
Abstract
Modifications to highly conserved developmental gene regulatory networks are thought to underlie morphological diversification in evolution and contribute to human congenital malformations. Relationships between gene expression and morphology have been extensively investigated in the limb, where most of the evidence for alterations to gene regulation in development consists of pre-transcriptional mechanisms that affect expression levels, such as epigenetic alterations to regulatory sequences and changes to cis-regulatory elements. Here we report evidence that alternative splicing (AS), a post-transcriptional process that modifies and diversifies mRNA transcripts, is dynamic during limb development in two mammalian species. We evaluated AS patterns in mouse (Mus musculus) and opossum (Monodelphis domestica) across the three key limb developmental stages: the ridge, bud, and paddle. Our data show that splicing patterns are dynamic over developmental time and suggest differences between the two mammalian taxa. Additionally, multiple key limb development genes, including Fgf8, are differentially spliced across the three stages in both species, with expression levels of the conserved splice variants, Fgf8a and Fgf8b, changing across developmental time. Our data demonstrates that AS is a critical mediator of mRNA diversity in limb development and provides an additional mechanism for evolutionary tweaking of gene dosage.
Similar content being viewed by others


Introduction
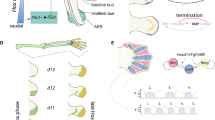
Understanding how development confers both robustness and evolvability is a major aim of many developmental studies1,2,3. In this regard, the tetrapod limb has been a great model to investigate the relationship between developmental mechanisms and skeletal morphology. Of particular interest is how gene expression is regulated to generate morphological diversity in the limb. The tetrapod limb is composed of three primary modules, the stylopod, zeugopod, and autopod, which are organized along proximodistal, anteroposterior, and dorsoventral axes. While these modules are conserved across species, the size, shape, and number of elements within each module varies among tetrapod taxa (Fig. 1A).

Tetrapod limb development (A) Isolated forelimbs from neonatal mouse (left) and chick (right) stained for bone (red) and cartilage (blue). Note differences in digit number, length, and orientation relative to the zeugopod. (B–D) Early limb development is characterized by 3 key stages: ridge, bud, and paddle. (B) The limb is initiated at precise locations along the body axis where retinoic acid (RA), T-Box transcription factors (TBX), and Fgf10 induce the expression of Fgf8 in the distal ectoderm (ridge stage). (C) In the bud stage, genes regulating limb patterning and proliferation are initiated downstream of SHH (turquoise) and AER-Fgfs (pink). (D) In the paddle stage, regionalization of limb modules is mediated by Hox patterning genes: Meis1/2 (stylopod, lavender), Hoxa11 (zeugopod, blue), and Hoxa13 (autopod, pink).
Tetrapod limb development proceeds through several key stages (Fig. 1B–D;4,5,6). In the ridge stage, limbs initiate at precise locations along the anterior–posterior axis extending from the flank, where Fgf10 induces Fgf8 expression distally in the apical ectodermal ridge (AER). In the bud stage, Fgf8 and other fibroblast growth factors secreted from the AER induce Sonic hedgehog (Shh) expression in the zone of polarizing activity (ZPA). Integrated signals from the AER and ZPA further induce a conserved set of gene regulatory networks (GRNs) that are critical for patterning and proliferation of the limb modules (Fig. 1C;4). In the paddle stage, the restricted regional expression of Hox genes defines limb module domains (Fig. 1D).
GRNs underlying these early developmental stages are highly conserved among tetrapods, while gene expression patterns at later developmental stages are more divergent7,8,9,10. It is widely hypothesized that modifications to these early patterning GRNs, specifically alterations to gene expression (spatial, temporal, level), have a significant role in generating morphological diversity6,7,8,9. Alterations to gene expression have been shown to result from changes to cis-regulatory elements such as promoters and enhancers11,12,13,14,15. Additionally, differences in epigenetic modification of regulatory elements of key patterning genes have also been shown to underlie differences in limb morphology16. Together, these and other studies indicate that transcriptional regulation of gene dosage (mRNA levels) early in limb development is a critical mediator of variation in limb morphology.
Alternative splicing (AS) is a post-transcriptional process where exons (and in some cases introns) from the same precursor-mRNA (pre-mRNA) transcript are joined in different combinations to yield distinct, but related, mRNA transcripts17. AS results in the production of different isoforms of a specific pre-mRNA molecule, which potentially impacts gene dosage in several different ways. First, splicing can affect the function of a protein through the inclusion or exclusion of key domains18,19. Second, AS can regulate protein levels through the inclusion of sequences that contribute to nonsense-mediated mRNA decay (NMD), transcript turnover, and/or mRNA localization20. Together, these data suggest several mechanisms through which modification of AS could impact gene dosage within GRNs, and thus contribute to the regulation of limb morphogenesis.
Although the role of AS in tissue patterning has not been widely investigated, it has been shown to be a key mediator of cell differentiation and fate decisions21. Splicing has been shown to affect lineage decisions in stem cell differentiation and reprogramming22,23, haematopoiesis24, myogenesis25, adipogenesis26, and neurogenesis27,28,29,30. AS has also been implicated in tissue function and identity31, and differences in splicing contribute to evolutionary differences in tissue identity32. The role of AS in tissue morphogenesis is less clear, as few studies have investigated splicing during embryogenesis31. Here, we compare patterns of AS in limb development of two different mammalian species, the house mouse (Mus musculus) and the gray short-tailed opossum (Monodelphis domestica).
Relative to mice and other eutherian mammals, marsupial neonates are precocial and must crawl into their mother’s pouch to continue development. Their accelerated forelimb development is mediated, in part, by a heterochronic shift in gene expression during development33. Both fore- and hindlimb fields are specified early in opossum compared to mouse, and Shh expression in the ZPA occurs prior to the outgrowth of the limb bud33. Interestingly, there is no AER in the developing limbs of Monodelphis domestica, only clusters of disorganized cells in the distal ectoderm34. Fgf signaling is retained in this region with a similar expression pattern, though at a slightly earlier stage compared to mouse33,34.
Our analysis in mouse and opossum limbs indicates a significant role for mRNA processing in limb development. We find that splicing patterns change over developmental time and differ between the two taxa. Different isoforms resulting from AS are predicted to contribute to both NMD of transcripts and distinct protein variants. Our data indicate the importance of post-transcriptional mechanisms contributing to gene dosage, which suggests an additional layer of complexity and modularity in developmental GRNs that may have been essential to the evolution of morphological variation.
Methods
Data collection and processing
Publicly available RNA-Seq data for mouse (Mus musculus)35 and opossum (Monodelphis domestica)36 limbs at the ridge, bud, and paddle stages (GSE71390 and GSE136445, respectively) were used to evaluate splicing. The amount of data differed between species but was relatively consistent within species (SupTable 1). Read quality was checked through FastQC37 and reads were trimmed to 70 bp in mouse replicates and 75 bp for opossum replicates using rMATS v3.2.538. Trimmed FASTQ files were aligned to mouse and opossum Ensembl reference genomes (GRCm39.107, ASM229v1.107, respectively) using STAR v2.7.10a39. Resulting BAM files were sorted by chromosomal coordinate and reads assigned to genomic features using FeatureCounts in the Subread package v1.6.2.40. Resulting read counts were used for differential expression analysis (DEA) following TMM normalization in EdgeR41. Differential expression analysis was conducted between the ridge and bud, and between the bud and paddle stages in both mouse and opossum. Statistically significant differential expression events were determined using a false discovery rate (FDR) cutoff of 0.05 and an absolute fold change > 1.5. Volcano plots were constructed with the R package ggplot242.
Differential splicing analysis and visualization
Differential splicing analysis was performed through rMATS v4.1.038 using default settings, except the detection of novel splice sites (–novelSS) was included. As with the DEA, differential splicing was performed between the ridge and bud and between the bud and paddle stages in both the mouse and opossum. A meta-file with the calculated delta percent spliced in (DPSI) values, p-value, and q-value from each analysis was generated through Jutils v1.043. Statistical significance of splicing events was determined using a q-value < 0.05. Heatmaps with Z-score normalization of the significant DPSI values for replicates of each stage for both species were created with Jutils v1.0 with default settings. Sashimi plots were generated with ggsashimi44, using BAM files aligned to the house mouse and opossum reference genomes (GRCm39.107 and ASM229v1.107, respectively) with a minimum read coverage of 10 except for mouse genes Fgf8, Fgfr2 and Fgfr3 which was reduced to 5. All other figures and plots were created in R45 with the ggplot2 package42 using the meta-file created by Jutils.
Spry1 alignment
Mouse Spry1 coding sequence was aligned to the opossum ortholog via NCBI Blastn.
Gene enrichment analysis
At each stage comparison in each species, genes with significant differential splicing events (q-value < 0.05) were extracted and combined to carry out a gene set enrichment analysis in DAVID46 using default settings. Significant functional enrichments were determined by a Benjamini–Hochberg FDR cutoff of 0.05 in the molecular function, cellular component, biological process, and KEGG pathway categories. Plots of the gene enrichment analysis results were done via the ggplot2 package in R.
Results
Splicing is dynamic during limb development
To investigate alternative splicing (AS) patterns in development, we took advantage of previously published RNA-seq data from the house mouse (Mus musculus) and gray short-tailed opossum (Monodelphis domestica) forelimbs35,36. These datasets consisted of samples isolated from ridge, bud, and paddle stages (see Fig. 1). The frequency and distribution of different categories of splicing in ridge, bud, and paddle stages were evaluated using replicate multivariate analysis of transcript splicing (rMATS) to calculate the ratio of reads including or excluding exons or introns, referred to as the percent spliced in index (PSI). AS events in these data were categorized by rMATS as skipped exons (SE), alternative 5′ or 3′ splice sites (A5SS and A3SS), mutually exclusive exons (MXE), and retained introns (RI; Fig. 2A). Clustering of splicing events compared between replicates based on the PSI at each developmental stage revealed that splicing patterns at each stage are distinct for both the mouse and the opossum (SupFig. 1). These data also indicate that developmental replicates are well matched. In mouse, 39.5% of genes are alternatively spliced in one or more of the 3 stages across limb development, while in opossum 76.7% of genes are alternatively spliced. In both mouse and opossum, we found that splicing is dynamic in early limb development, with changes in the genes, frequency, and types of splicing through development (SupFig. 2). Shifts in the distribution of PSI values are particularly notable in mutually exclusive exons (MXEs) and retained intron (RI) events. In mice, MXE events have high PSI values in the ridge stage then become more evenly distributed in the bud stage. In opossum, PSI values for RI are much lower on average than PSI values for other splicing events, however, both the total number of RI events and their average PSI value increase over developmental time. PSI distribution in RI events is also highly dynamic in the mouse. Additionally, the distribution of splicing event types for both species changes over developmental time (Fig. 2b). Most notably, the number of retained intron (RI) events decreases in mouse over developmental time, yet increases in the opossum over developmental time, shifting the distribution of RI events for each species (Fig. 2b; SupFig. 3).

Patterns of alternative splicing change over developmental time. (A) Overview of splicing types and calculation of changes in event frequency as defined in rMATS, which are presented as ΔPSI (delta percent spliced in). (B) Proportion of each event type between each stage for mouse (top) and opossum (bottom). (C) Box plots of the absolute values of ΔPSI by event type for mouse (top) and opossum (bottom) between the ridge and bud (left), and bud and paddle (right).
Next, we wanted to determine how AS impacts mRNA composition over developmental time. To do so, differential splicing was calculated between the ridge and bud stages, and bud and paddle stages for both mouse and opossum (Fig. 2). Differential splicing was determined by the “delta percent spliced in” (DPSI) value of each AS event, which represents the difference in the ratio of isoforms present between stages. In the mouse, there are 167 significant differential splicing events between the ridge and bud stages and 146 events between the bud and paddle stages (Table 1). In the opossum, there are 1265 significant differential splicing events between the ridge to bud stages and 1325 between the bud to paddle stages (Table 1). However, the absolute difference in PSI (DPSI) was much higher in mouse across all splicing types compared to opossum, while opossum exhibited a greater range of DPSI (Fig. 2C). Specifically, the mean absolute DPSI of all splicing events in the mouse was 0.347 in the ridge to bud stage and 0.380 in the bud to paddle stage while the opossum had a mean absolute DPSI of 0.153 in the ridge to bud stage, and 0.145 in the bud to paddle stage (Table 1).
It should be noted that differences between species are likely influenced by sequence coverage (Sup Table 1). With greater coverage, we might expect finding more differential splicing events of smaller effect (DPSI) due to potential increased discovery power particularly for low DPSI. To accommodate for this uncertainty, when looking at the highest DPSI that one expects to find with moderate coverage, we find a comparable number of events (see SupFig. 5). Notably, of all the splicing events with absolute DPSI above 0.6, mouse had nearly as many events (43) as opossum (48).
Splicing contributes to increased proteomic diversity
To determine potential consequences of AS, we first evaluated how differential splicing relates to changes in differential gene expression (DGE; Fig. 3; SupFig. 4). To visualize differential gene expression, volcano plots were generated using ggplot242; (Fig. 3A,B; SupFig. 4). Among differentially expressed genes, we noted that key members of the Fgf signaling pathway as well as Shh are differentially expressed in both mouse and opossum in the bud to paddle stage (Fig. 3A,B). In mouse, both Fgf8 and Shh are downregulated from the bud to paddle stage, while several other Fgf ligands are upregulated (Fig. 3A). We then quantified the number of genes between the ridge and bud, and bud and paddle stages that were either differentially expressed, differentially spliced, or both (Fig. 3C,D). In both species in both stage comparisons, there are more genes that are differentially expressed than differentially spliced (Fig. 3C,D). Further, we found that there are significantly more differentially spliced genes that are also differentially expressed than expected in both the mouse ridge to bud and bud to paddle comparisons (Fisher’s test p-value = 0.0337 and 0.0017, respectively) (Fig. 3C). Conversely, the opossum had fewer differentially spliced genes that are also differentially expressed than expected in both stage comparisons (Fisher’s test p-value = 5.405e−07 and 4.545e−07, respectively) (Fig. 3D). When filtering for the top differentially spliced genes (DPSI > 0.5), there was no significant difference between the observed versus expected number of genes that are also differentially expressed. However, there were 6 and 3 genes that were both differentially spliced and differentially expressed in the mouse ridge to bud and bud to paddle comparisons at this threshold, respectively, and 3 genes in this overlap in the opossum bud to paddle comparison (SupTable 2). Opossum ridge to bud comparison had no differentially spliced genes that were also differentially expressed at this threshold.

Several differentially spliced genes are also differentially expressed. Volcano plots of the differentially expressed genes between the bud and paddle stages in (A) mouse and (B) opossum. Significantly downregulated genes are shown in green and significantly upregulated genes are shown in purple. Significance was determined by a − log10(FDR) > 1.3 (horizontal line) and either a logFC > 0.6 or a logFC < − 0.6 (vertical lines). (C, D) Venn diagrams showing the relationship between differentially expressed genes (blue), genes that are differentially spliced (orange), and genes exhibiting both differential expression and splicing (white) in the ridge to bud stage (left), and bud to paddle stage (right) in (C) mouse and (D) opossum. The percentage of differentially spliced genes that are also differentially expressed are shown below. Only genes used in both the differential gene expression and differential splicing analyses were kept. Ridge-Bud differential gene expression is shown in supplementary Fig. 4.
Splicing impacts essential limb development genes
To investigate the impact of splicing on limb patterning GRNs, we created a rank plot of all significant splicing events for each species between each stage (Fig. 4A,B). We assigned a rank to each splicing event based on the absolute DPSI value of each event (SupTable 3). The rank of 1 is assigned to the splicing event with the lowest absolute DPSI and numbers increase as the absolute DPSI increases. Some genes may be ranked more than once if they exhibit more than one differential splicing event. While the top ranked genes differed between stage comparisons and species, various genes involved in limb development, in particular Fgf signaling pathway members, were recurrently differentially spliced (Fig. 4A,B).

Splicing targets genes involved in RNA and protein binding. (A, B) Rank plots of the significant absolute delta PSI [abs(ΔPSI)] values for the ridge-bud (left) and bud-paddle (right) in the (A) mouse and (B) opossum. The genes with the top 5 abs(ΔPSI) values are shown in red and genes involved in the Fgf signaling pathway are shown in blue. (C, D) Gene ontology analysis of significantly differentially spliced genes at both stage comparisons for (C) mouse and (D) opossum.
To investigate pathways significantly impacted by splicing during limb development, we ran a gene ontology (GO) enrichment analysis using all differentially spliced genes (Fig. 4C,D). In both species, differentially spliced genes showed significant enrichment in the categories of RNA binding and nuclear speckles, nuclear domains associated with splicing factors (Fig. 4C, D). Additionally, several other notable pathways were identified in the opossum involving mRNA processing via the spliceosome as well as specific components of the splicing machinery (Fig. 4D). Furthermore, differentially spliced genes in the opossum show enrichment in processes that could contribute to regulation of mRNA levels such as the ubiquitin protein ligase binding, RNA degradation, ubiquitin mediated proteolysis, endocytosis, and RNA degradation (Fig. 4D).
Fgf8 is alternatively spliced in mammalian limb development
Our analysis identified several members of the Fgf pathway, including Fgf8 and Spry1, as being differentially spliced in early limb development (Fig. 4A; Table 2). Notably, members of the Fgf pathway are among the most highly spliced genes (SupFig. 5). Fgf8 exhibits 3 significant splicing events in mouse between ridge and bud stages, with ranks of 55 (0.217; A3SS), 144 (0.583; SE), and 149 (0.633; SE) out of 167 total events (Fig. 4A). These events describe the change in inclusion of 33 bp at the 5′ end of exon 1D, the skipping of exon 1C for the Fgf8a isoform, and the skipping of exon 1C for the Fgf8b isoform, respectively (Fig. 5A). Notably, Fgf pathway members exhibit many splicing events in both species at both developmental stage comparisons (Fig. 4A).

Fgf8 splice variants are present in both mouse and opossum limbs. (A) Splicing of Fgf8 produces two key splice variants, Fgf8a and Fgf8b, which differ by the exclusion or inclusion of 33 basepairs of the exon 1D. (B, C) Sashimi plot of (B) mouse and (C) opossum Fgf8 exons 1b–1d at the ridge (yellow), bud (blue) and paddle (green) stages. Reads spanning exon junctions are shown by the bridges with read counts indicated. Fgf8a and Fgf8b are present in both mouse and opossum limbs at the ridge and bud stages.
To further investigate Fgf8 splicing, we generated sashimi plots, which visualize the number of reads (peaks) that span specific exon-exon junctions that contribute to AS of a gene (Fig. 5A,B). The Fgf8 gene has two major conserved splice variants: Fgf8a and Fgf8b47,48. These two variants differ by the inclusion of 33 base pairs at the 5′ end of exon 1D in Fgf8b (Fig. 5A). When comparing the bud with ridge stage in mouse (Fig. 5B), a relatively smaller peak is observed around exon 1D in the ridge and bud stages indicating reduced inclusion, which is also reflected in the rMATs data reporting a negative change in PSI from ridge to bud (Table 2). When comparing paddle with ridge or bud stage, there is also an overall reduction in peaks reflecting a reduction in overall gene expression, which is consistent with the canonical downregulation of Fgf8 in the paddle stage49, also detected in our differential expression analysis (Fig. 3C). Additionally, Fgf8e and Fgf8f., which also occur in humans, are identified in our dataset (Fig. 5B). Notably, we also find the Fgf8a and Fgf8b splice variants in the opossum (Fig. 5C), which have not previously been reported in this species. However, no differential splicing from ridge to bud or bud to paddle was observed in the opossum. Nonetheless, overall expression of Fgf8 is reduced from bud to paddle stage as observed in mouse.
Our data also indicates that Spry1 is differentially spliced in the mouse between the bud and paddle stages, while no differential splicing was observed in opossum (Fig. 4A,B). Therefore, we also generated sashimi plots of mouse Spry1 and the opossum Spry1 ortholog to visualize the skipped exon splicing event seen in our analysis (SupFig. 6A,B). Alternative splicing of mouse Spry1 involves the inclusion or exclusion of the second exon (SupFig. 6A). No event was found in the opossum Spry1 ortholog, likely due to the lack of the first two exons in the opossum gene annotation (Sup Fig. 6B). To confirm the absence of the first two exons of Spry1 in opossum, we aligned the nucleotide (SupFig. 6C) and protein (not shown) sequences. These data indicate that the opossum Spry1 ortholog coding sequence has an 80% sequence identity to the mouse and begins at position 625 of the mouse sequence. The protein sequences have 89% identity.
Discussion
AS is differentially regulated during limb development
Several mechanisms regulating gene expression at the transcriptional level have been identified to have important roles in tetrapod limb development and evolution. Here, we identify high levels of post-transcriptional mRNA processing in limb development. We also find that alternative splicing patterns change over developmental time and differ between mouse and opossum. Different isoforms resulting from AS are predicted to contribute to both NMD of transcripts and distinct protein variants in the developing limb. Our data indicate the importance of post-transcriptional mechanisms contributing to gene dosage, which may also have been a target for evolutionary modification.
Splicing as a mediator of morphological evolution
A prevailing hypothesis in evo-devo in general3, and limb development specifically6,7,8,9, is that alterations to cis-regulatory sequences of developmental regulatory genes is the central mechanism underlying the evolution of morphological diversity. A compelling aspect of this hypothesis is the modularity imposed by increasing complexity of cis-regulation3. Interestingly, comparisons of tissue-specific gene expression across vertebrates has shown conservation of gene expression despite extensive alterations to cis-regulatory sequences, which was argued to imply that changes in gene expression levels are unlikely to sufficiently explain phenotypic diversity among vertebrates50,51. An alternative, but also not mutually exclusive hypothesis, is that modifications to AS patterns in development contribute to species-specific phenotypes. Barbosa-Morais and colleagues (2012) have shown that splicing patterns have diverged more rapidly in vertebrate evolution than have alterations to gene expression levels. AS is also modular in that alterations to splicing can occur as cis-directed changes to splice recognition sites as well as through tissue-specific expression of trans-acting ribonucleoproteins18. The importance of AS to limb morphology is reflected in limb anomalies in spliceosomopathies52,53. Notably, changes in protein isoforms resulting from a single novel splice variant can cause major morphological changes, as has recently been shown for the Tbxt gene and tail loss54. Together, these data suggest that AS is an evolutionarily versatile process that can facilitate adaptation and morphological diversification.
AS is a key mediator of gene dosage
AS results in the production of different isoforms of a specific pre-mRNA molecule, which potentially impacts gene dosage at several levels. First, AS can regulate protein levels through the inclusion of sequences contributing to nonsense-mediated mRNA decay, transcript turnover, and/or mRNA localization20. Second, splicing can also affect the function of a protein through the inclusion or exclusion of key domains. For example, the exclusion of exons containing disorder domains are frequently observed among species-classifying splicing events18,19. Disordered domains lack stable 3D structures, allowing them to undergo induced fit structural changes55. The exclusion of disordered domains thus alters protein–protein and protein-DNA interactions. Additionally, exon exclusion may alter the ability of a protein to be phosphorylated, or have other modifications that affect protein function, especially of signaling factors56. Together, these data suggest several levels at which evolutionary modification of AS could have impacted gene dosage within GRNs regulating limb morphogenesis. Our data complements other recent analyses that suggest that post-transcriptional regulation of mRNA has important impacts on gene expression57,58.
A specific example that has important implications for limb development is the alternative splicing of Fgf8. Our data indicate that both isoforms, Fgf8a and Fgf8b, are present during limb development, with Fgf8a being more abundant in both mouse and opossum (Fig. 5A,B). This is relevant because the two protein isoforms, FGF8a and FGF8b, have distinct ERK signaling activity59, which is mediated, in part, by the inclusion of additional amino acids in FGF8b that increase its receptor binding affinity60. Further, evidence suggests that signaling from these isoforms initiates different downstream gene expression59,61. Therefore, exclusion of the 5′ end of exon 1D in Fgf8 is a mechanism that tweaks Fgf8 signaling activity. Our data indicate that alternative splicing of Fgf8 is differentially regulated during development in mouse, with the relative contribution of Fgf8a decreasing from the ridge to bud stage (Fig. 5B). Since FGF8a is the less efficient isoform, a developmental shift in the relative proportion of this isoform is predicted to have less of an impact on Fgf signaling than would a reduction in Fgf8b. Thus, differences in total Fgf8 expression (mRNA levels) may not adequately reflect differences in Fgf8 signaling during limb development.
Fgf signaling is tightly regulated at many different levels, including ligands, receptors, modulators, intracellular transducers, and gene regulatory targets62,63, and mutations impacting Fgf signaling activity have been associated with limb anomalies such as polydactyly64. It is therefore interesting that we also identified splice variants of Fgfr1, 2, and 3 in the opossum and Fgfr1 and Fgfr2 in the mouse (Fig. 4). Alternative splicing of the Fgfrs regulates binding affinity to different Fgf ligands and are mediators of cell-type specificity63,65. Finally, Fgf pathway members are among the genes with the largest DPSI, suggesting substantial splicing differences between developmental stages. These data additionally suggest that differential regulation of AS during development modifies Fgf signaling, or the effective dosage of Fgf8 during limb development.
Post-transcriptional regulation provides functional diversity in RBPs
Our data show that RNA binding proteins (RBPs), including proteins within the major spliceosome, are among the most enriched functions of differentially spliced genes (Fig. 4C,D). This is consistent with previous studies indicating that evolution of trans-acting RBPs play a critical role in generating proteome diversity18. Why would RNA binding proteins (or any protein category) exhibit selection for differential splicing? One possibility is that it serves as a mechanism to modify dosage as argued for Fgf8 above, where two protein isoforms may perform the same function but at different efficiencies. A second possibility is that AS allows for diversity in function, for example via the inclusion or exclusion of disordered domains, which is particularly important for RBPs66. Previous work has shown that cis-mediated species-specific differences in splicing of RBPs preferentially affect disordered regions rather than nucleic acid binding domains18,19. This is relevant to AS because disordered domains lack stable 3D structures, which allows them to undergo induced fit structural changes, where inclusion or exclusion of disordered domains enhances variability of protein–protein interactions55. RBPs are critical for AS, but are also heavily involved in many aspects of post-transcriptional gene regulation57. Therefore, selection for AS within RBP gene families may have been particularly important for tweaking gene expression via post-transcriptional mechanisms19.
Bioinformatics and big data in studies of development
One challenge in computational biology that is also reflected in our analysis is determining how to maximize large amounts of sequencing data while minimizing false positives due to noise. This is often explored by setting multiple thresholds during the analysis pipeline, such as filtering based on read coverage or fold change differences. In the context of differential AS, setting a minimum DPSI threshold can be used to reduce false positives and capture large differences in splicing. However, this potentially increases false negatives by eliminating splicing events with subtle yet relevant changes between samples or excluding genes with low expression. Here, we present splicing events regardless of DPSI, but nonetheless highlight that Fgf pathway members are among the genes with the largest DPSI (SupFig. 5), suggesting our approach detects biologically relevant signals. However, we acknowledge that differences between the species are affected, at some level, by differences in the datasets and require further validation.
Splicing differences between species
Our study detected a substantial difference in the total number of splicing events in each limb development stage comparison between mouse and opossum. This could be due to differences in sequence coverage, wherein the greater depth in sequencing for the opossum (> 3 × higher sequencing depth) likely increased the sensitivity in detecting transcription, alternative splicing, and differentiation among samples (SupTable 1) For example, we detected only 40% of genes being spliced among the three limb developmental stages in mouse compared to 77% for opossum. We also detected ten times more differential splicing among stages in opossum, however the majority of these events had small values of DPSI compared to events detected in mouse. In the mouse ridge-bud comparison, 6% of the events have abs(DPSI) values < 0.1, with the bud-paddle comparison having 4%. In the opossum, 50% and 53%, respectively, have abs(DPSI) values < 0.1. When we reach high abs(DPSI) values, the total number of differentially spliced events for both species is nearly equal (e.g., 43 for mouse and 48 for opossum at abs(DPSI) > 0.6), with mouse having more differential splicing at abs(DPSI) > 0.75 (13 vs. 12). Therefore, while we may be able to detect more subtle splicing changes (i.e., smaller isoform ratio differences) in opossum versus mouse, the species have a similar propensity for alternative splicing with large isoform ratio differences through limb development.
Data availability
The data used in this manuscript are publicly available RNA-Seq data for mouse (Mus musculus) [33] and opossum (Monodelphis domestica) [34] limbs at the ridge, bud, and paddle stages (GSE71390 and GSE136445, respectively).
References
Schwab, D. B., Casasa, S. & Moczek, A. P. On the reciprocally causal and constructive nature of developmental plasticity and robustness. Front. Genet. 9, 735 (2018).
Waddington, C. H. Canalization of development and genetic assimilation of acquired characters. Nature 183(4676), 1654–1655 (1959).
Carroll, S. B. Evo-devo and an expanding evolutionary synthesis: A genetic theory of morphological evolution. Cell 134(1), 25–36 (2008).
Zeller, R., Lopez-Rios, J. & Zuniga, A. Vertebrate limb bud development: Moving towards integrative analysis of organogenesis. Nat. Rev. Genet. 10(12), 845–858 (2009).
Allard, P. & Tabin, C. J. Achieving bilateral symmetry during vertebrate limb development. Semin. Cell Dev. Biol. 20(4), 479–484 (2009).
Zhu, M. & Tabin, C. J. The role of timing in the development and evolution of the limb. Front. Cell Dev. Biol. 11, 1135519 (2023).
Petit, F., Sears, K. E. & Ahituv, N. Limb development: A paradigm of gene regulation. Nat. Rev. Genet. 18(4), 245–258 (2017).
Gehrke, A. R. & Shubin, N. H. Cis-regulatory programs in the development and evolution of vertebrate paired appendages. Semin. Cell Dev. Biol. 57, 31–39 (2016).
Sears, K. et al. Timing the developmental origins of mammalian limb diversity. Genesis 56(1), 66 (2018).
Cooper, K. L. et al. Patterning and post-patterning modes of evolutionary digit loss in mammals. Nature 511(7507), 41–45 (2014).
Wray, G. A. The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 8(3), 206–216 (2007).
Schneider, I. & Shubin, N. H. The origin of the tetrapod limb: from expeditions to enhancers. Trends Genet. 29(7), 419–426 (2013).
Andrey, G. et al. A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science 340(6137), 1234167 (2013).
Kvon, E. Z. et al. Progressive loss of function in a limb enhancer during snake evolution. Cell 167(3), 633e11-642e11 (2016).
Booker, B. M. et al. Bat accelerated regions identify a bat forelimb specific enhancer in the HoxD locus. PLoS Genet. 12(3), e1005738 (2016).
Yakushiji-Kaminatsui, N. et al. Similarities and differences in the regulation of HoxD genes during chick and mouse limb development. PLoS Biol. 16(11), e3000004 (2018).
Nilsen, T. W. & Graveley, B. R. Expansion of the eukaryotic proteome by alternative splicing. Nature 463(7280), 457–463 (2010).
Barbosa-Morais, N. L. et al. The evolutionary landscape of alternative splicing in vertebrate species. Science 338(6114), 1587–1593 (2012).
Gueroussov, S. et al. Regulatory expansion in mammals of multivalent hnRNP assemblies that globally control alternative splicing. Cell 170(2), 324e23-339e23 (2017).
Braunschweig, U. et al. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 24(11), 1774–1786 (2014).
Fiszbein, A. & Kornblihtt, A. R. Alternative splicing switches: Important players in cell differentiation. Bioessays 39(6), 66 (2017).
Pritsker, M. et al. Diversification of stem cell molecular repertoire by alternative splicing. Proc. Natl. Acad. Sci. USA 102(40), 14290–14295 (2005).
Ohta, S. et al. Global splicing pattern reversion during somatic cell reprogramming. Cell Rep. 5(2), 357–366 (2013).
Chen, L. et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science 345(6204), 1251033 (2014).
Bland, C. S. et al. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Res. 38(21), 7651–7664 (2010).
Mei, B. et al. Only the large soluble form of preadipocyte factor-1 (Pref-1), but not the small soluble and membrane forms, inhibits adipocyte differentiation: Role of alternative splicing. Biochem. J. 364(Pt 1), 137–144 (2002).
Dillman, A. A. et al. mRNA expression, splicing and editing in the embryonic and adult mouse cerebral cortex. Nat. Neurosci. 16(4), 499–506 (2013).
Fiszbein, A. et al. Alternative splicing of G9a regulates neuronal differentiation. Cell Rep. 14(12), 2797–2808 (2016).
Mosca, S. et al. Human NDE1 splicing and mammalian brain development. Sci. Rep. 7, 43504 (2017).
Su, C. H. & Tarn, W. Y. Alternative splicing in neurogenesis and brain development. Front. Mol. Biosci. 5, 12 (2018).
Baralle, F. E. & Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 18(7), 437–451 (2017).
Merkin, J. et al. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 338(6114), 1593–1599 (2012).
Keyte, A. L. & Smith, K. K. Developmental origins of precocial forelimbs in marsupial neonates. Development 137(24), 4283–4294 (2010).
Doroba, C. K. & Sears, K. E. The divergent development of the apical ectodermal ridge in the marsupial Monodelphis domestica. Anat. Rec. 293(8), 1325–1332 (2010).
Sears, K. E. et al. The relationship between gene network structure and expression variation among individuals and species. PLoS Genet. 11(8), e1005398 (2015).
Onimaru, K. et al. Developmental hourglass and heterochronic shifts in fin and limb development. Elife 10, 66 (2021).
Andrews, S., FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Shen, S. et al. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 111(51), E5593–E5601 (2014).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29(1), 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30(7), 923–930 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1), 139–140 (2010).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (ed. S.-V., 2016).
Yang, G. et al. Jutils: A visualization toolkit for differential alternative splicing events. Bioinformatics 37(22), 4272–4274 (2021).
Garrido-Martin, D. et al. ggsashimi: Sashimi plot revised for browser- and annotation-independent splicing visualization. PLoS Comput. Biol. 14(8), e1006360 (2018).
Team, R.C., R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing (2022).
Sherman, B. T. et al. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50(W1), W216–W221 (2022).
Crossley, P. H. & Martin, G. R. The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development 121(2), 439–451 (1995).
Sunmonu, N. A., Li, K. & Li, J. Y. Numerous isoforms of Fgf8 reflect its multiple roles in the developing brain. J. Cell Physiol. 226(7), 1722–1726 (2011).
Moon, A. M. & Capecchi, M. R. Fgf8 is required for outgrowth and patterning of the limbs. Nat. Genet. 26(4), 455–459 (2000).
Chan, E. T. et al. Conservation of core gene expression in vertebrate tissues. J. Biol. 8(3), 33 (2009).
Necsulea, A. et al. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 505(7485), 635–640 (2014).
Beauchamp, M. C. et al. Spliceosomopathies and neurocristopathies: Two sides of the same coin?. Dev. Dyn. 249(8), 924–945 (2020).
Drake, K.D., et al. Minor spliceosome disruption causes limb growth defects without altering patterning. bioRxiv p. 2020.03.16.994384 (2020).
Xia, B. et al. On the genetic basis of tail-loss evolution in humans and apes. Nature 626(8001), 1042–1048 (2024).
Tompa, P. et al. Intrinsically disordered proteins: Emerging interaction specialists. Curr. Opin. Struct. Biol. 35, 49–59 (2015).
Singh, P. et al. The role of alternative splicing and differential gene expression in cichlid adaptive radiation. Genome Biol. Evol. 9(10), 2764–2781 (2017).
Corbett, A. H. Post-transcriptional regulation of gene expression and human disease. Curr. Opin. Cell Biol. 52, 96–104 (2018).
Franks, A., Airoldi, E. & Slavov, N. Post-transcriptional regulation across human tissues. PLoS Comput. Biol. 13(5), e1005535 (2017).
Sato, T. & Nakamura, H. The Fgf8 signal causes cerebellar differentiation by activating the Ras-ERK signaling pathway. Development 131(17), 4275–4285 (2004).
Olsen, S. K. et al. Structural basis by which alternative splicing modulates the organizer activity of FGF8 in the brain. Genes Dev. 20(2), 185–198 (2006).
Fletcher, R. B., Baker, J. C. & Harland, R. M. FGF8 spliceforms mediate early mesoderm and posterior neural tissue formation in Xenopus. Development 133(9), 1703–1714 (2006).
Ornitz, D. M. & Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 4(3), 215–266 (2015).
Zhang, X. et al. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J. Biol. Chem. 281(23), 15694–700 (2006).
Handschuh, K. et al. ESCRT-II/Vps25 constrains digit number by endosome-mediated selective modulation of FGF-SHH signaling. Cell Rep. 9(2), 674–687 (2014).
Warzecha, C. C. et al. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol. Cell 33(5), 591–601 (2009).
Calabretta, S. & Richard, S. Emerging roles of disordered sequences in RNA-binding proteins. Trends Biochem. Sci. 40(11), 662–672 (2015).
Author information
Authors and Affiliations
Contributions
S.D. and F.M. performed the data analyses. All authors contributed to experiment design and interpretation. S.D., F.M., and JLF prepared figures. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Driscoll, S., Merkuri, F., Chain, F.J.J. et al. Splicing is dynamically regulated during limb development. Sci Rep 14, 19944 (2024). https://doi.org/10.1038/s41598-024-68608-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68608-z
- Springer Nature Limited