the expression patterns of these model genes in normal pancreatic cell lines and PC cell lines. We found the mRNA levels of DCBLD2, DKK1, DSG2, KRT17, and LGR5 were higher in at least one of the PC cell lines, while the opposite was true for LOXL2 (Fig. 9A).

Validation of the expression traits of model genes. (A) The qRT-PCR results demonstrated the expression levels of six model genes in pancreatic cancer cell lines and normal pancreatic cell lines. (B) Immunohistochemical images and the mean optical density (MOD) values of pancreatic tumors and normal tissues. (*p < 0.05; **p < 0.01; ***p < 0.001; ns, non-significant).
Considering that LGR5 has been widely demonstrated to stain significantly higher in PC tissues than in normal pancreatic tissues42,43, we performed immunohistochemical analyses of only the remaining five target genes. The levels of DCBLD2, DKK1, DSG2, KRT17, and LOXL2 proteins were determined by immunohistochemistry, and the MOD values were assessed. Figure 9B shows representative images of DCBLD2, DKK1, DSG2, KRT17, and LOXL2 protein levels, respectively. Similarly, the MOD values of DCBLD2, DKK1, DSG2, KRT17, and LOXL2 were significantly higher in PC tissues compared with the controls (Fig. 9B).
Discussion
PC is a type of gastrointestinal cancer that has a high fatality rate1. Over the last few years, improved treatments have improved the treatment and prognosis of PC patients, but the survival rate remains low. The Eph/ephrin signaling pathway is extensively involved in tumor-related biological functions. Intracellular signaling is triggered in a bidirectional way after ligand–receptor contact. Eph receptors and ephrin ligands are well-known for their roles in carcinogenesis, cancer development, and neovascularization in a variety of cancers. In addition, researches have shown that they could be potential novel anticancer targets44. However, there was a lack of a comprehensive study of the Eph/ephrin signaling pathway in PC.
In our study, we started by looking into genetic mutations in EPGs in different cancers. Then, we analyzed the changes in Eph/ephrin pathway expression and determined whether these genes were present as protective or risk genes in various forms of tumor. Results showed that the majority of EPGs were found to be risk genes in various cancers. Then we investigated the relationship between EPGs and PC, and classified 930 samples into 3 clusters according to the expression levels of EPGs. We found that C2 with high Eph/ephrin scores had a significantly worse prognosis than C1 and C3, which again suggests that the Eph/ephrin pathway may play an oncogenic role in PC.
Adjuvant chemotherapy after surgical resection is usually the preferred treatment option for early stage PC, and its therapeutic efficacy has been widely recognized45. However, the complexity and heterogeneity of TME in PC always leads to the development of therapeutic resistance and differential response to therapy in different patients. As a result, we confirm the sensitivity of several of the most commonly used medications in PC therapy. We found that the different clusters had distinct medication sensitivities, implying that patients could receive a more tailored treatment plan based on their Eph/ephrin-related gene expression patterns.
The immune cells within the TME perform a vital function in tumorigenesis, so we investigated the relationship between Eph/ephrin pathway score and ICI. We discovered that Eph/ephrin pathway score was positively correlated with macrophage infiltration, parainflammation, and APC co-stimulation, but negatively correlated with Tfh cells. TME is an intricate assembly of the tumor, immune, stromal, and extracellular components46. We found that C1 with a poorer prognosis may have a higher immune abundance. Furthermore, we found that ICGs are more highly expressed in the C1. The C1 cluster's strong immune cell infiltration may be a local compensatory phenomenon of active ICGs. Despite the high level of anticancer immunity in C1, high expression of ICG in C1 may inhibit immune cell function and prevent the body from generating an effective antitumor immune response, which could lead to tumor immune escape and a poor prognosis.
Molecular typing aims to classify tumors into different subtypes based on their molecular characteristics, helping to understand tumor heterogeneity and providing a basis for personalized treatment. However, molecular typing still has some shortcomings; it cannot accurately predict the prognosis of a specific patient, whereas prognostic models can be used as tumor-specific biomarkers to predict a patient's risk score and prognosis. Thus, we developed a prognostic signature comprised of 6 genes (i.e., LOXL2, DCBLD2, DKK1, DSG2, LGR5, and KRT17). LOXL2 has been shown to enhance tumor progression, invasion, and metastasis through both cell-autonomous and non-cell-autonomous mechanisms in many tumor entities47. DCBLD2 is intimately linked to tumor invasion and migration and is overexpressed in lung, colorectal, and glioblastoma malignancies48,49,50. A member of the DKK family, DKK1 is linked to aggressive characteristics and a worse prognosis in patients with PC51. It has been shown that DKK1 is associated with the accumulation of bone marrow-derived suppressor cells in PC and contributes to the suppression of anti-tumor T cell responses52. The dysregulation of DSG2 is thought to be associated with poor prognosis in various tumors53,54,55,56. In addition, it has been found that DSG2 deficiency promotes the tumorigenic behavior of PC cells57. LGR5 is considered as a marker for adult stem cells58. It is associated with tumorigenesis and metastasis and is a marker of predictive response to platinum-based drug treatment59. KRT17 belongs to the type I keratin family and is involved in the development of or associated with bad prognosis in various tumors60,61,62,63,64,65.
With the use of our signature, PC patients in all cohorts may be successfully divided into two subgroups, with the high-risk group having a worse prognosis than the low-risk group. Our signature has a high predictive performance, as indicated by the ROC curves. Following a thorough investigation of the quantity of immune cell infiltration, it was shown that the low-risk group had higher levels of various anti-tumor immune cells, such as B cells66, and T cells CD4+67. Because ICGs are so important in immunotherapy, we then studied the differences in ICGs expression between two subgroups. After combining differences in all sets, we found that CD70, TNFSF4, CD80, CD274, and PDCD1LG2 were upregulated in the high-risk subgroup, while CD40LG, JAK2, and CD160 were reversed. It is well known that cancer cells are capable of activating immunological checkpoint pathways with immunosuppressive properties68. Thus, these ICGs are expected to act as potential efficient therapeutic targets. Finally, we verified the expression of the model genes by qRT-PCR and immunohistochemistry. The experimental validation results were generally consistent with the results of our bioinformatics analysis. However, LOXL2 was downregulated in qRT-PCR results but upregulated in immunohistochemistry. We believe that this discrepancy may be caused by a variety of factors, including tumor heterogeneity, differences in the tumor microenvironment, epigenetic modifications, cell culture conditions, and differences in post-transcriptional regulatory mechanisms. The combined effect of these factors may lead to significant differences in gene expression patterns between cell lines cultured in vitro and in situ tumor tissues.
This is the first report to synthesize the role of Eph/ephrin signaling pathway in PC using bioinformatics and experimental validation. The shortcoming is that although we elucidated the relationship between Eph/ephrin signaling and PC prognosis, biological behaviors, tumor immune microenvironment, and drug sensitivity by constructing Eph/ephrin signaling-associated molecular subtypes and prognostic models, the dataset of the present study was retrospective and was obtained from publicly available databases. Therefore, this research should be validated in prospective clinical studies with large samples. Also because of the small sample size in the C3 subtype and the majority of the sample being low-risk patients, we only supplemented the analysis of the survival performance of the prognostic model in the C1 and C2 subtypes. Meanwhile, the lack of theoretical basis for dividing the sample according to the median risk score is based only on previous similar studies, and patients with very similar risk scores may be categorized differently depending on whether their values are slightly greater or less than the median, so further exploration is still needed for the cutoff values for high and low risk. Finally, more mechanistic experiments are needed to further investigate the role of Eph/ephrin signaling in the occurrence and development of PC.
Conclusion
Our research identified six genes (i.e., LOXL2, DCBLD2, DKK1, DSG2, LGR5, and KRT17) with the potential to serve as a novel biomarker of the progression and prognosis of PC by integrating a series of bioinformatics techniques. Based on Eph/ephrin scores, PC patients could be divided into 3 subgroups (Eph/ephrin-inactive, Eph/ephrin-normal, and Eph/ephrin-active) with distinct prognosis, immune characteristics, and drug sensitivity. Our study shed fresh light on the prognosis and therapy of PC, as well as provided the theoretical groundwork for future Eph/ephrin signaling pathway research.
Data availability
The datasets analysed during the current study are available in the TCGA (https://portal.gdc.cancer.gov/), GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE57495; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE28735; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE62452), ICGC (https://dcc.icgc.org), ArrayExpress (https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-6134), and GTEx (https://gtexportal.org) databases.
References
Vincent, A., Herman, J., Schulick, R., Hruban, R. H. & Goggins, M. Pancreatic cancer. Lancet 378, 607–620. https://doi.org/10.1016/S0140-6736(10)62307-0 (2011).
Halbrook, C. J., Lyssiotis, C. A., Pasca di Magliano, M. & Maitra, A. Pancreatic cancer: Advances and challenges. Cell 186, 1729–1754. https://doi.org/10.1016/j.cell.2023.02.014 (2023).
Chiorean, E. G. & Coveler, A. L. Pancreatic cancer: Optimizing treatment options, new, and emerging targeted therapies. Drug Des. Devel. Ther. 9, 3529–3545. https://doi.org/10.2147/DDDT.S60328 (2015).
Kullander, K. & Klein, R. Mechanisms and functions of Eph and ephrin signalling. Nat. Rev. Mol. Cell Biol. 3, 475–486. https://doi.org/10.1038/nrm856 (2002).
Murai, K. K. & Pasquale, E. B. ’Eph’ective signaling: Forward, reverse and crosstalk. J. Cell Sci. 116, 2823–2832. https://doi.org/10.1242/jcs.00625 (2003).
Pasquale, E. B. Eph-ephrin promiscuity is now crystal clear. Nat. Neurosci. 7, 417–418. https://doi.org/10.1038/nn0504-417 (2004).
Papadakos, S. P., Dedes, N., Gkolemi, N., Machairas, N. & Theocharis, S. The EPH/Ephrin system in pancreatic ductal adenocarcinoma (PDAC): From pathogenesis to treatment. Int. J. Mol. Sci. https://doi.org/10.3390/ijms24033015 (2023).
Adams, R. H. & Klein, R. Eph receptors and ephrin ligands. Essential mediators of vascular development. Trends Cardiovasc. Med. 10, 183–188. https://doi.org/10.1016/s1050-1738(00)00046-3 (2000).
Zelinski, D. P., Zantek, N. D., Stewart, J. C., Irizarry, A. R. & Kinch, M. S. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 61, 2301–2306 (2001).
Fox, B. P. & Kandpal, R. P. Invasiveness of breast carcinoma cells and transcript profile: Eph receptors and ephrin ligands as molecular markers of potential diagnostic and prognostic application. Biochem. Biophys. Res. Commun. 318, 882–892. https://doi.org/10.1016/j.bbrc.2004.04.102 (2004).
Berclaz, G. et al. Loss of EphB4 receptor tyrosine kinase protein expression during carcinogenesis of the human breast. Oncol. Rep. 9, 985–989 (2002).
Kinch, M. S., Moore, M. B. & Harpole, D. H. Jr. Predictive value of the EphA2 receptor tyrosine kinase in lung cancer recurrence and survival. Clin. Cancer Res. 9, 613–618 (2003).
Nakamura, R. et al. EPHA2/EFNA1 expression in human gastric cancer. Cancer Sci. 96, 42–47. https://doi.org/10.1111/j.1349-7006.2005.00007.x (2005).
Kataoka, H. et al. Expression profile of EFNB1, EFNB2, two ligands of EPHB2 in human gastric cancer. J. Cancer Res. Clin. Oncol. 128, 343–348. https://doi.org/10.1007/s00432-002-0355-0 (2002).
Guan, M., Xu, C., Zhang, F. & Ye, C. Aberrant methylation of EphA7 in human prostate cancer and its relation to clinicopathologic features. Int. J. Cancer 124, 88–94. https://doi.org/10.1002/ijc.23890 (2009).
Landen, C. N., Kinch, M. S. & Sood, A. K. EphA2 as a target for ovarian cancer therapy. Expert Opin. Ther. Targets 9, 1179–1187. https://doi.org/10.1517/14728222.9.6.1179 (2005).
Batlle, E. et al. EphB receptor activity suppresses colorectal cancer progression. Nature 435, 1126–1130. https://doi.org/10.1038/nature03626 (2005).
Iiizumi, M. et al. EphA4 receptor, overexpressed in pancreatic ductal adenocarcinoma, promotes cancer cell growth. Cancer Sci. 97, 1211–1216. https://doi.org/10.1111/j.1349-7006.2006.00313.x (2006).
Hafner, C., Becker, B., Landthaler, M. & Vogt, T. Expression profile of Eph receptors and ephrin ligands in human skin and downregulation of EphA1 in nonmelanoma skin cancer. Mod. Pathol. 19, 1369–1377. https://doi.org/10.1038/modpathol.3800660 (2006).
Zhuang, G. et al. Elevation of receptor tyrosine kinase EphA2 mediates resistance to trastuzumab therapy. Cancer Res. 70, 299–308. https://doi.org/10.1158/0008-5472.CAN-09-1845 (2010).
Huang, F. et al. Identification of candidate molecular markers predicting sensitivity in solid tumors to dasatinib: Rationale for patient selection. Cancer Res. 67, 2226–2238. https://doi.org/10.1158/0008-5472.CAN-06-3633 (2007).
Wang, X. D. et al. Identification of candidate predictive and surrogate molecular markers for dasatinib in prostate cancer: Rationale for patient selection and efficacy monitoring. Genome Biol. 8, R255. https://doi.org/10.1186/gb-2007-8-11-r255 (2007).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. https://doi.org/10.1093/bioinformatics/bts034 (2012).
Chen, D.-T. et al. Prognostic fifteen-gene signature for early stage pancreatic ductal adenocarcinoma. PloS One 10, e0133562. https://doi.org/10.1371/journal.pone.0133562 (2015).
Zhang, G. et al. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer. Clin. Cancer Res. 19, 4983–4993. https://doi.org/10.1158/1078-0432.CCR-13-0209 (2013).
Zhang, G. et al. DPEP1 inhibits tumor cell invasiveness, enhances chemosensitivity and predicts clinical outcome in pancreatic ductal adenocarcinoma. PloS One 7, e31507. https://doi.org/10.1371/journal.pone.0031507 (2012).
Yang, S. et al. A novel MIF signaling pathway drives the malignant character of pancreatic cancer by targeting NR3C2. Cancer Res. 76, 3838–3850. https://doi.org/10.1158/0008-5472.CAN-15-2841 (2016).
Puleo, F. et al. Stratification of pancreatic ductal adenocarcinomas based on tumor and microenvironment features. Gastroenterology 155, 1999-2013.e1993. https://doi.org/10.1053/j.gastro.2018.08.033 (2018).
Zhang, J. et al. The International Cancer Genome Consortium Data Portal. Nat. Biotechnol. 37, 367–369. https://doi.org/10.1038/s41587-019-0055-9 (2019).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. 102, 15545–15550. https://doi.org/10.1073/pnas.0506580102 (2005)
Sepulveda, J. L. Using R and bioconductor in clinical genomics and transcriptomics. J. Mol. Diagn. https://doi.org/10.1016/j.jmoldx.2019.08.006 (2020).
Chen, C. et al. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. https://doi.org/10.1016/j.molp.2020.06.009 (2020).
Cui, Y. et al. Determination and characterization of molecular heterogeneity and precision medicine strategies of patients with pancreatic cancer and pancreatic neuroendocrine tumor based on oxidative stress and mitochondrial dysfunction-related genes. Front. Endocrinol. (Lausanne) 14, 1127441. https://doi.org/10.3389/fendo.2023.1127441 (2023).
Sun, J., Chen, F. & Wu, G. Role of NF-κB pathway in kidney renal clear cell carcinoma and its potential therapeutic implications. Aging 15, 11313–11330. https://doi.org/10.18632/aging.205129 (2023).
Xie, D., Wang, S., Jiang, B., Li, G. & Wu, G. The potential value of the Purinergic pathway in the prognostic assessment and clinical application of kidney renal clear cell carcinoma. Aging 16, 246–266. https://doi.org/10.18632/aging.205364 (2024).
Maeser, D., Gruener, R. F. & Huang, R. S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. https://doi.org/10.1093/bib/bbab260 (2021).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612. https://doi.org/10.1038/ncomms3612 (2013).
Guo, C. et al. The landscape of gene co-expression modules correlating with prognostic genetic abnormalities in AML. J. Transl. Med. 19, 228. https://doi.org/10.1186/s12967-021-02914-2 (2021).
Shen, H. & Laird, P. W. Interplay between the cancer genome and epigenome. Cell 153, 38–55. https://doi.org/10.1016/j.cell.2013.03.008 (2013).
Binnewies, M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550. https://doi.org/10.1038/s41591-018-0014-x (2018).
Ieguchi, K. & Maru, Y. Eph/Ephrin signaling in the tumor microenvironment. Adv. Exp. Med. Biol. 1270, 45–56. https://doi.org/10.1007/978-3-030-47189-7_3 (2021).
Kuraishi, Y. et al. Correlation of clinicopathological features and leucine-rich repeat-containing G-protein-coupled receptor 5 expression in pancreatic ductal adenocarcinoma. Pathol. Res. Pract. 215, 152623. https://doi.org/10.1016/j.prp.2019.152623 (2019).
Kamakura, M. et al. LGR5 expression and clinicopathological features of the invasive front in the fat infiltration area of pancreatic cancer. Diagn. Pathol. 17, 21. https://doi.org/10.1186/s13000-022-01203-w (2022).
Xi, H. Q., Wu, X. S., Wei, B. & Chen, L. Eph receptors and ephrins as targets for cancer therapy. J. Cell Mol. Med. 16, 2894–2909. https://doi.org/10.1111/j.1582-4934.2012.01612.x (2012).
Guenther, M. et al. The impact of adjuvant therapy on outcome in UICC stage I pancreatic cancer. Int. J. Cancer 151, 914–919. https://doi.org/10.1002/ijc.34044 (2022).
Belli, C. et al. Targeting the microenvironment in solid tumors. Cancer Treat Rev. 65, 22–32. https://doi.org/10.1016/j.ctrv.2018.02.004 (2018).
Alonso-Nocelo, M. et al. Macrophages direct cancer cells through a LOXL2-mediated metastatic cascade in pancreatic ductal adenocarcinoma. Gut 72, 345–359. https://doi.org/10.1136/gutjnl-2021-325564 (2023).
Feng, H. et al. EGFR phosphorylation of DCBLD2 recruits TRAF6 and stimulates AKT-promoted tumorigenesis. J. Clin. Investig. 124, 3741–3756. https://doi.org/10.1172/JCI73093 (2014).
Pagnotta, S. M. et al. Ensemble of gene signatures identifies novel biomarkers in colorectal cancer activated through PPARγ and TNFα signaling. PloS One 8, e72638. https://doi.org/10.1371/journal.pone.0072638 (2013).
Koshikawa, K. et al. Significant up-regulation of a novel gene, CLCP1, in a highly metastatic lung cancer subline as well as in lung cancers in vivo. Oncogene 21, 2822–2828 (2002).
Liu, D.-J. et al. The role of Dickkopf-1 as a potential prognostic marker in pancreatic ductal adenocarcinoma. Cell Cycle 16, 1622–1629. https://doi.org/10.1080/15384101.2017.1356510 (2017).
D’Amico, L. et al. Dickkopf-related protein 1 (Dkk1) regulates the accumulation and function of myeloid derived suppressor cells in cancer. J. Exp. Med. 213, 827–840. https://doi.org/10.1084/jem.20150950 (2016).
Qin, S. et al. DSG2 expression is correlated with poor prognosis and promotes early-stage cervical cancer. Cancer Cell Int. 20, 206. https://doi.org/10.1186/s12935-020-01292-x (2020).
Yang, T. et al. DSG2 expression is low in colon cancer and correlates with poor survival. BMC Gastroenterol. 21, 7. https://doi.org/10.1186/s12876-020-01588-2 (2021).
Xu, S. et al. Negative expression of DSG1 and DSG2, as prognostic biomarkers, impacts on the overall survival in patients with extrahepatic cholangiocarcinoma. Analyt. Cell. Pathol. (Amsterdam) 9831646, 2020. https://doi.org/10.1155/2020/9831646 (2020).
Han, C. P. et al. Desmoglein-2 overexpression predicts poor prognosis in hepatocellular carcinoma patients. Eur. Rev. Med. Pharmacol. Sci. 22, 5481–5489. https://doi.org/10.26355/eurrev_201809_15808 (2018).
Hütz, K., Zeiler, J., Sachs, L., Ormanns, S. & Spindler, V. Loss of desmoglein 2 promotes tumorigenic behavior in pancreatic cancer cells. Mol. Carcinog. 56, 1884–1895. https://doi.org/10.1002/mc.22644 (2017).
Song, Y. et al. Predicting tumor repopulation through the gene panel derived from radiation resistant colorectal cancer cells. J. Transl. Med. 21, 390. https://doi.org/10.1186/s12967-023-04260-x (2023).
Iżycka, N. et al. Cancer stem cell markers-clinical relevance and prognostic value in high-grade serous ovarian cancer (HGSOC) based on the cancer genome atlas analysis. Int. J. Mol. Sci. https://doi.org/10.3390/ijms241612746 (2023).
Kim, C. Y. et al. Proteomic analysis reveals overexpression of moesin and cytokeratin 17 proteins in colorectal carcinoma. Oncol. Rep. 27, 608–620. https://doi.org/10.3892/or.2011.1545 (2012).
Chivu-Economescu, M. et al. Knockdown of KRT17 by siRNA induces antitumoral effects on gastric cancer cells. Gastr. Cancer 20, 948–959. https://doi.org/10.1007/s10120-017-0712-y (2017).
Dong, M., Dong, Z., Zhu, X., Zhang, Y. & Song, L. Long non-coding RNA MIR205HG regulates KRT17 and tumor processes in cervical cancer via interaction with SRSF1. Exp. Mol. Pathol. 111, 104322. https://doi.org/10.1016/j.yexmp.2019.104322 (2019).
Sarlos, D. P., Yusenko, M. V., Peterfi, L., Szanto, A. & Kovacs, G. Dual role of KRT17: Development of papillary renal cell tumor and progression of conventional renal cell carcinoma. J. Cancer 10, 5124–5129. https://doi.org/10.7150/jca.32579 (2019).
Wang, Z. et al. Overexpression of KRT17 promotes proliferation and invasion of non-small cell lung cancer and indicates poor prognosis. Cancer Manag. Res. 11, 7485–7497. https://doi.org/10.2147/CMAR.S218926 (2019).
Li, D. et al. KRT17 functions as a tumor promoter and regulates proliferation, migration and invasion in pancreatic cancer via mTOR/S6k1 pathway. Cancer Manag. Res. 12, 2087–2095. https://doi.org/10.2147/CMAR.S243129 (2020).
Sarvaria, A., Madrigal, J. A. & Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell Mol. Immunol. 14, 662–674. https://doi.org/10.1038/cmi.2017.35 (2017).
Tay, R. E., Richardson, E. K. & Toh, H. C. Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 28, 5–17. https://doi.org/10.1038/s41417-020-0183-x (2021).
Darvin, P., Toor, S. M., Sasidharan Nair, V. & Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 50, 1–11. https://doi.org/10.1038/s12276-018-0191-1 (2018).
Author information
Authors and Affiliations
Contributions
This study's design, analysis, and paper preparation benefited from the efforts of Jifeng Liu, Qihang Yuan, and Xu Chen. Yao Yang and Yunshu Zhang were responsible for the collection of clinical samples and experimental analysis. Tong Xie contributed to the revision of the manuscript. Shuang Li and Bing Qi were involved in the design and data analysis. Prof. Dong Shang proposed the study conceptualization, reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, J., Yuan, Q., Chen, X. et al. Prognostic and therapeutic value of the Eph/Ephrin signaling pathway in pancreatic cancer explored based on bioinformatics. Sci Rep 14, 17650 (2024). https://doi.org/10.1038/s41598-024-68385-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68385-9
- Springer Nature Limited