
Abstract
Evolution of unisexual flowers involves extreme changes in floral development. Spinach is one of the species to discern the formation and evolution of dioecy. MADS-box gene family is involved in regulation of floral organ identity and development and in many other plant developmental processes. However, there is no systematic analysis of MADS-box family genes in spinach. A comprehensive genome-wide analysis and transcriptome profiling of MADS-box genes were undertaken to understand their involvement in unisexual flower development at different stages in spinach. In total, 54 MADS-box genes found to be unevenly located across 6 chromosomes and can be divided into type I and type II genes. Twenty type I MADS-box genes are subdivided into Mα, Mβ and Mγ subgroups. While thirty-four type II SoMADSs consist of 3 MIKC*, and 31 MIKCC -type genes including sixteen floral homeotic MADS-box genes that are orthologous to the proposed Arabidopsis ABCDE model of floral organ identity determination, were identified in spinach. Gene structure, motif distribution, physiochemical properties, gene duplication and collinearity analyses for these genes are performed in detail. Promoters of both types of SoMADS genes contain mainly MeJA and ABA response elements. Expression profiling indicated that MIKCc genes exhibited more dynamic and intricate expression patterns compared to M-type genes and the majority of type-II genes AP1, SVP, and SOC1 sub-groups showed female flower-biased expression profiles, suggesting their role in carpel development, while PI showed male-biased expression throughout flower developmental stages, suggesting their role in stamen development. These results provide genomic resources and insights into spinach dioecious flower development and expedite spinach improvement.
Similar content being viewed by others

Introduction
MADS-box gene family holds significant involvement across various stages of plant development1. Prior investigations have shed light on the crucial role played by the MADS-box family in numerous developmental processes, including the promotion of vegetative development, determination of flower organ identity, regulation of flowering time, facilitation of seed and fruit development, as well as the formation of pollen and embryo sac1,2,3. This gene family is characterized by the MADS domain of about 60 amino acids sequence, involved in DNA dimerization and binding based on CC[A/T]6GG (CArG boxes) consensus sequence and present at the N-terminal region of proteins4. This term “MADS” is acquired from four MADS-box family members in (i) yeast MCM1 genes (Mini Chromosome Maintenance1), (ii) Arabidopsis AG (AGAMOUS), (iii) snapdragon DEF (Deficiens), and (iv) humans SRF genes (Serum response factor)5,6,7,8. The phylogenetic classification of the MADS-box gene family reveals two distinct types: type I (SRF-like) and type II (MEF2-like), based on their conserved domains9. In plants, type I genes exhibit shorter lengths and simpler structures, consisting solely of the MADS domain. Further classification assigns them into three clades: Mα, Mβ, and Mγ. In contrast, type II genes demonstrate greater complexity, characterized by the presence of the MADS (M) domain, Intervening (I) domain, Keratin (K) domain, and a highly variable C-terminal (C) domain. These type II genes are commonly referred to as MIKC-type genes2,9. The MIKC-type genes can be further divided into two subgroups, MIKCc and MIKC*, based on the variations in their intervening regions10. Investigations on Arabidopsis thaliana have reported the sub-classification of MIKCc-type genes into 12 groups, guided by their phylogenetic relationships9. Over the extensive evolutionary timeline of this gene family, duplication events of varying degrees have occurred, followed by sub-functionalization, leading to the functional diversification of MADS-box genes11.
Flowering represents a pivotal process in plants, requiring the coordinated interaction of multiple genes to perceive environmental and developmental cues and make critical decisions. MADS-box genes could be involved in the regulation of various plant developmental aspects12,13,14,15, but initially, these genes were discovered as floral organ identity genes in Arabidopsis thaliana and Antirrhinum majus16. These genes were summarized into ABC model that later expanded to ABCDE model. The "ABCDE model" originated from studies on mutants displaying floral organ identity defects, elucidating the co-determination of floral organ identities by class A, B, C, D, and E genes11. In Arabidopsis, the process of sepal development is influenced by the class A [AP1(APETALA1)] and class E [SEP1/2/3/4 (SEPALLATA1/2/3/4)] protein complex, while petal development involves the collaboration of class A, class B, and class E protein complexes. Stamen development is regulated by the class B [AP3(APETALA3) and PI (PISTILLATA)], class C [AG (AGAMOUS)], and class E protein complex, while carpel development is controlled by the class C and class E protein complex. Ovule development, on the other hand, is modulated by class D genes [SHP1/2(SHATTERPROOF1/2), STK(SEEDSTICK)]17,18. The class E proteins are known to form tetramer protein complexes and interact with type A, B, and C proteins19.
Additionally, there are several other type II MADS-box genes responsible for governing flower initiation and flowering time. Noteworthy examples include FLC (FLOWERING LOCUS C), SOC1 (SUPPRESSOR OF CONSTANS1), SVP (SHORT VEGETATIVE PHASE), MAF1/FLM (MADS AFFECTING FLOWERING), and AGL15 (AGAMOUS-LIKE 15), AGL18, AGL24. These genes play crucial roles in integrating various flowering signals, such as those from photoperiod and vernalization pathways, among others12,20,21. These genes function as essential components in the intricate regulatory network governing the timing of flowering. Floral development ABCDE model has been widely tested on a collection of flowering plants and found to be preserved on genomic and functional levels22,23. However, a few anomalies to the conventional Antirrhinum and Arabidopsis model have been illustrated in the mechanism involved in the evolution of floral morphologies.
Spinach, known as Spinacia oleracea (2n = 2 × = 12), displays dioecious characteristics, although there are certain androdioecious populations. In case of unisexual flowers, the termination of stamens or carpels happens during the early stages of primordia formation, leading to a complete absence of stamens in female flowers and carpels in male flowers24. Utilizing virus-induced gene silencing (VIGS), the B-class PISTILLATA and APETALLA3 genes expression was suppressed in male spinach plants. This led to the development of mosaic individuals displaying homeotic changes, including the conversion of stamen into carpel and the occurrence of gynoecia in the typically non-existent fourth whorl25, indicated the role of B class genes in stamen determination and also for the suppression of female fourth whorl organs. These B-class masculinizing factors highlighted the significance of MADS-box family genes in unisexual flower development in spinach. Completion of a high-quality spinach YY genome provided an opportunity to investigate the comprehensive analysis of the MADS-box genes underlying unisexual flower initiation and development in spinach. Our study encompassed whole genome identification, phylogenetic relationships, examination of gene structures, conserved motifs, physical location, protein–protein interaction, promoter analysis, and expression patterns at different flower developmental stages of these genes. Through our investigation, we aimed to enhance our understanding of the evolutionary aspects and functional roles of MADS-box genes in spinach, thereby establishing a fundamental basis for future research on the mechanisms governing flowering development and regulation in spinach.
Material and methods
Identification of MADS–box genes in spinach
Spinach high-quality YY genome sequenced by “FAFU and UIUC-SIB Joint Center for Genomics and Biotechnology, National Sugarcane Engineering Technology Research Center, Fujian Provincial Key Laboratory of Haixia Applied Plant Systems Biology, Fujian Agriculture and Forestry University, Fuzhou, Fujian 350002, China” was used in this study26. Arabidopsis and Oryza sativa MADS-box proteins were obtained from TIAR (https://www.arabidopsis.org/) and phytozome (https://phytozome.jgi.doe.gov/pz/portal.html), respectively9,27. To maximize the MADS-box gene number in spinach genome, two methods were applied. Firstly, Hidden Markov Model profile of two domains (i) SFR-domain (PF00319) and (ii) MEF2-domain (PF09047), retrieved from Pfam database were used against spinach protein database, using the HMMER version 3.0 software28. All of the proteins with an e-value lower than 0.001 were selected. Secondly, BLASTp searches (with parameters query-coverage > 40, identity > 50, evalue 0.000001) were used with Arabidopsis and rice MADS-box proteins as query against spinach protein database. Redundant sequences were removed by aligning them using Clustal-Omega and DNAMAN. Finally, non-redundant and non-repetitive MADS-box protein sequences from these both searches were further verified for conserved domain using PfamScan (https://www.ebi.ac.uk/Tools/pfa/pfamscan/) and Batch CD-Search (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi).
Physiochemical characterization, phylogenetic analysis, and subcellular localization of MADS-box genes in spinach
The physiochemical protein properties like isoelectric point (PI), molecular weight (MW), coding DNA sequence (CDS) length, amino acid (A.A) length, were predicted by ExPASy using ProtParam tool (https://web.expasy.org/protparam/)29.
To understand the phylogenetic relationship and classifying these genes into different subgroups, multiple alignment was performed based on Arabidopsis, rice, and spinach MADS-box protein sequences using MUSCLE30 with default parameters. Alignment quality was checked, and low quality aligned regions (e.g., regions with many gaps or uncertain base calls), were trimmed using alignment-editing software like Gblocks. Additionally, observe the alignment manually to avoid any excessive loss of biological information31. After that, RaxML program was used for phylogenetic tree analyses under maximum likelihood method, and tree was visualized using iTOL (http://itol.embl.de/)32,33. Genes were further confirmed based on best-blast hits against Arabidopsis MADS-box genes.
MADS-box proteins subcellular localization was predicted by using two programs, CELLO v2.5 (http://cello.life.nctu.edu.tw/), and WoLFPSORT (http://www.genscript.com/wolf-psort.html). The final selected result was based on the high confidence predictions from these two programs.
Gene structure and motif distribution of MADS-box genes
To analyze structural divergence, gene structure and their encoded proteins conserved motifs were investigated. MADS-box genes structural organizations were retrieved from spinach gff3 files26. The conserved motifs were investigated by an online tool “The MEME Suite version 5.1.0” (http://meme-suite.org/). The analysis parameters were chosen as follows: (i) width of motif between > 6 and < 200 a.a., (ii) maximum motifs no. as10, (iii) background model as 0-order model of sequences. The acquired motifs were annotated with an online SMART program (http://smart.embl-heidelberg.de.). All the parameters were visualized using TBtools with Gene structure view (Advance) option34.
Chromosomal localization, gene duplication and collinearity analysis
Chromosome distribution and localization parameters of spinach MADS-box genes were obtained from genomic and annotation files. Physical location was mapped on the chromosomes using TBtools34.
The occurrence of tandem and segmental or whole-genome duplications (WGD) offers insights into the evolution of gene families and the advancement of genomes. MADS-box genes showing homology and having only a single intervening gene on the same chromosome were categorized as tandem duplicates, whereas homologs found on different chromosomes were classified as segmental duplications. MADS-box duplicated genes were identified efficiently based on Blast search with identity > 80% and query coverage > 70%, and visualized for their location on chromosomes using TBtools. The number for synonymous substitutions per synonymous site (Ks) and nonsynonymous substitutions per nonsynonymous site (Ka) were acquired using the straightforward KaKs calculator within TBtools software. Subsequently, the Ks values acquired for each pair of genes were converted into divergence times measured in millions of years as T = Ks/2λ, with λ = 6.5 × 10−935. This calculation was based on an assumed substitution rate (λ) of 6.1 × 10−9 substitutions per site per year, specifically for eudicots36.
MCScan toolkit was used to analyze the collinearity of spinach, Amarantus (A.hypochondriacus), quinoa (C.quinoa), Beet (B.vulgaris) and Arabidopsis (A. thaliana). MADS-box proteins and plot collinear genes on the chromosomes34.
Protein–protein network interaction of SoMADS-proteins
The protein–protein interaction network of MADS-box proteins were predicted and constructed based on Arabidopsis homologous proteins using an online tool STRING (https://string-db.org/). The parameters for STRING tool were set as follows; network type-full STRING network; line thickness indicates the strength of data support; the meaning of network edges-evidence; the minimum required interaction score was set as medium to high confidence parameter (0.65).
Cis-regulatory elements from the promoter region of MADS-box genes
Plant CARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) was used to find out the phytohormones related cis-acting elements from the genomic DNA sequences of the promoter region 2000 bp upstream regions of the MADS-box genes start site (–ATG–).
Plant material, and expression analysis at different stages of flower development
Expression pattern of MADS-box genes was observed from five different stages of flower development in male and female spinach plant. These developmental stages include; stage1 (flower bud size = 0.2–0.5 mm): two opposite sepal primordia established in the periphery of the meristem (male, female), stage2 (flower bud size = 0.5–1 mm): two extra sepals form within the first whorl in space between initial sepal primordia; sepals expands laterally around the outer whorl and distally to surround the floral meristem (female), stage3: sepal primordia grow, four stamen primordia form in the periphery of the central dome; sepal covers the central dome and dome begin to differentiate into an ovary (female), stage4: stamen primordia develop to distinct anthers; central region of ovary form pistil and ovule differentiate in the ovary (female), stage5: anthers mature; ovule mature, stigma extends out of sepal closure (female).These stages were grouped into “early stage” (S1–S2) and “mature stage” (S3–S5). Raw transcriptome data files of these five different developmental stages (S1–S5) of male (M) and female (F) flowers were downloaded from previous study BioProject number PRJNA72492326. After removing low-quality reads using Trimmomatic37, clean reads were mapped to the reference YY genome using STAR aligner38. Mapping reads referring to each transcript were assembled and FPKM values were calculated using StringTie39. The dynamic expression pattern was presented by R package “pheatmap” based on FPKM numeric values of different samples. Expression pattern comparisons were made at each corresponding stage (S1 through S5) between the two sex types (F, female; M, male) (i.e., FS1 compared to MS1) and between early and mature stages of flower development.
qRT-PCR analysis
Samples were collected at same stages of flower development as used for RNA-sequencing but from different plant samples. The primers for expression analysis of MADS-box genes were designed by SnapGene. SYBR® Premix Ex Taq II (TaKaRa Bio Inc., Japan) was used to detect expression levels of target genes. All qPCR assays were carried out in a CFX- 96-well Real-Time System (BioRad, USA). The reaction mixture consisted of 10 μl SYBR Premix Ex Taq II, 2 μl of diluted cDNA, and 1 μM of each primer in a final volume of 20 μl. The qPCR protocol consisted of an initial thermal cycling step of 95 °C for 30 s, followed by 40 cycles of denaturation at 95 °C for 5 s and annealing with a temperature 60 °C for 60 s. All samples were repeated three times. GAPDH was used as a control gene for data normalization using the 2−∆∆ct method. All the primers are listed in Supplementary Data, Table S1.
Results
Identification and physiochemical properties of MADS-box genes in spinach
For MADS-box gene identification, we analyzed spinach genome using HMM-profile of MADS-domains (SRF-type-I (PF00319) and MEF2-type-II (PF09047)) and BLASTp searches using query of 102 Arabidopsis and 73 rice MADS-box protein sequences. Redundant and low-quality sequences with no start and stop codon were removed to improve sequence quality. Finally, 54 unique MADS-box proteins were confirmed after conserved domain analysis and serially named SoMADS1 through SoMADS54. Most of the MADS-box proteins contained MADS- MEF2 like 57% (31 genes), 35% (19 genes) contained MADS-SRF like domains and 7% (4 genes) contained no MEF2 like or SRF-like but have K-domain (Figs. 1, 2B).

Multi-sequence alignment and domain analysis of SoMADS proteins. Black box represents the conserved MADS domain.

Gene structure and distribution pattern of conserved motifs and domains in spinach. (A) Distribution of conserved motifs identified using MEME. The motifs (1–10) are depicted in different colors, and the gene names and respective groups are displayed on the left side of the figure. The ruler at the bottom indicates the amino acid length of the sequences. (B) Distribution of conserved domain in MADS-box genes using CDD-search. (C) Gene structures of MADS-box genes with full-length coding sequences. The exon–intron structure of SoMADS genes was predicted using GSDS. Introns are represented by lines, while exons are indicated by yellow boxes. The size of the introns and exons can be measured using the bottom scale.
Physiochemical properties showed that MADS-box genes varied in ORF length, ranging from 241 to 1612 bp with an encoded protein ranging from 80 a.a. to 537 a.a. The predicted PI for the proteins ranged from 4.65 to 11.03. These proteins had a molecular mass of 8–58 KDa (Table 1). Subcellular localization of spinach MADS-box proteins was predicted and shown in Table 1. Most of the proteins were predicted to localize in the nucleus. Whereas, SoMADS4, 7, 25, 29, 33, 46, 51, 52, 53, 54 were predicted in nucleus and cytoplasm, SoMADS10 and 16 in cytoplasm, SoMADS39 and 45 in the chloroplast, SoMADS23, 40 in mitochondria, and SoMADS26, 34 in both nucleus and chloroplast, SoMADS28, 50 in chloroplast and mitochondria, suggesting that these genes may require for coordinated expression in multiple organelles.
Multiple-sequence alignment, phylogenetic analysis of MADS-box genes
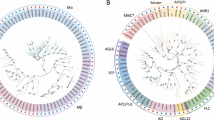
Multiple sequence alignment and evolutionary tree analyses of identified spinach MADS-box genes were performed. The multiple sequences analysis between AtMADS, OsMADS and SoMADS genes presented highly identical and conserved region (MADS-box domain), indicating the importance of this domains for MADS-box gene function. To assess the evolutionary relationship, this alignment was used to construct phylogenetic tree, and was further confirmed based on best blast-hits. Thirty-four (62.96%) out of 54 genes were considered as type-II, while 20 (37%) genes as type-I. Both type-I and type-II genes were further classified into detailed sub-groups. Nine out of 20 type-I genes were named as Mα subgroup, while 3 as Mβ and 8 as Mγ subgroup. Thirty-four type-II genes were divided into 31 MIKCc and 3 MIKC* subgroups. Based on the Arabidopsis MIKCc type-II genes classification, 31 MIKCc type-II genes were further grouped into 11 subfamilies; TT16 (1), AGL12-like (1), SEP (5), SOC1 (2), AP1/CAL/FUL (6), SVP (4), PI (2), AG/SHP (1), ANR1 (5) and AGL15/18 (3) (Table 1; Fig. 3). SoMADS7 was not categorized in phylogenetic tree and was classified as AP3 based on best-blast hit in Arabidopsis and further confirmed through NCBI blast search.

A phylogenetic tree of MADS-box genes is presented, featuring Arabidopsis, rice and spinach. The tree was constructed using the maximum likelihood method with RaxML, program and visualized using iTOL. The tree was categorized into 15 subfamilies, using Arabidopsis and rice as reference. Each sub-family is depicted in distinct colors.
Gene structure and conserved motif analysis
To further understand the development and functions of SoMADS genes, the conserved motifs and exon–intron pattern were analyzed. The relationship between 54 MADS-box genes was investigated through the construction of a phylogenetic tree using the Maximum likelihood method and grouped into type-I and type-II (Fig. 2A). Genomic structural analysis of MADS-box genes revealed that the number of exons varied from 1 to 13 (Fig. 2C). Type-I genes have simpler structure with 1 exon and no intron, however, type-II genes have complex structure comparing with type-I with exon no. 2–13. Most of the type-II members have 7 or 8 exons. These results showed most of MADS-box genes within a group have similar gene structures (same exon no.) with different intron length (Fig. 2C).
For conserved motif distribution analysis, all 54 identified MADS-box proteins were subjected to MEME software. In total, 10 motifs were identified and annotated by SMART program (Fig. 2A). Motif 1 represents MADS-domain, motif 2 K-box, while motif 4 represents coil-coil domain. Motif 1 was the most conserved MADS-domain in all MADS-box proteins except four genes (SoMADS6, 7, 9, 16) which having motif 2 (K-domain) and were consistent with multiple alignment results (Fig. 1). K-box domain was identified in most of type-II gene, but not in any type-I genes, consistent with previous studies. Overall, motif analysis showed that proteins from the same sub-family shared similar motif distribution (Fig. 2A). In conclusion, after analyzing the composition of conserved motifs, gene structure, phylogenetic relationships, the results may indicate that MADS-box genes have highly conserved amino acids and genes within a group may have the same functions.
Genomic distribution, gene duplication and collinearity analysis of MADS-box genes in spinach
All spinach MADS-box genes were distributed on six chromosomes. Sixteen (29.6%) MADS-box genes were mapped to the sex chromosome-chromosome 1 (Chr1) (7-type-II, 9-type-I), followed by 13 (24.1%) in Chr5 (9-typeII, 4-type-I), 11 (20.4) in Chr4 (7-typeII, 4-type-I), 6 (11.1%) in Chr6 (5-typeII, 1-type-I), 5 (9.3%) in Chr3 (5-typeII), and 3 (5.6%) in Chr2 (1-typeII, 2-type-I) (Table 1; Fig. 4A). The gene duplication events were analyzed. The type-II duplicated gene pair include only AP1/CAL/FUL SoMADS4/SoMADS5, while gene pairs SoMADS47/SoMADS48, SoMADS53/SoMADS54, SoMADS51/SoMADS52, and SoMADS39/SoMADS40 were identified as tandem duplicates, and are of type-I. The synonymous rate (Ks), non-synonymous rate (Ka), and Ka/Ks of these duplicates were calculated, and duplication time was speculated using the values of Ks (Table 2). The Ka/Ks values of all these duplicates are less than 1, indicating that purifying selection occurred in these duplicates and gene pairs may have similar functions. The Ks of four segment duplicates ranges from 0.07 to 0.51. Thus, the divergent time ranges from 5.4 to 39.3 million years ago.

(A) Distribution of MADS-box genes on six chromosomes in spinach genome. The marks on the left hand represent the location of these genes on their chromosome. Tandem duplications are indicated by black lines, (B) Collinear analysis of MADS-box genes in spinach and its close relatives. Gray lines in the background represent collinear relationships throughout the genome, and the blue lines mainly represent the collinear MADS-box gene pairs.
We next investigated the orthologous relationship of MADS-box genes between spinach and other related species, with MCScanX in TBtools as shown in Fig. 4B and Table S2. A total of 25, 55, 30, and 17 ortholog MADS-box gene pairs were respectively identified between spinach and Amarantus (A.hypochondriacus), quinoa (C.quinoa), Beet (B.vulgaris) and Arabidopsis (A. thaliana). Spinach had the most MADS-box ortholog pairs with C.quinoa, followed by B. vulgaris. Most of the collinear genes pairs belong to type-II (AP1/CAL/FUL; AG; SEP and SOC1) MADS-box genes, while few belong to type I genes. Type I MADS-box genes had faster birth and death compared with type II genes, which could further explain the different pattern of type I and type II genes in collinearity analysis.
Gene ontology (GO) enrichment analysis
GO analysis indicated that MADS-box genes are mainly associated with regulation of metabolic and biological process, developmental processes, and reproduction in addition to three molecular functions, namely, nucleic acid binding transcription factor activity, binding, and translation regulation activity (Fig. 5).

GO-enrichment analysis of MADS-box genes in spinach.
Expression profiling of MADS-box genes in spinach
We observed the expression profile of 54 MADS-box genes at five different stages of male and female flower development and were grouped into two categories. Stage1 and 2 were considered as early stage (mainly floral bud) and stage 3, 4, and 5 were considered as later stage (reproductive organ differentiation and maturation) of flower development in both male and female sex types. Genes with FPKM < 1 in all tissues tested were removed. Thirty-three out of 54 genes showed expression (FPKM > 1) and expression pattern observed was as follows (Fig. 6).

The red and blue colors indicate the expression levels of SoMADS genes from high to low, The tissues cover early stages of flower development (FS1: female stage1; FS2: female stage 2; MS1: male stage 1; MS2: male stage 2) and mature stages of flower development (FS3: female stage 3; FS4: female stage 4; FS5: female stage 5; MS3: male stage 3; MS4: male stage 4; MS5: male stage 5). Blue box represents male-biased expression, and red box represents female-biased expression.
ABCDE-model genes
A-class genes AP1/CAL/FUL SoMADS2, 4, 5, and 6 were found to have female-biased expression throughout flower development, however, SoMADS1 expressed at early stage (stage 1) of female flower development. B-class group is comprised of APETALA3 (AP3) and PISTILLATA (PI). In our results, three orthologous of B-class genes have been reported. SoMADS8 is the same PISTILLATA gene reported in previous study (Accession no. GQ120478)40. While SoMADS9 is newly identified in our analysis. SoMADS7 (AP3) is different from what was reported previously (Accession no. GQ120477)40, it showed male-biased expression at stage1 but more especially at male flower maturation stages (stage 4 and 5). Both PISTILATTA genes SoMADS8 and 9 were expressed throughout male flower development. While there was no detectable transcript expression for these genes in any of the female flower development stage. There is another homeotic gene sub-group closely related to B-class, named the B-sister class (TT16). In our study, one B-sister gene SoMADS10 expressed during (early-stage 2nd) and especially later stage (ovule differentiate and maturation stage 3–5) of female flower, and negligible expression was detected in male flower. No D-class, SEEDSTICK (STK) related gene was identified in YY genome. Five genes were associated with the E-class, namely, SoMAD12, 13, 14, 15, and 16. SEPETELLA genes SoMADS14, 15, 16 display female-biased expression throughout flower developmental stages, while SoMADS12,13 showed male-biased expression at stage 1 of male flower development suggest their role in stamen induction (Fig. 6). Further, we chosen seven SoMADS genes (SoMADS8, SoMADS9 (PI), SoMADS7 (AP3), SoMADS22 (SVP), SoMADS2, SoMADS5, SoMADS4 (AP1/CAL/FUL), SoMADS15.16 (SEP) with significant differences in expression pattern between male and female at either early, mature or throughout flower developmental stages for qRT-PCR to validate the RNA-seq data (Fig. 7). The expression patterns of the seven SoMADS-box genes were tested and basically consistent with the RNA-seq data, suggesting that the RNA-seq data were reliable.

Expression level of SoMADS2,5,4 (AP1/CAL/FUL), SoMADS8.9 (PI), SoMADS7 (AP3), SoMADS15,16 (SEP) and SoMADS22 (SVP) at five different stages of male and female flower development. QRT-PCR data was analyzed using the 2^(−ΔΔCT) method. The results are presented as mean ± standard error (SE) with a sample size of 3 (n = 3). Left side scale (black color) represents qPCR results, while right side scale (brown color) represents RNA-seq result.
Flowering-time and other MADS-box genes
Two representative genes, SOC1 and SVP, play critical and antagonistic roles in the floral transition. In this study, four SVP (SoMADS19, 20, 21 22) one SOC1 (SoMADS18) showed female-bias expression at stage 1 of flower development, while SVP SoMADS20, 21, 22 continue their expression at later stage suggest their role in female flower development. AGL15 represses floral transition, together with its close paralog AGL18 in Arabidopsis. In spinach SoMADS24 (AGL15 ortholog) and SoMADS26 (AGL18 ortholog) expressed at later stage of male flower. Three MIKC* genes (SoMADS 32, 33, 34) showed male-bias expression at later stage (4th and 5th stage of anther maturation). Five genes of ANR1 showed no significant expression in any developmental stages of both sex types except for SoMADS27 at later stage of male flower (Figs. 2, 4). Type-I genes (Mα, Mβ, Mγ) didn’t show any expression in our study except for SoMADS35, 46 (Mβ), SoMADS41, 43 (Mα) at 3rd and 4th stage (anther differentiation) of male flower indicate their role in stamen development. Overall, transcripts of type-II SoMADS were more abundant than type-I.
Cis-regulatory element and expression pattern analysis under hormone treatment
Plant hormones play significant roles in both flowering and flower development processes. To gain deeper insights into how spinach MADS-box genes respond to phytohormones, an analysis of cis-acting elements was conducted. This analysis focused on the promoter region, specifically the 2-kb region upstream of the transcription start codon, across all 54 genes. These cis-elements were categorized into six overarching groups based on their interactions with various plant hormones, including auxin (TGA element, AuxRR-core), abscisic acid (ABRE), ethylene (ERE), gibberellin (P-box, GARE-motif, TATC-box), methyl jasmonate-MeJA (CGTCA-motif, TGACG-motif), and salicylic acid (TCA-element, SARE). Among these categories, ABRE (37.5%) was the most prevalent, followed by MeJA-responsive elements TGACG-motif (15%) and CGTCA-motif (15%), as illustrated in Fig. 8C.

Enrichment of phytohormones related cis-elements in promoter region of MADS-box genes. (A) Type-II genes (B) Type-I genes (C) Ration of all phytohormones related cis-elements found in 54 SoMADS (D) qPCR analysis of five genes under control and different hormones (ABA, Auxin, GA and JA) treatments in male flower(MF), female flower (FF), male leaf (ML), and female leaf (FL) samples. Different colors show the candidate genes chosen for the analysis.
Type I genes contained up to eight ABA response elements, up to five ethylene response elements, and up to ten MeJA response elements, while one type-I SoMADS45 has 24 MeJA response elements suggest that it might strongly affected by JA. Type II genes possessed up to nine ABA response elements,up to ten MeJA response elements, and up to four ethylene response elements (Fig. 8). AP1/CAL/FUL (SoMADS1, 4, 5) SEP (SoMADS13,14), ANRI (SoMADS27,31), MIKC* (SoMADS34) had the most (7–9) ABA response elements. PI (SoMADS8), SVP (SoMADS21), AGL18 (SoMADS25), and ANRI (SoMADS30) contained most (8–10) MeJA response elements, suggesting these genes might strongly responsive to jasmonic acid signals. However, only few (around 3–4) auxin, SA and GA response elements were present in the promoter of MADS-box genes. The detailed distribution of cis-elements in all SoMADS genes is provided in heatmap (Fig. 8).
To see whether, these cis-elements in SoMADS genes are really responsive to hormone signaling, we treated the spinach plant with 30uM of GA, JA, ABA and IAA, respectively and collected samples of flower (mature flower before pollination), leaves (fresh leaves at the top of plant) tissues from male and female sex types for qPCR analysis. We chose five candidate genes (AP1/CAL/FUL SoMADS 4,5; PI SoMADS8, SEP SoMADS14; and ANRI SoMADS 27), to observe their expression in these tissues Fig. 8D. SoMADS4 and 5 showed increased expression in flower under ABA, IAA in male and female, more significant increase was observed in female flower. SoMADS4 also showed increased expression in female leaves under IAA treatment. SoMADS8 showed significantly higher expression in female and male flower under JA, GA and ABA treatment. SoMADS14 showed higher expression in female leaves than male under ABA and IAA treatment. SoMADS27showed higher expression in leaves in both male and female under IAA treatment. This shows that MADS-box genes are responsive to hormones, but the change in expression is sex-biased. More detailed study of MADS-box genes under hormone treatment should be conducted in future.
Protein–protein interaction of SoMADS
To see whether these MADS-box protein interact with each other, we conducted the protein–protein interaction network analysis of MADS-box proteins based on known Arabidopsis proteins. SoMADS portions having higher homologous similarity with Arabidopsis proteins were selected as STRING proteins and these were same as our best-blast hits for these genes classification. Among all, 25 SoMADS proteins (nodes) associated with known Arabidopsis proteins showed interaction (Fig. 9). SoMADS7 homologous to AtAP3 protein had interaction with API homologous proteins SoMADS5, PI SoMADS9, AG SoMADS11 and SEP SoMADS15, 16 proteins. SoMADS9 showed homology with AtPI protein and interacts with AP3 homologous protein SoMADS7, and AG homologous SoMADS11. SoMADS18 showed homology with SOC1 protein and interacts with AP1 homologous SoMADS5, 6, SEP SoMADS16, SVP SOMADS22. SoMADS5 showed homology with AP1 protein, showed interaction with AP3 SoMADS7, SEP SoMADS15, 16, SOC1 SoMADS18, and SVP SoMADS19, 22. Arabidopsis proteins belonging to different groups may have diverse functions but proteins which have strong interaction may form a dimer to perform similar functions as in Arabidopsis. The higher the interaction coefficient, the thicker the line between proteins and vice versa. All interaction of SoMADS proteins based on Arabidopsis homologues can be seen in (Fig. 9; Supplementary Data, Table S3).

Protein–protein interaction of SoMADS based on Arabidopsis orthologs. The parameters including the number of nodes: 25, average node degree: 2.56, and avg. local clustering coefficient: 0.421.
Discussion
Overview of MADS-box in spinach
MADS-box transcription factors play important roles in flowering and floral organ development in plants41,42. The identification and evolutionary analysis of MADS-box gene family have been intensively studied in many plant species, including Arabidopsis (108), rice (75), Brassica (160), soybean (163), maize (75), Populus trichocarpa (105), Pyrus bretschneideri (95), Malus × domestica (147) etc.9,27,43,44,45,46,47,48. When contrasted with these species, certain plants such as pineapple (with 48 members) and bamboo (with 42 members) exhibit a comparatively reduced count of MADS-box genes49,50. In our study, comparatively small number (54) of MADS-box genes were identified in spinach. Previous studies showed gene duplication events and whole-genome duplications (WGDs) are the causes for MADS-box genes expansion51,52. Genome triplication event (γ) had been reported during the evolutionary history of spinach genome and found to be shared by core eudicots53. However, no recent WGD (ρ) event was occurred in spinach, while, recent WGD has been observed in Populus trichocarpa with 105 MADS-box genes46,54.
MADS-box genes are categorized into two distinct classes: type-I and type-II, each with distinct evolutionary origins. Within the type-II genes, the MIKCc cluster stands out for its plant-specific nature and its significant contributions to floral organogenesis55. However, flowering plants widely differ in the number of MIKCc family genes9,27,43,44. Based on phylogenetic tree analysis, spinach contains 31 MIKCc type-II MADS-box genes that could further be categorized into 11 subfamilies. In addition, type-II SoMADS genes have complex structure with average of 7–8 exons, while type-I genes having only one exons. The similar phenomena have also been observed in rice27, maize45, soybean44, B. rapa43, Brachypodium56, and other species, which can be interpreted as a divergence between type I and type II MADS genes concerning their inclinations for intron deletion or acquisition, highlighting a conserved evolutionary pattern within the plant kingdom. SoMADS genes sharing the same group displayed analogous motif arrangements, indicating shared ancestral evolutionary origins. The uniformity in terms of intron count, motif composition, and phylogenetic relationships serves as evidence that the classification of these genes was accurate. Most of the MADS-box genes were distributed on Chr-1 but none of them were the part of sex-determination region (Chr1:145.31–162.73 Mb)26. Duplication analysis revealed that most of the duplicated gene pairs belong to type-I. Collinearity analysis showed spinach exhibited the highest number of MADS-box ortholog pairs with quinoa, followed by B. vulgaris and A.hypochondriacus attributed to their close evolutionary relationship. It was determined that quinoa and spinach, part of the Chenopodiaceae subfamily, underwent divergence approximately 16 million years ago. Furthermore, they both share a common ancestor with A. hypochondriacus, with divergence occurring around 25 million years ago57. The majority of collinear gene pairs belonged to type-II MADS-box genes (AP1/CAL/FUL, AG, SEP, and SOC1), with fewer belonging to type I genes. Studies showed that type-I genes have experienced faster birth-and-death evolution than type-II genes in the Arabidopsis and rice lineages, potentially explaining the distinct patterns observed in gene duplication and collinearity analysis58.
Through promoter analysis of MADS-box genes in spinach, a numerous potential ABA and JA response elements were identified (Fig. 8). The role of ABA in promoting flowering and flower formation has been evidenced in litchi and apple studies59,60, which contrasts with observations in Pharbitis nil, where ABA has been found to inhibit flowering61. Notably, JA signaling has demonstrated the ability to stimulate anther filament elongation, trigger stomium opening during anthesis, and facilitate the generation and release of viable pollen62. The male fertility of JA mutants can be restored through the application of exogenous jasmonic acid63. Hormone-responsive cis-elements in SoMADS suggest that these genes might be responsive to phytohormones at flower induction and development stages.
MIKCc genes might contribute towards unisexual flower development in spinach
Type-I SoMADS-genes were not expressed, while MIKCc showed diverse expression during female and male flower development stages suggest that MIKCc type genes play important roles in the growth and development of reproductive tissues.
The well-known ABCDE model plays a crucial role in determining the specific floral organ identities. Within this model, the E-class SEP proteins hold significant importance as they contribute to petal and stamen identities by facilitating interactions between class-A and -B proteins and between class-B and -C proteins, respectively16,64. Across various species, SEP-like genes exhibit varying degrees of sub-functionalization and neo-functionalization65. In Arabidopsis, a decrease in SEP activity results in abnormal ovule development, mirroring the effects observed in stk shp1 shp2 triple mutants66. Expression profiling of E-class (SEP) SoMADS14, 15, 16 genes showed significant female-biased expression throughout flower development indicate their role in carpel induction, differentiation and ovule maturation. A-class AP1/CAL designated as floral meristem and sepal/petal identity genes in Arabidopsis67, while FUL has a wide range of functions; e.g. in the flower organ specification and carpel development68. A total of six AP1/CAL/FUL: homologous genes (SoMADS1- 6) were detected and four of them (SoMADS2, 4, 5, 6) showed relatively higher expression in female flower throughout their developmental stages. In Arabidopsis, the AP1 protein engages in dimerization with flowering time-associated genes SVP (and/or SOC1), resulting in the inhibition of B and C type gene expression at early flower development69,70. The distinct levels of expression seen in B and C class genes have been linked to the development of unisexual flowers in dioecious Spinacia oleracea and monoecious Quercus suber L.40,71. In situ hybridization study in spinach revealed that B-class genes (AP3, PI) expressed in male than female parts of flower40, and suppression of their expression in male spinach resulted not only switch from staminate to pistillate flowers, but also the formation of gynoecia in the normally absent fourth whorl in contrast with Arabidopsis where only organ identity changes. However, the AP3 identified in YY genome is different from what was identified in previous studies, and not significantly differentially expression at early stage of flower development (Fig. 7)40. Comparing the transcriptome data of spinach sex-types at different developmental stages and co-expression analysis with reference to candidate YY-specific sex determinant (NRT1/PTR6.4) in sex-determination region on sex chromosome reveal that B-class genes might function as an important intermediate for stamen/carpel differentiation and might regulated by JA/GA26. PISTILLATA gene SoMADS8 promoter found to have enriched MeJA cis-element suggest that it might strongly regulated by JA. In dioecious persimmons, SVP and SOC1 promote gynoecium development and suppress androecium under the control of the homeodomain transcription factor, MeGI that can determine floral sexuality72. Transgenic Arabidopsis revealed a significantly negative correlation between MeGI/SVP and PI in the androecium72. In this study, SVP (SoMADS19, 20, 21, 22), SOC1 (SoMADS18) along with AP1/CAL/FUL (SoMADS2, 4, 5, 6) showed female-biased expression from early stage of flower development, in contrast with B-class PISTILLATA genes (SoMADS8,9) which showed male-specific expression throughout flower development indicate that these genes (AP1, SVP, SOC1) might encode important repressors of B-class genes during sex organ differentiation and might act downstream of sex-determination gene. And this hypothesis also strengthen by protein–protein interaction analysis in which most of the AP1, SVP, SOC1, PI and SEP related proteins showed interaction based on Arabidopsis homologues (Fig. 9). Based on the expression profile we proposed a simple model for MADS-box function in unisexual flower development in spinach (Fig. 10), and this need to be verified in future.

MADS-box model might involve in unisexual flower development in spinach.
Conclusion
The role of the MADS-box gene family in determining the identity and specification of flower organs has been extensively documented in eudicots. This study identified a total of 54 genes within the spinach YY genome. A comparison of phylogenetic relationship among spinach, rice, and Arabidopsis categories spinach SoMADS into 34 type II and 20 type I genes. Expression profiles of these MADS-box genes at different flower developmental stages between male and female sex types suggested that most of A-class (AP1/CAL/FUL), E-class (SEP) and flowering-time genes (SVP, SOC1) has female-biased expression, while B-class (PI) has male-biased expression. As sexual dimorphism in spinach does not stem from the homeotic transformation of pre-existing organs; instead, it arises from the distinct initiation and development patterns of the third and fourth whorl primordia. There is a likelihood that the control of sex determination has its origins upstream of the ABCDE genes that are responsible for floral organ identity. In the scenario where sex determination regulation initiates upstream, it becomes evident that the MADS-box genes play a crucial role as central integrating points within the regulatory cascade, and the functional characterization of the identified key genes in this study need to be performed in near future for better understanding of fundamental biological process of unisexual flower development in spinach.
Data availability
The transcriptome reads sequences are publically available at NCBI BioProject, under the accession number: PRJNA724923 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA724923/).
References
Theißen, G., Melzer, R. & Ruümpler, F. MADS-domain transcription factors and the floral quartet model of flower development: Linking plant development and evolution. Development 143, 3259–3271 (2016).
Smaczniak, C., Immink, R. G. H., Angenent, G. C. & Kaufmann, K. Developmental and evolutionary diversity of plant MADS-domain factors: Insights from recent studies. Development 139, 3081–3098 (2012).
Fatima, M. et al. Expression profiling of MADS-box gene family revealed its role in vegetative development and stem ripening in S. spontaneum. Sci. Rep. 10, 1–14 (2020).
Pellegrini, L., Tan, S. & Richmond, T. J. Structure of serum response factor core bound to DNA. Nature 376, 490–498 (1995).
Yanofsky, M. F. et al. The protein encoded by the Arabidopsis homeotic gene agamous resembles transcription factors. Nature 346, 35–39 (1990).
Norman, C., Runswick, M., Pollock, R. & Treisman, R. Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell 55, 989–1003 (1988).
Passmore, S., Maine, G. T., Elble, R., Christ, C. & Tye, B. K. Saccharomyces cerevisiae protein involved in plasmid maintenance is necessary for mating of MATα cells. J. Mol. Biol. 204, 593–606 (1988).
Sommer, H. et al. Deficiens, a homeotic gene involved in the control of flower morphogenesis in Antirrhinum majus: The protein shows homology to transcription factors. EMBO J. 9, 605–613 (1990).
Pařenicová, L. et al. Molecular and phylogenetic analyses of the complete MADS-Box transcription factor family in Arabidopsis new openings to the MADS world. Plant Cell 15, 1538–1551 (2003).
Henschel, K. et al. Two ancient classes of MIKC-type MADS-box genes are present in the Moss Physcomitrella patens. Mol. Biol. Evol. 19, 801–814 (2002).
Theißen, G. & Saedler, H. Floral quartets. Nature 409, 469–471 (2001).
Hartmann, U. et al. Molecular cloning of SVP: A negative regulator of the floral transition in Arabidopsis. Plant J. 21, 351–360 (2000).
Searle, I. et al. The transcription factor FLC confers a flowering response to vernalization by repressing meristem competence and systemic signaling in Arabidopsis. Genes Dev. 20, 898–912 (2006).
Ferrándiz, C., Liljegren, S. J. & Yanofsky, M. F. Negative regulation of the SHATTERPROOF genes by FRUITFULL during Arabidopsis fruit development. Science 289, 436–438 (2000).
Nesi, N. et al. The transparent testa 16 locus encodes the arabidopsis bsister mads domain protein and is required for proper development and pigmentation of the seed coat. Plant Cell 14, 2463–2479 (2002).
Pelaz, S., Ditta, G. S., Baumann, E., Wisman, E. & Yanofsky, M. F. B and C floral organ identity functions require SEPALLATTA MADS-box genes. Nature 405, 200–203 (2000).
Weigel, D. & Meyerowitz, E. M. The ABCs of floral homeotic genes. Cell 78, 203–209 (1994).
Ehlers, K. et al. The MADS box genes ABS, SHP1, and SHP2 are essential for the coordination of cell divisions in ovule and seed coat development and for endosperm formation in Arabidopsis thaliana. PLoS ONE 11, e0165075 (2016).
Pan, Z. J. et al. Flower development of Phalaenopsis orchid involves functionally divergent SEPALLATA-like genes. New Phytol. 202, 1024–1042 (2014).
Samach, A. et al. Distinct roles of constans target genes in reproductive development of Arabidopsis. Science 288, 1613–1616 (2000).
Michaels, S. D. & Amasino, R. M. FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 11, 949–956 (1999).
Ren, Z. et al. Genome-wide identification of the MIKC-type MADS-Box gene family in Gossypium hirsutum L. unravels their roles in flowering. Front. Plant Sci. 8, 384 (2017).
Mendoza, L., Thieffry, D. & Alvarez-Buylla, E. R. Genetic control of flower morphogenesis in Arabidopsis thaliana: A logical analysis. Bioinformatics 15, 593–606 (1999).
Sherry, R. A., Eckard, K. J. & Lord, E. M. Flower development in dioecious Spinacia oleracea (Chenopodiaceae). Am. J. Bot. 80, 283 (1993).
Sather, D. N., Jovanovic, M. & Golenberg, E. M. Functional analysis of B and C class floral organ genes in spinach demonstrates their role in sexual dimorphism. BMC Plant Biol. 10, 46 (2010).
Ma, X. et al. The spinach YY genome reveals sex chromosome evolution, domestication, and introgression history of the species. Genome Biol. 23, 1–30 (2022).
Arora, R. et al. MADS-box gene family in rice: Genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genom. 8, 242 (2007).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 39, W29 (2011).
Gasteiger, E. et al. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook 571–607 (Humana Press, 2005).
Edgar, R. C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577 (2007).
Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312 (2014).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016).
Chen, C. et al. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202 (2020).
Juretic, N., Hoen, D. R., Huynh, M. L., Harrison, P. M. & Bureau, T. E. The evolutionary fate of MULE-mediated duplications of host gene fragments in rice. Genome Res. 15, 1292–1297 (2005).
Lynch, M. & Conery, J. S. The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155 (2000).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Pfent, C., Pobursky, K. J., Sather, D. N. & Golenberg, E. M. Characterization of SpAPETALA3 and SpPISTILLATA, B class floral identity genes in Spinacia oleracea, and their relationship to sexual dimorphism. Dev. Genes Evol. 215, 132–142 (2005).
Teo, Z. W. N., Zhou, W. & Shen, L. Dissecting the function of MADS-Box transcription factors in orchid reproductive development. Front. Plant Sci. 10, 1474 (2019).
Castelán-Muñoz, N. et al. MADS-box genes are key components of genetic regulatory networks involved in abiotic stress and plastic developmental responses in plants. Front. Plant Sci. 10, 853 (2019).
Duan, W. et al. Genome-wide analysis of the MADS-box gene family in Brassica rapa (Chinese cabbage). Mol. Genet. Genom. 290, 239–255 (2015).
Fan, C.-M. et al. Genome-wide expression analysis of soybean MADS genes showing potential function in the seed development. PLoS ONE 8, e62288 (2013).
Zhao, Y. et al. Whole-genome survey and characterization of MADS-box gene family in maize and sorghum. Plant Cell. Tissue Organ Cult. 105, 159–173 (2011).
Leseberg, C. H., Li, A., Kang, H., Duvall, M. & Mao, L. Genome-wide analysis of the MADS-box gene family in Populus trichocarpa. Gene 378, 84–94 (2006).
Wang, R. et al. Genome-wide identification of the MADS-box transcription factor family in pear (Pyrus bretschneideri) reveals evolution and functional divergence. PeerJ 5, e3776 (2017).
Velasco, R. et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 42, 833–839 (2010).
Zhang, X., Fatima, M., Zhou, P., Ma, Q. & Ming, R. Analysis of MADS-box genes revealed modified flowering gene network and diurnal expression in pineapple. BMC Genom. 21, 1–16 (2020).
Zhang, Y., Tang, D., Lin, X., Ding, M. & Tong, Z. Genome-wide identification of MADS-box family genes in moso bamboo (Phyllostachys edulis) and a functional analysis of PeMADS5 in flowering. BMC Plant Biol. 18, 1–18 (2018).
Paterson, A. H., Bowers, J. E. & Chapman, B. A. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. U. S. A. 101, 9903–9908 (2004).
Yu, J. et al. The genomes of Oryza sativa: A history of duplications. PLoS Biol. 3, e38 (2005).
Xu, C. et al. Draft genome of spinach and transcriptome diversity of 120 Spinacia accessions. Nat. Commun. 8, 1–10 (2017).
Rodgers-Melnick, E. et al. Contrasting patterns of evolution following whole genome versus tandem duplication events in Populus. Genome Res. 22, 95–105 (2012).
Guan, H. et al. Genome-wide identification and expression analysis of MADS-box family genes in litchi (Litchi chinensis sonn.) and their involvement in floral sex determination. Plants 10, 2142 (2021).
Wei, B. et al. Genome-Wide Analysis of the MADS-Box Gene Family in Brachypodium distachyon. PLoS ONE 9, e84781 (2014).
Zou, C. et al. A high-quality genome assembly of quinoa provides insights into the molecular basis of salt bladder-based salinity tolerance and the exceptional nutritional value. Cell Res. 27, 1327 (2017).
Nam, J. et al. Type I MADS-box genes have experienced faster birth-and-death evolution than type II MADS-box genes in angiosperms. Proc. Natl. Acad. Sci. U. S. A. 101, 1910–1915 (2004).
Li, W. F. et al. Exogenous ABA and its inhibitor regulate flower bud induction of apple cv. ‘Nagafu No. 2′ grafted on different rootstocks. Trees 35, 609–620 (2021).
Cui, Z., Zhou, B., Zhang, Z. & Hu, Z. Abscisic acid promotes flowering and enhances LcAP1 expression in Litchi chinensis Sonn. South Afr. J. Bot. 88, 76–79 (2013).
Frankowski, K. et al. Ethylene, auxin, and abscisic acid interactions in the control of photoperiodic flower induction in Pharbitis nil. Biol. Plant. 58, 305–310 (2014).
Wilson, Z. A., Song, J., Taylor, B. & Yang, C. The final split: The regulation of anther dehiscence. J. Exp. Bot. 62, 1633–1649 (2011).
Song, S. et al. The Jasmonate-ZIM domain proteins interact with the R2R3-MYB transcription factors MYB21 and MYB24 to affect jasmonate-regulated stamen development in Arabidopsis. Plant Cell 23, 1000–1013 (2011).
Honma, T. & Goto, K. Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 409, 525–529 (2001).
Acri-Nunes-Miranda, R. & Mondragón-Palomino, M. Expression of paralogous SEP-, FUL-, AG- and STK-like MADS-box genes in wild-type and peloric Phalaenopsis flowers. Front. Plant Sci. 5, 76 (2014).
Favaro, R. et al. MADS-Box protein complexes control carpel and ovule development in Arabidopsis. Plant Cell 15, 2603–2611 (2003).
Alejandra Mandel, M., Gustafson-Brown, C., Savidge, B. & Yanofsky, M. F. Molecular characterization of the Arabidopsis floral homeotic gene APETALA1. Nature 360, 273–277 (1992).
Litt, A. & Irish, V. F. Duplication and diversification in the APETALA1/FRUITFULL floral homeotic gene lineage: Implications for the evolution of floral development. Genetics 165, 821–833 (2003).
Liu, C. et al. Direct interaction of AGL24 and SOC1 integrates flowering signals in Arabidopsis. Development 135, 1481–1491 (2008).
Gregis, V., Sessa, A., Dorca-Fornell, C. & Kater, M. M. The Arabidopsis floral meristem identity genes AP1, AGL24 and SVP directly repress class B and C floral homeotic genes. Plant J. 60, 626–637 (2009).
Sobral, R. & Costa, M. M. R. Role of floral organ identity genes in the development of unisexual flowers of Quercus suber L. Sci. Rep. 7, 1–15 (2017).
Yang, H. W., Akagi, T., Kawakatsu, T. & Tao, R. Gene networks orchestrated by MeGI: A single-factor mechanism underlying sex determination in persimmon. Plant J. 98, 97–111 (2019).
Funding
RNA-seq, qPCR experiment and publication fee were supported by startup fund from Natural Science Foundation of Fujian Province, China (Grant No. 2018J01606).
Author information
Authors and Affiliations
Contributions
M.F. and R.M. conceived the project and designed experiments. M.F., M.X. carried out the experiment. M.F. analyzed the data and wrote the manuscript. R.M. and J.Z. revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fatima, M., Ma, X., Zhang, J. et al. Genome-wide analysis of MADS-box genes and their expression patterns in unisexual flower development in dioecious spinach. Sci Rep 14, 18635 (2024). https://doi.org/10.1038/s41598-024-68965-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68965-9
- Springer Nature Limited