
Abstract
Prostate cancer, one of the most prevalent malignancies among men worldwide, is intricately linked with androgen signaling, a key driver of its pathogenesis and progression. Understanding the diverse expression patterns of androgen-responsive genes holds paramount importance in unraveling the biological intricacies of this disease and prognosticating patient outcomes. In this study, utilizing consensus clustering analysis based on the expression profiles of androgen-responsive genes, prostate cancer patients from the TCGA database were stratified into two distinct subtypes, denoted as C1 and C2. Notably, the C1 subtype demonstrates a significant upregulation of certain genes, such as CGA and HSD17B12, along with a shorter progression-free survival duration, indicating a potentially unfavorable prognosis. Further analyses elucidated the immune infiltration disparities, mutation landscapes, and gene functional pathways characteristic of each subtype. Through integrated bioinformatics approaches and machine learning techniques, key genes such as BIRC5, CENPA, and MMP11 were identified as potential therapeutic targets, providing novel insights into tailored treatment strategies. Additionally, single-cell transcriptome analysis shed light on the heterogeneous expression patterns of these genes across different cell types within the tumor microenvironment. Furthermore, virtual screening identified candidate drugs targeting the BIRC5 receptor, offering promising avenues for drug development. Collectively, these findings deepen our understanding of prostate cancer biology, paving the way for personalized therapeutic interventions and advancing the quest for more effective treatments in prostate cancer management.
Similar content being viewed by others


Introduction
Prostate cancer is the second most common cancer in men and the fifth leading cause of death worldwide, and its incidence is closely related to age1. As a complex disease, its development and progression are influenced by a variety of genetic and environmental factors, making it a significant challenge in the field of oncology2,3. Despite advances in diagnostic methods, the variability in how the disease progresses—ranging from indolent to aggressively metastatic forms—poses unique challenges for treatment and management4. Early detection through prostate-specific antigen testing has improved outcomes, yet the heterogeneity of the disease often leads to variable responses to conventional therapies, underlining the need for more personalized approaches to treatment5,6.
Androgens, such as testosterone, play a pivotal role in the normal development and functioning of the prostate gland but are also implicated in the pathogenesis of prostate cancer7,8. These hormones exert their effects by binding to the androgen receptor (AR), which then activates a cascade of genetic expressions that promote cellular growth and survival9. In the context of prostate cancer, the AR pathway is a critical driver of tumor growth and progression, making it a primary target for therapeutic intervention10,11. androgen deprivation therapy (ADT), which inhibit the action of testosterone or the AR itself, have been standard treatments and provide significant benefits in many cases12,13,14. However, resistance to these therapies frequently develops, leading to disease progression and the need for alternative strategies15,16,17.
The current treatment landscape for prostate cancer includes a range of options from surgery and radiation therapy to hormone therapy and immunotherapy18,19,20,21. While these treatments can be effective, particularly in the early stages of the disease, they often come with significant side effects and complications22. Additionally, the development of resistance to hormone therapy is a major hurdle, particularly in advanced stages6,23. This has prompted ongoing research into more effective and less invasive therapeutic strategies that can address the diverse biological pathways involved in prostate cancer progression, including immune evasion, cell cycle regulation, and genetic mutations.
This research utilizes bioinformatics methods to delve deeper into the molecular intricacies of prostate cancer, particularly the differential expression of androgen-responsive genes. By employing advanced analytical techniques such as consensus clustering, gene set enrichment analysis, and machine learning, this study identifies novel subtypes and potential therapeutic targets within prostate cancer. This stratification allows for a more nuanced understanding of the disease’s biological diversity, potentially leading to the development of targeted therapies that can improve patient outcomes and provide personalized treatment strategies. Through these insights, the research aims to contribute to the evolving landscape of precision medicine in oncology, addressing critical gaps in current treatment modalities and offering new avenues for therapeutic intervention.
Methods
Data collection
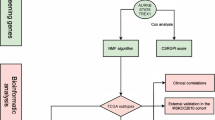
Transcriptomic data and associated clinical information for prostate cancer samples, downloaded from the TCGA database, encompass a total of 554 samples. This dataset includes 502 samples of prostate cancer tissues and 52 samples of normal tissues. This valuable resource facilitates the investigation of the molecular mechanisms underlying prostate cancer and contributes to the advancement of clinical treatment strategies. The dataset GSE137829 was downloaded from the GEO database, which includes single-cell transcriptome data from six prostate cancer samples. The entire analysis workflow is illustrated in Fig. 1.

Schematic representation of the complete analysis workflow.
Genetic analysis of male hormones
In this study, we focused on genes associated with male hormones retrieved from the Genecard database, specifically CGA, CYP17A1, HSD17B12, HSD17B3, HSD3B1, HSD3B2, LHB, POMC, SRD5A1, SRD5A2, and SRD5A3. Raw data normalization was performed using the normalizeBetweenArrays function from the limma package in R to standardize data across the dataset. Subsequently, we extracted the expression profiles of these genes and conducted cluster analysis using the ConsensusClusterPlus package. The analysis was configured with a maximum of 9 clusters, 50 repetitions, and utilized the "PAM" clustering algorithm with "euclidean" distance metric. After evaluating the consistency scores using the calcICL function, we determined that 2 clusters best represented the underlying data structure.
Clinical correlation analysis
Progression-Free Survival (PFS) is a crucial indicator in prostate cancer research. To compare PFS between clusters C1 and C2, we used Kaplan–Meier survival curves. We also stratified target genes into high and low expression groups to construct survival curves, assessing the impact on patient prognosis. Additionally, we examined clinical parameters such as age, race, and Gleason score to identify differences between C1 and C2.
Tumor mutational burden analysis
We obtained gene mutation data for prostate cancer patients from the TCGA database and processed it with the maftools package, visualizing genes with high mutation frequencies. We created waterfall plots for both groups C1 and C2, showcasing the mutation profiles of the 20 most common genes, and generated interaction plots to illustrate mutation relationships.
Immunoinfiltration analysis
Initially, the prepared transcriptome data from groups C1 and C2 are processed through the estimate function to analyze the estimation of tumor purity. The scores are then visualized using a violin plot. Subsequently, the CIBERSORT function is executed to analyze the proportions of 22 characteristic genes of immune cells, thereby evaluating the proportion of various immune cells in each sample. Upon completion of the analysis, visualization tools can be employed to display the results of the proportions of different immune cell types in different samples through a box plot.
Drug susceptibility analysis
We utilize the pRRophetic R package to conduct a drug sensitivity analysis between two groups of samples, and construct a statistical model to predict the response of tumor biological specimens to chemotherapy drugs. Initially, functions within the pRRophetic package will be employed to estimate the half-maximal inhibitory concentration (IC50) of the drug. During this process, we set a filter condition of p < 0.001 to ensure the statistical significance of the results.
Gene set enrichment analysis (GSEA)
Transcriptome data files for groups C1 and C2 have been prepared and pathway enrichment analysis has been conducted using the GSEA4.3.2 software. During the analysis, the c2.cp.kegg_medicus.v2023.2.Hs.symbols gene set database was selected as a reference. To ensure the reliability of the results, we set the screening criteria: NES score greater than 1.5, and FDR q-val less than 0.05.
Analysis of differentially expressed genes (DEGs)
In the analysis of DEGs between the two groups of samples, the Limma software package was utilized to identify genes with significant differences. By setting a threshold of an absolute log fold change (|LogFC|) greater than 0.8 and a p-value less than 0.05, genes with statistically significant differences were able to be filtered out.
Weighted gene co-expression network analysis (WGCNA)
In the process of conducting a WGCNA analysis between groups C1 and C2, the initial step involves the reading and organization of data, with a focus on the top 25% of genes exhibiting the greatest fluctuation for the analysis. This is followed by the clustering of samples and the generation of a sample clustering diagram. Any samples below the cut line in the sample tree are then removed. Upon the preparation of clinical data, a heatmap of the sample clustering is created. The optimal power value is selected and a scatter plot of power values is generated. The data is then transformed into an adjacency matrix and a TOM matrix is calculated. Gene clustering is performed and a gene clustering diagram is generated. Dynamic modules are identified and module clustering is conducted. A heatmap of module genes is created, and the correlation between the module and trait data is calculated, followed by the creation of a correlation heatmap. Finally, the correlation and its P-value between gene module membership and gene trait significance are calculated.
Go and KEGG enrichment analysis
We employ the clusterProfiler package to perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis. Initially, we conduct DEGs and WGCNA, and then use the intersection of the two as the gene list. Subsequently, we utilize functions within the clusterProfiler package to identify the enrichment of these genes in GO and KEGG pathways. Finally, we visualize the results using bar and bubble charts.
Protein–protein interaction analysis
The intersection genes obtained from the above analysis are input into the STRING database for Protein–Protein Interaction (PPI) analysis. When uploading the gene list, we select the “Multiple proteins” option for batch upload. Upon completion of the analysis, the results are exported and opened in Cytoscape software for further analysis. Within Cytoscape, we utilize built-in network centrality algorithms, such as MCC, Degree, and Closeness, to identify and select the top 10 hub genes in the network.
Machine learning
We prepared expression data of 21 genes from PPI analysis and PFS clinical data. We applied Random Forest (RF), Support Vector Machine-Recursive Feature Elimination (SVM-RFE), and Least Absolute Shrinkage and Selection Operator (Lasso) methods. In the RF analysis, we trained the model and obtained the importance score for each gene, selecting the gene with the highest score. In the SVM-RFE analysis, we selected the feature number with the highest accuracy and the smallest error. In the Lasso analysis, we trained the model and obtained the coefficient for each gene, selecting the gene with a non-zero coefficient as the key gene. Finally, we compared the key genes obtained from the RF, SVM-RFE, and Lasso analyses, selecting the genes that were chosen in all methods as the final key genes.
Immune checkpoint correlation analysis
We conducted a t-test to analyze differential expression of immune checkpoint genes between groups C1 and C2, visualized using a box plot. Furthermore, we examined the Pearson correlation between the differentially expressed genes identified by machine learning and the immune checkpoint genes. The outcomes were presented via a correlation heatmap, unveiling potential associations between them.
The Human Protein Atlas analysis
We utilized the immunohistochemical staining image data from the HPA database to evaluate the distribution differences of proteins in normal and cancerous tissues. Specifically, we conducted immunohistochemical analysis on the expression levels of BIRC5, CENPA, and MMP11 proteins in normal prostate tissue, low-grade prostate cancer, and high-grade prostate cancer tissues.
Single-cell analysis
In the processing of the GSE137829 dataset, quality control is initially performed on the single-cell transcriptome data from the six included prostate cancer samples, adhering to the criteria of nFeature_RNA > 500, percent.mt < 20%, and nCount_RNA > 1000. Subsequently, the Harmony package is utilized for data integration and dimensionality reduction to eliminate batch effects. Cluster analysis is conducted to identify distinct cell populations, and these populations are annotated via marker genes to ascertain their biological characteristics. Visualization of the enriched cell populations is carried out for specific genes, namely BIRC5, CENPA, and MMP11. Thereafter, these cell populations undergo GSEA to understand the potential roles of these genes in prostate cancer.
Virtual screening
We identified the crystal structure of BIRC5 protein (4a0i) from the Protein Data Bank (PDB) and constructed a simulation box. Using Autodock Vina software, we performed molecular docking with 1662 energy-minimized natural products, filtering potential therapeutic drugs based on binding affinity values.
Results
Analysis of androgenic subgroups in prostate cancer
In the realm of prostate cancer research, the elucidation of expression patterns of androgen-responsive genes is pivotal for a comprehensive understanding of the disease’s biological attributes and for prognosticating patient outcomes. Utilizing consensus clustering analysis predicated on the expression patterns of androgen-responsive genes, we stratified prostate cancer patients within the TCGA database into two distinct subtypes, designated as C1 and C2 (Fig. 2A,B). The C1 subtype encompasses 174 cases, while the C2 subtype comprises 328 cases. A salient characteristic of the C1 subtype is the markedly elevated expression of genes such as CGA, HSD17B12, HSD17B3, LHB, POMC, SRD5A1, and SRD5A3 compared to the C2 subtype (Fig. 2C). Conversely, patients within the C2 subtype demonstrate a significantly enhanced expression of POMC and SRD5A2 genes relative to the C1 subtype (Fig. 2C). Importantly, the progression-free survival duration of patients within the C2 subtype significantly surpasses that of patients within the C1 subtype, intimating a potential association of the C2 subtype with a more favorable prognosis (Fig. 2D). The results revealed no significant differences in terms of age and ethnicity between the two groups (Fig. 2E). However, a notable distinction was observed in the Gleason scores. The proportion of patients with high Primary and Secondary Gleason grades in Group C1 was greater than that in Group C2 (Fig. 2E).

Stratification of prostate cancer patients into two distinct subtypes based on the expression patterns of androgen-responsive genes. (A,B) Consensus clustering analysis of prostate cancer patients within the TCGA database resulted in the identification of two distinct subtypes, C1 and C2. The C1 subtype includes 174 cases, while the C2 subtype consists of 328 cases. (C) Differential analysis of androgen-responsive gene expression between C1 and C2 subtypes. (D) Analysis of progression-free survival between C1 and C2 subtypes. (E) Sankey diagram representing the clinical correlation analysis between C1 and C2 patients. (C) primarily employs the t-test, while (D) utilizes the logrank test. ** for p < 0.01, and *** for p < 0.001.
Analysis of androgenic subtype immune infiltration, mutation load, drug sensitivity, and GSEA
We employed the ESTIMATE package to assess the extent of immune infiltration between the C1 and C2 subgroups, revealing a superior degree of immune infiltration in the C2 subtype relative to the C1 subtype (Fig. 3A). Further stratification using CIBERSORT determined the proportion of diverse immune cells within each tissue, indicating an elevated presence of B cells naïve, T cells CD4 memory resting, Monocytes, and Mast cells activated in the C2 group compared to the C1 group (Fig. 3B). Conversely, the C1 group showed a higher prevalence of Macrophages M1 and Macrophages M2 (Fig. 3B). We procured tumor mutation data from the TCGA database and conducted an in-depth analysis of gene mutations within prostate cancer tissues. Significant mutations were observed in genes such as TP53, SPOP, TTN, FOXA1, and MUC16 within these tissues (Fig. 3C). A waterfall chart illustrated the mutation frequency of each gene within the C1 and C2 subgroups, revealing that TP53 had the highest mutation frequency in the C1 group at 18%, while SPOP had the highest mutation frequency in the C2 group at 12%. Overall, the C1 group demonstrated a higher gene mutation frequency relative to the C2 group (Fig. 3D). The interrelation between gene mutations within prostate cancer was visualized using a heatmap (Fig. 3E). GSEA enrichment analysis results indicated significant enrichment of cell cycle and DNA repair-related signaling pathways within the C1 group (Fig. 3F). Additionally, we extrapolated the sensitivity of the two groups to various anti-tumor drugs using IC50 values, based on the transcription data of the C1 and C2 groups (Supplementary Fig. S1).

Comparative analysis of immune infiltration, gene mutations, and pathway enrichment between C1 and C2 subgroups in prostate cancer. (A) Assessment of immune infiltration between the C1 and C2 subgroups using the ESTIMATE package. (B) Stratification of diverse immune cells within each tissue using CIBERSORT. (C) In-depth analysis of gene mutations within prostate cancer tissues, highlighting significant mutations in genes. (D) A waterfall chart illustrating the mutation frequency of each gene within the C1 and C2 subgroups. (E) Heatmap visualization of the interrelation between gene mutations within prostate cancer. (F) GSEA enrichment analysis results indicating a significant enrichment of cell cycle and DNA repair-related signaling pathways within the C1 group.
Results of DEGs and WGCNA analysis
The DEGs analysis was conducted on the C1 and C2 subtypes of prostate cancer, which are characterized by androgen-related genes. We set a threshold of |LogFC|> 0.8 and a P-value less than 0.05. The comparison between the C2 and C1 subtypes revealed a total of 656 DEGs, with 586 showing upregulation and 70 showing downregulation (Fig. 4A,B). Subsequently, the top quartile of genes, based on expression variability, were selected for WGCNA analysis. The soft-thresholding power of 10 was employed to construct a scale-free network, revealing complex gene interactions (Fig. 4C). Dynamic module detection was utilized within this network to identify gene clusters, potentially representing gene groups acting in concert during disease progression (Fig. 4D). In this study, 17 modules were identified, each containing at least 100 genes. The identification of these modules aids in understanding the biological differences between the prostate cancer subtypes. Among these modules, the MEmagenta module was found to be significantly positively correlated with the C1 subtype (Fig. 4E). Therefore, the genes within this module were selected for further analysis. This analysis will contribute to a deeper understanding of the pathogenesis of prostate cancer and may provide potential targets for future therapeutic strategies.

DEGs and WGCNA analysis of C1 and C2 subtypes in prostate cancer. (A) Volcano plot of DEGs between C1 and C2 subtypes. (B) Heatmap of DEGs between C1 and C2 subtypes. (C) Soft-thresholding selection. (D) Cluster dendrogram of highly connected genes in the key module. (E) Cluster dendrogram of modules with highly connected genes.
Gene set functional pathway and PPI analysis results
Through an in-depth analysis of the intersection of differentially expressed genes and WGCNA module-related genes, we successfully identified 37 common genes (Fig. 5A). Subsequently, we conducted GO and KEGG enrichment analyses on these 37 genes. The GO analysis revealed the crucial role of these genes in the process of cellular mitosis, especially in the positioning of the spindle and centrosome, which are vital structures in cell division (Fig. 5B). The KEGG analysis emphasized the significant role of these genes in pyrimidine metabolism and the P53 signaling pathway, pathways that play a central role in cell cycle regulation and tumor suppression (Fig. 5C). Finally, we performed a PPI analysis on these genes, revealing an interaction network of nodes including 21 proteins. Through the analysis methods of MCC, Degree, and Closeness, we further identified 10 key hub genes, including MYBL2, MKI67, CENPF, KIF20A, TOP2A, UBE2C, CDC20, CDK1, BIRC5, and TROAP (Fig. 5D). These hub genes may play a central role in cell proliferation, differentiation, or disease progression.

Comprehensive analysis of common genes and their functional implications. (A) Venn diagram of the intersection of DEGs and WGCNA module-related genes. (B) GO analysis results of the intersected gene set. (C) KEGG analysis results of the intersected gene set. (D) PPI analysis results of the intersected gene set.
Machine learning screening for potential therapeutic targets
In the pursuit of identifying pivotal genes within the PPI data, we employed methodologies such as RF, SVM-RFE, and Lasso. The random forest algorithm was implemented with a set of 500 trees. The error rate exhibited a variation in correlation with the number of trees. We discerned the optimal quantity of trees and conducted a screening of genes based on their significance scores. Consequently, we selected the top 7 genes that demonstrated an importance exceeding 7.5 (Fig. 6A). In the context of SVM-RFE, we utilized k-fold cross-validation to select feature variables. The generalization error was estimated through an array of feature combinations, ultimately leading to the selection of the top 16 features exhibiting the smallest error as feature genes. The variable combinations and their corresponding accuracy and error are depicted in Fig. 6B. During the Lasso analysis, the model was trained and a coefficient was obtained for each gene. During the Lasso analysis, we identified key genes with a non-zero coefficient, resulting in the discovery of a total of 7 such genes. (Fig. 6C). By intersecting the results from these three analytical methods, we identified three key genes: BIRC5, CENPA, and MMP11 (Fig. 6D). These genes were found to be significantly overexpressed in the C1 subtype of prostate cancer compared to the C2 subtype (Fig. 6E). Furthermore, the genes BIRC5 and CENPA were significantly overexpressed in prostate cancer relative to normal tissue, while MMP11 exhibited a trend towards overexpression (Fig. 6F–H). Patients with high gene expression had a significantly reduced progression-free survival period (Fig. 6F–H). Additionally, the expression of these three genes was associated with a higher risk in various types of cancer, including ACC, KIRC, KIRP, LGG, LIHC, and LUAD (Fig. 6I). From a protein level perspective, we analyzed these three proteins in normal prostate, high-grade, and low-grade prostate cancer tissues using the HPA database. The immunohistochemistry results revealed that these three proteins exhibited higher expression in both high-grade and low-grade prostate cancer tissues compared to normal prostate tissues (Supplementary Fig. S2).

Identification and analysis of key genes using RF, SVM-RFE, and Lasso methodologies. (A) Selection of top 7 genes with importance exceeding 7.5 using the Random Forest algorithm. (B) Selection of top 16 features with the smallest error as feature genes using SVM-RFE. (C) Identification of 7 key genes with non-zero coefficients using Lasso analysis. (D) Intersection of results from the three analytical methods. (E) Significant overexpression of these genes in the C1 subtype of prostate cancer compared to the C2 subtype. (F–H) Differential expression and clinical prognosis analysis of BIRC5, CENPA, and MMP11 genes between prostate cancer and normal tissues. (I) Association of the expression of these genes with a higher risk in various types of cancer.
Immune cell infiltration, tumor mutational burden and immune checkpoint correlation analysis
We conducted a Spearman correlation analysis on the expression levels of three genes, BIRC5, CENPA, and MMP11, in prostate cancer tissues, as well as the infiltration levels of 21 types of immune cells in each tissue. In Fig. 7A, we present the relationships between these genes and the infiltration levels of 21 different immune cells in the form of a heatmap. Notably, there is a significant positive correlation between BIRC5, CENPA, and MMP11 and M2 macrophages and Tregs cells, suggesting their potential roles in the immune microenvironment of prostate cancer. Furthermore, by comparing the expression differences of immune checkpoint-related genes between Group C1 and Group C2, researchers can gain further insights into the immune escape mechanisms of prostate cancer (Fig. 7B). For instance, genes such as PDCD1LG2, SIGLEC15, PDCD1, and CD28 are significantly more expressed in Group C2 than in Group C1, while genes like TNFRSF18 and LDHA are significantly more expressed in Group C1 than in Group C2. We further explored the correlations between these three genes and immune checkpoint genes, which are presented in the form of a heatmap in Fig. 7C. We also conducted a correlation analysis on these three genes and the tumor mutation burden in prostate cancer tissues, revealing a significant positive correlation (Fig. 7D). These findings provide us with a new perspective for a deeper understanding of the immune microenvironment in prostate cancer.

Correlation analysis of gene expression and immune cell infiltration in prostate cancer. (A) Heatmap presenting the relationships between the expression levels of BIRC5, CENPA, and MMP11 and the infiltration levels of 21 different immune cells. (B) Comparison of the expression differences of immune checkpoint-related genes between Group C1 and Group C2. (C) Heatmap of the correlations between BIRC5, CENPA, MMP11, and immune checkpoint genes. (D) Correlation analysis of BIRC5, CENPA, MMP11, and the tumor mutation burden in prostate cancer tissues.
Prostate cancer single cell analysis results
Single-cell transcriptome analysis is a powerful tool that can reveal the heterogeneity of different cell types within the tumor microenvironment. We obtained single-cell transcriptome data samples of prostate cancer from the GEO database and conducted an in-depth analysis. After data quality control, dimensionality reduction, and clustering, we categorized these samples into several groups, including epithelial cells, T cells, fibroblasts, myofibroblasts, myeloid cells, endothelial cells, mast cells, and B cells, and visualized them using UMAP plot (Fig. 8A). The marker genes of each cell group were visualized in the form of a bubble chart (Fig. 8B). We also visualized the three key genes screened out by machine learning in various cell groups. The results showed that BIRC5 and CENPA are mainly expressed in epithelial cells, while MMP11 is mainly expressed in fibroblasts (Fig. 8C–E). In addition, we conducted GSEA on the cell groups to explore the signaling pathways involved by these three key genes. The results showed that the BIRC5 gene mainly participates in the P53 signaling pathway, CENPA is mainly related to the cell cycle, and MMP11 mainly participates in steroid hormone biosynthesis (Fig. 8F–H).

Single-cell transcriptome analysis and visualization of key genes in prostate cancer. (A) UMAP plot visualizing the categorization of single-cell transcriptome data samples into various cell groups. (B) Bubble chart representing the marker genes of each cell group. (C–E) Visualization of the expression of three key genes (BIRC5, CENPA, and MMP11) screened out by machine learning in various cell groups. (F–H) GSEA of the cell groups to explore the signaling pathways involved by these three key genes.
Virtual screening of potential drugs targeting the BIRC5 receptor
The protein encoded by BIRC5 is known as Survivin, an inhibitor of apoptosis that is typically expressed during embryonic development and in proliferating cells in adults. In cancer cells, the expression of Survivin is often elevated, making it a promising target for anti-cancer drugs. In PPI screening, the BIRC5 gene is selected as the receptor protein for drug screening due to its characteristic as a central node. In our study, we downloaded the Survivin protein structure with the identifier 4A0I from the PDB database for analysis. Initially, we protonated the protein to better simulate its state under physiological pH conditions (Fig. 9A). During the virtual screening process, we established a docking box centered on the ligand’s location and screened 1662 natural product molecules. The affinity of these molecules was sorted by their binding affinities to identify potential compounds with high affinity for Survivin (Fig. 9B). Among the screened compounds, Polyphyllin VI, Narirutin, Lanatoside C, momordin-Ic, and Gracillin demonstrated hydrogen bond binding with specific amino acid residues of Survivin, including ASN119, GLN56, and LYS122 (Fig. 9C). The presence of these hydrogen bonds may play a key role in the stability and activity of the compounds, enhancing intermolecular interactions. This provides us with a window to understand how these compounds bind to Survivin and how they might affect its function.

Virtual screening and interaction analysis of potential compounds with Survivin. (A) The Survivin protein structure (PDB ID: 4A0I). (B) Binding affinity of potential compounds with high affinity. (C) Hydrogen bond interactions of selected compounds (Polyphyllin VI, Narirutin, Lanatoside C, momordin-Ic, and Gracillin) with specific amino acid residues of Survivin.
Discussion
In the domain of prostate cancer research, the exploration of androgen-related mechanisms has provided critical insights into the pathogenesis and progression of the disease, particularly given its dependence on androgen signaling24,25,26. Current therapeutic regimens predominantly aim to disrupt these androgen pathways, typically through the use of ADT27,28. However, the emergence of castration-resistant prostate cancer (CRPC) underscores a significant shortfall in these strategies, highlighting the urgent need for novel therapeutic targets that can provide more durable control over the disease progression29,30.
In this study, we employed a novel approach of classifying patients with prostate cancer from the TCGA database into two subtypes, C1 and C2, based on the differential expression patterns of androgen-related genes. Notably, genes associated with androgen regulation, such as CGA, HSD17B12, LHB, SRD5A1, and SRD5A3, exhibited significantly higher expression in the C1 group31,32,33,34,35. This stratification into distinct subtypes provides a more nuanced understanding of the correlation between androgen levels and tumor behavior, as well as patient prognosis. Interestingly, patients within the C2 subtype not only demonstrated a more robust immune response but also had a lower incidence of key genetic mutations. These factors are often indicative of a better response to existing treatments and potentially longer disease-free survival. Our findings suggest an intrinsic link between the immune landscape of prostate tumors and androgen levels, a hypothesis that could potentially revolutionize approaches to immunotherapy in prostate cancer. This refined classification and its implications underscore the importance of considering androgen levels in therapeutic strategies for prostate cancer.
Moreover, the application of machine learning to identify critical genes associated with these subtypes offers promising new avenues for therapeutic intervention. Genes such as BIRC5, CENPA, and MMP11, identified in the study as significantly overexpressed in more aggressive prostate cancers, open up opportunities for targeted therapies. For instance, BIRC5 (Survivin), known for its role in inhibiting apoptosis and promoting cell proliferation, could be an ideal target for drugs designed to induce cell death in tumor cells36,37. Similarly, targeting CENPA, a histone variant involved in chromosome segregation during cell division, could disrupt the proliferative capacity of cancer cells, addressing the issue of rapid tumor growth38,39. The implications of these findings extend beyond simply identifying new drug targets; they also enhance our understanding of the tumor microenvironment and its interaction with systemic therapies. The potential synergy between targeted therapies against these genes and existing treatments like ADT or chemotherapy could lead to more comprehensive treatment protocols that address multiple aspects of tumor biology simultaneously. Additionally, the drug screening results presented in this study, which suggest possible inhibitors for these key proteins, lay the groundwork for future preclinical and clinical trials that could one day lead to more effective and personalized treatment options for prostate cancer patients.
In conclusion, this study not only reinforces the importance of androgen signaling in prostate cancer but also highlights the potential for integrating advanced bioinformatics and machine learning tools to unearth new therapeutic targets and strategies. By moving towards a more tailored treatment approach based on specific genetic and molecular profiles, we can hope to improve the efficacy of treatments, reduce the occurrence of resistance, and ultimately enhance the prognosis for patients suffering from this challenging disease. Future research should aim to validate these potential targets in preclinical settings, ensuring that treatments continue to evolve in line with our deepening understanding of the disease.
Data availability
All TCGA molecular and clinical data sets used in this study are publicly available and can be found here: https://portal.gdc.cancer.gov/projects/TCGA-PRAD. The GSE137829 dataset used in this study is publicly available and can be found here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137829. The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
References
Rawla, P. Epidemiology of prostate cancer. World J. Oncol. 10(2), 63–89. https://doi.org/10.14740/wjon1191 (2019).
Seibert, T. M. et al. Genetic risk prediction for prostate cancer: Implications for early detection and prevention. Eur. Urol. 83(3), 241–248. https://doi.org/10.1016/j.eururo.2022.12.021 (2023).
Kearns, J. T., Helfand, B. T. & Xu, J. Moving prostate cancer polygenic risk scores from research towards clinical practice. Eur. Urol. Focus 8(4), 913–915. https://doi.org/10.1016/j.euf.2022.08.011 (2022).
Wang, G., Zhao, D., Spring, D. J. & DePinho, R. A. Genetics and biology of prostate cancer. Genes Dev. 32(17–18), 1105–1140. https://doi.org/10.1101/gad.315739.118 (2018).
Carlsson, S. V. & Vickers, A. J. Screening for prostate cancer. Med. Clin. N. Am. 104(6), 1051–1062. https://doi.org/10.1016/j.mcna.2020.08.007 (2020).
Jamroze, A., Chatta, G. & Tang, D. G. Androgen receptor (AR) heterogeneity in prostate cancer and therapy resistance. Cancer Lett. 518, 1–9. https://doi.org/10.1016/j.canlet.2021.06.006 (2021).
van Winden, L. J. & van Rossum, H. H. Testosterone analysis in prostate cancer patients. Adv. Clin. Chem. 108, 73–104. https://doi.org/10.1016/bs.acc.2021.07.007 (2022).
Kobayashi, T., Inoue, T., Kamba, T. & Ogawa, O. Experimental evidence of persistent androgen-receptor-dependency in castration-resistant prostate cancer. Int. J. Mol. Sci. 14(8), 15615–15635. https://doi.org/10.3390/ijms140815615 (2013).
Westaby, D. et al. A new old target: Androgen receptor signaling and advanced prostate cancer. Annu. Rev. Pharmacol. Toxicol. 62, 131–153. https://doi.org/10.1146/annurev-pharmtox-052220-015912 (2022).
Feng, K. et al. Emerging proteins involved in castration-resistant prostate cancer via the AR-dependent and AR-independent pathways (review). Int. J. Oncol. https://doi.org/10.3892/ijo.2023.5575 (2023).
Merkens, L. et al. Aggressive variants of prostate cancer: Underlying mechanisms of neuroendocrine transdifferentiation. J. Exp. Clin. Cancer Res. 41(1), 46. https://doi.org/10.1186/s13046-022-02255-y (2022).
Schaeffer, E. M. et al. Prostate cancer, Version 4.2023, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 21(10), 1067–1096. https://doi.org/10.6004/jnccn.2023.0050 (2023).
Dai, C., Dehm, S. M. & Sharifi, N. Targeting the androgen signaling axis in prostate cancer. J. Clin. Oncol. 41(26), 4267–4278. https://doi.org/10.1200/jco.23.00433 (2023).
Desai, K., McManus, J. M. & Sharifi, N. Hormonal therapy for prostate cancer. Endocr. Rev. 42(3), 354–373. https://doi.org/10.1210/endrev/bnab002 (2021).
Snow, O. et al. Androgen receptor plasticity and its implications for prostate cancer therapy. Cancer Treat. Rev. 81, 101871. https://doi.org/10.1016/j.ctrv.2019.05.001 (2019).
Sun, R. et al. Androgen receptor variants confer castration resistance in prostate cancer by counteracting antiandrogen-induced ferroptosis. Cancer Res. 83(19), 3192–3204. https://doi.org/10.1158/0008-5472.Can-23-0285 (2023).
Cheng, Q. et al. Pre-existing castration-resistant prostate cancer-like cells in primary prostate cancer promote resistance to hormonal therapy. Eur. Urol. 81(5), 446–455. https://doi.org/10.1016/j.eururo.2021.12.039 (2022).
Wasim, S., Lee, S. Y. & Kim, J. Complexities of prostate cancer. Int. J. Mol. Sci. https://doi.org/10.3390/ijms232214257 (2022).
Cha, H. R., Lee, J. H. & Ponnazhagan, S. Revisiting immunotherapy: A focus on prostate cancer. Cancer Res. 80(8), 1615–1623. https://doi.org/10.1158/0008-5472.Can-19-2948 (2020).
Sridaran, D. et al. Prostate cancer immunotherapy: Improving clinical outcomes with a multi-pronged approach. Cell Rep. Med. 4(10), 101199. https://doi.org/10.1016/j.xcrm.2023.101199 (2023).
Evans, A. J. Treatment effects in prostate cancer. Mod. Pathol. 31(S1), S110-121. https://doi.org/10.1038/modpathol.2017.158 (2018).
Philippou, Y., Sjoberg, H., Lamb, A. D., Camilleri, P. & Bryant, R. J. Harnessing the potential of multimodal radiotherapy in prostate cancer. Nat. Rev. Urol. 17(6), 321–338. https://doi.org/10.1038/s41585-020-0310-3 (2020).
Ge, R. et al. Epigenetic modulations and lineage plasticity in advanced prostate cancer. Ann. Oncol. 31(4), 470–479. https://doi.org/10.1016/j.annonc.2020.02.002 (2020).
Erdmann, É. et al. Androgen receptor-mediated transcriptional repression targets cell plasticity in prostate cancer. Mol. Oncol. 16(13), 2518–2536. https://doi.org/10.1002/1878-0261.13164 (2022).
Maranto, C. et al. Stat5 induces androgen receptor (AR) gene transcription in prostate cancer and offers a druggable pathway to target AR signaling. Sci. Adv. 10(9), eadi2742. https://doi.org/10.1126/sciadv.adi2742 (2024).
Kim, W. K. et al. Aberrant androgen action in prostatic progenitor cells induces oncogenesis and tumor development through IGF1 and Wnt axes. Nat. Commun. 13(1), 4364. https://doi.org/10.1038/s41467-022-32119-0 (2022).
Nigro, M. C. et al. Current androgen receptor antagonists under investigation for resistant prostate cancer. Expert Rev. Anticancer Ther. 22(2), 191–202. https://doi.org/10.1080/14737140.2022.2020651 (2022).
Yu, E. M. & Aragon-Ching, J. B. Advances with androgen deprivation therapy for prostate cancer. Expert Opin. Pharmacother. 23(9), 1015–1033. https://doi.org/10.1080/14656566.2022.2033210 (2022).
Cai, M. et al. Current therapy and drug resistance in metastatic castration-resistant prostate cancer. Drug Resist. Update 68, 100962. https://doi.org/10.1016/j.drup.2023.100962 (2023).
Baratchian, M. et al. H3K9 methylation drives resistance to androgen receptor-antagonist therapy in prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 119(21), e2114324119. https://doi.org/10.1073/pnas.2114324119 (2022).
Kemiläinen, H. et al. The hydroxysteroid (17β) dehydrogenase family gene HSD17B12 is involved in the prostaglandin synthesis pathway, the ovarian function, and regulation of fertility. Endocrinology 157(10), 3719–3730. https://doi.org/10.1210/en.2016-1252 (2016).
Bièche, I. et al. CGA gene (coding for the alpha subunit of glycoprotein hormones) overexpression in ER alpha-positive prostate tumors. Eur. Urol. 41(3), 335–341. https://doi.org/10.1016/s0302-2838(02)00020-9 (2002).
Yang, X. et al. Homozygous nonsense mutation Trp28X in the LHB gene causes male hypogonadism. J. Assist. Reprod. Genet. 35(5), 913–919. https://doi.org/10.1007/s10815-018-1133-5 (2018).
Yazawa, T. et al. Profiles of 5α-reduced androgens in humans and eels: 5α-dihydrotestosterone and 11-ketodihydrotestosterone are active androgens produced in eel gonads. Front. Endocrinol. (Lausanne) 12, 657360. https://doi.org/10.3389/fendo.2021.657360 (2021).
Stiles, A. R. & Russell, D. W. SRD5A3: A surprising role in glycosylation. Cell 142(2), 196–198. https://doi.org/10.1016/j.cell.2010.07.003 (2010).
Fäldt Beding, A., Larsson, P., Helou, K., Einbeigi, Z. & Parris, T. Z. Pan-cancer analysis identifies BIRC5 as a prognostic biomarker. BMC Cancer 22(1), 322. https://doi.org/10.1186/s12885-022-09371-0 (2022).
Li, F., Aljahdali, I. & Ling, X. Cancer therapeutics using survivin BIRC5 as a target: What can we do after over two decades of study?. J. Exp. Clin. Cancer Res. 38(1), 368. https://doi.org/10.1186/s13046-019-1362-1 (2019).
Liao, J. et al. CENPA functions as a transcriptional regulator to promote hepatocellular carcinoma progression via cooperating with YY1. Int. J. Biol. Sci. 19(16), 5218–5232. https://doi.org/10.7150/ijbs.85656 (2023).
Wang, Q. et al. CENPA promotes clear cell renal cell carcinoma progression and metastasis via Wnt/β-catenin signaling pathway. J. Transl. Med. 19(1), 417. https://doi.org/10.1186/s12967-021-03087-8 (2021).
Funding
This work was funded by the Natural Science Foundation of Hunan Province of China (No. 2022JJ40249) and Scientific Research Project of Hunan Provincial Health Commission (No. 202204053845).
Author information
Authors and Affiliations
Contributions
L.H.,Y.X., S.J., T.D., Z.X. conceived the article and wrote the manuscript. H.S. reviewed and integrated it with additional data and references. All the remaining authors contributed with comments, adding data and references. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, L., Xie, Y., Jiang, S. et al. Insights into immune microenvironment and therapeutic targeting in androgen-associated prostate cancer subtypes. Sci Rep 14, 18036 (2024). https://doi.org/10.1038/s41598-024-68863-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68863-0
- Springer Nature Limited