
Abstract
The existing diagnostic methods of epilepsy have great limitations, and more reliable and less difficult diagnostic methods are needed. We collected serum samples of adult patients with first-diagnosed epilepsy (EPs) and seizure control patients (EPRs) for non-targeted metabolomics detection and found that they were both significantly altered, with increased expression of nicotine addiction, linoleic acid metabolism, purine metabolism, and other metabolic pathways. The diagnostic model based on 4 differential metabolites achieved a diagnostic efficiency of 99.4% in the training cohort and 100% in the validation cohort. In addition, the association analysis of oral flora, serum metabolism, and clinical indicators also provided a new angle to analyze the mechanism of epilepsy. In conclusion, this study characterized the serum metabolic characteristics of EPs and EPRs and the changes before and after epilepsy control based on a large cohort, demonstrating the potential of metabolites as non-invasive diagnostic tools for epilepsy.
Similar content being viewed by others

Introduction
Epilepsy is one of the most common brain diseases, which is characterized by repetitive, episodic, and transient central nervous system dysfunction caused by excessive discharge of brain neurons1,2. Epilepsy affects more than 70 million people worldwide and affects people of all ages with high mortality and disability rates. The World Health Organization has listed it as one of the neuropsychiatric diseases that need to be treated and prevented urgently3,4. The diagnosis of epilepsy requires comprehensive judgment based on the patient's medical history, clinical manifestations and signs, electroencephalogram, neuroimaging, and genetic testing. Because of the complex etiology and seizure types of epilepsy, the choice of antiepileptic drugs is also very difficult. How to diagnose and treat epilepsy more timely and accurately has always been a hot spot and major difficulty in the field of international epilepsy research.
Metabolomics plays an important role in the study of systems biology, which has the advantages of high throughput, high sensitivity, and high precision, and can combine the changes of endogenous metabolites of organisms with the physiological and pathological changes of diseases. In recent years, metabolomics research has developed rapidly, and it is often used in the field of medical research to screen potential biomarkers of diseases, type diagnosis, prognosis, and efficacy evaluation. Metabolites are the result of local or systemic cellular responses5. Because metabolites can cross the blood–brain barrier, they may to some extent indicate changes in the biology of the central nervous system6,7. Beamer et al. found that blood levels of adenosine and its breakdown products can indicate epilepsy8. Engelke et al.9 combined metabolomics with infrared ion spectroscopy and found that 6-oxoPIP can be used as a biomarker for the diagnosis of pyridoxine-dependent epilepsy (PDE-ALDH7A1). However, the metabolome of large epilepsy cohorts has not been characterized or studied.
Therefore, this study intends to reveal the changes in serum metabolomics of epileptic patients through a large cohort, clarify the material basis of epilepsy from the perspective of metabolomics, provide the biological basis and new ideas for further studies, and establish a diagnostic model by identifying differential potential biomarkers. In addition, we combined oral microbiome, serum metabolome, and clinical indicators to provide a broad theoretical basis for the early diagnosis and pathogenesis of epilepsy.
Results
The research protocol and flow chart
A total of 944 samples from Henan Province were prospectively included in this study for statistical analysis, including 509 tongue swabs (186 EPs, 22 EPRs, 301 HCs) and 435 serum samples (131 EPs, 22 EPRs, 282 HCs). We randomly selected the serum samples of 100 EPs and 200 HCs as a discovery cohort, identified different metabolites and their key metabolic pathways between the two groups, obtained the best potential biomarkers using random forest and fivefold cross-validation, and established a diagnostic model. Furthermore, we verified the diagnostic capability of the classifier with serum samples of 31 EPs and 82 HCs. We performed an association analysis of tongue flora, serum metabolites, and laboratory indicators for the same individual (100 EPs and 200 HCs). We also explored the serum metabolomics of 22 EPRs and 44 HCs (19 EPs and 19 EPRs). The specific research framework is shown in Fig. 1.

Research program. After strict inclusion and exclusion of 439 serum samples collected from Henan Province, 435 serum samples (282 HCs, 131 EPs, 22 EPRs) were analyzed. They were randomly divided into the discovery cohort and validation cohort, the former characterized metabolomic features, searched for markers, and established the prediction model by random forest method, while the latter conducted validation. In addition, Spearman correlation analysis was performed to explore the relationship between oral flora, serum metabolism, and clinical indicators among different populations. HCs, healthy controls; EPs, patients diagnosed with epilepsy; EPRs, patients whose seizures were under control; RFC, random forest classifier.
Basic information and clinical characteristics of participants
The basic information of 100 EPs and 200 HCs for serum samples was shown in Supplementary Table 1. There was no significant difference in sex or age between EPs and HCs (P > 0.05). Eps exhibited statistically significant differences in red blood cell (RBC) count, white blood cell (WBC) count, hemoglobin (Hb), albumin (ALB), uric acid (UA), estimated glomerular filtration rate (eGFR), and total bilirubin (TBIL) compared with HCs (P < 0.05), but these values were all within the normal range.
Characterization of serum metabolomics of EPs and establishment of diagnostic model
We explored serum metabolomic characteristics of EPs and HCs using untargeted metabolomic approaches. The principal component analysis (PCA) score graph (Supplementary Fig. 1A) showed a high degree of aggregation of quality control (QC), indicating good repeatability of QC and stability of the analysis system. In the discovery cohort, orthogonal partial least squares discrimination analysis (OPLS-DA) in multivariate statistical analysis showed that EPs and HCs were significantly separated (Fig. 2A). The intercept of Q2 in the permutation test was less than 0, indicating that the fitting effect of the OPLS-DA model was very good (Fig. 2B). Based on the identification and quantification of 1090 metabolites, 257 (23.58%) with intergroup differences were screened (variable importance in projection (VIP) > 1, P < 0.05) (Supplementary Table 2).

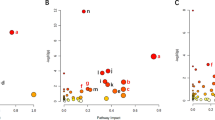
Characterization of serum metabolomics in EPs and HCs and the establishment of the diagnostic model. (A) The OPLS-DA score showed that the metabolites of the two groups (EPs, n = 100; HCs, n = 200) were significantly different. (B) The permutation test indicates that the model had a good fitting effect. (C) In the volcano plot, the dots on the left are differentially downregulated metabolites, and the dots on the right are differentially upregulated metabolites. The abscissa is the multiple of metabolite expression between the two groups (log2-fold change); the ordinate is the statistical test value of the difference in metabolite expression levels (-log10 (P value)). (D) A KEGG topology bubble diagram shows the relative influence of different metabolites on metabolic pathways between the two groups. Each bubble in the figure represents a KEGG pathway, and the horizontal axis represents the relative importance of metabolites in the pathway. The vertical axis shows the enrichment significance of metabolite-involved pathways. Bubble size is represented by Impact Value. The color represents the P value for pathway enrichment. (E) The POD value of EPs was significantly higher than that of HCs in the discovery cohort. (F) In the discovery cohort, the AUC was 99.4%. (G) In the validation cohort, the POD value of EPs (n = 31) was significantly higher than that of HCs (n = 82). (H) In the validation cohort, the AUC is 100%. OPLS-DA, Orthogonal Partial Least Squares Discrimination Analysis; KEGG, Kyoto Encyclopedia of Genes and Genomes; POD, probability of disease; AUC, area under the ROC curve; HCs, healthy controls; EPs, patients diagnosed with epilepsy. Centerline, median; box limits, upper and lower quartiles; circle or square symbol, mean; error bars, 95% CI.
According to multiples of changes in metabolite levels in EPs relative to HCs, a volcano plot (Fig. 2C) illustrated upregulated and downregulated metabolites. According to the heatmap (Supplementary Fig. 1F, Supplementary Table 2), expression levels of 117 metabolites, including malathion monocarboxylic acid, serylproline, aspartyl-threonine, and 7-Methyl-3-oxo-6-octenoyl-CoA were increased in EPs, whereas levels of 140 metabolites, such as Asp-Phe, phenylalanylphenylalanine, and artonol B decreased. Moreover, we found that these elevated differential metabolites were mainly fatty acyls and carboxylic acids and derivatives. A line chart depicts the expression trend of the two subclusters in all samples (Supplementary Fig. 1B), and the blue line represents the average expression of all metabolites in the subcluster.
KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment analysis (Supplementary Fig. 1D, Supplementary Table 3) was performed for different metabolites between groups. Of the 69 metabolic pathways that were enriched, 15 were statistically significant (P < 0.05). Among them, Nicotine addiction, Caffeine metabolism, Choline metabolism in cancer, and other metabolic pathways were significantly different. Furthermore, we analyzed the relative effects of different metabolites on metabolic pathways, and D-glutamine and D-glutamate metabolism, cutin, suberine and wax biosynthesis, caffeine metabolism, lysine biosynthesis, and alanine, aspartate and glutamate metabolism were the most affected (Fig. 2D).
In addition, we found four metabolite molecules that could most accurately distinguish epilepsy patients from healthy controls, and they were L-alpha-glutamyl-L-hydroxyproline, Vulgarone A, MG(0:0/14:1(9Z)/0:0) and Cis-3-Hexenyl phenylacetate, respectively (Supplementary Fig. 1C, E, Supplementary Table 4). Based on the optimal tag set, the POD (Probability of Disease) index of EPs in the queue was significantly higher than that of HCs (Fig. 2E), and the AUC (Area Under the Curve) reached 99.4% (95% CI: 98.24% to 100%, P < 0.0001) (Fig. 2F). Furthermore, 31 EPs and 82 HCs were used as the validation cohort to test the diagnostic capability of the model. The results showed that the POD index of EP was also higher than that of HC (Fig. 2G), with an AUC of 1 (95% CI: 100% to 100%, P < 0.0001) (Fig. 2H). These results indicate that specific serum metabolites can be used to diagnose epilepsy.
Association analysis of oral microecology, serum metabolome, and laboratory indicators in Eps and HCs
We matched tongue swabs and serum samples from the same individual and obtained 100 EPs and 200 HCs. In association analysis of oral microflora, metabolites, and clinical indicators (Fig. 3), there was a significant correlation among 21 OTUs (Operational Taxonomic Units), 10 metabolites, and 6 clinical indicators. In correlation analysis between clinical indicators and metabolomics (Supplementary Fig. 2A), ALB and TBIL correlated negatively with 7 metabolites (e.g., 7-methyl-3-oxo-6-octenoyl-CoA, malathion monocarboxylic acid) and positively with 3 metabolites (artonol B, Asp-Phe, and 10,11-dihydro-20-trihydroxy-leukotriene B4). In association analysis between clinical indicators and microbe species (Supplementary Fig. 2B, 21 OTUs (e.g., OTU113 (Prevotella nigrescens) and OTU22 (Lautropia mirabilis)) correlated negatively with RBCs, ALB, and TBIL. Spearman correlation analysis (Supplementary Fig. 2C) between 67 OTUs and 14 metabolites showed that 58 OTUs (e.g., OTU90 (Granulicatella elegans), OTU162 (Porphyromonas catoniae), and OTU85 (Corynebacterium matruchotii)) correlated positively with 11 metabolites (e.g., 10-hydroxycarbazepine, portulacaxanthin III, polyporusterone B, aspartyl-threonine) and negatively with Asp-Phe, artonol B and 10,11-dihydro-20-trihydroxy-leukotriene B4. In contrast, 5 OTUs, OTU12 (Peptostreptococcus stomatis) and OTU21 (Solobacterium moorei), correlated negatively with the above 11 metabolites and positively with the above three metabolites. These results suggest the correlation between oral microecology, serum metabolomics, and some clinical indicators.

Association analysis of the oral microecology, serum metabolome, and laboratory indicators in Eps. There are significant correlations among 6 laboratory indicators, 21 OTUs, and 10 metabolites. Red lines indicate negative correlations, blue lines indicate positive correlations, and the width of the lines represents the strength of the correlation (Spearman). The transparency of the lines represented the negative logarithm of the P-value of correlation, translucent lines meant (− lg P) > 5, and opaque lines meant (− lg P) > 10. The size of the points indicates the relative abundance of genera and metabolites. The colors of points display the different phyla of the microbiome. The circle represents the oral microbiome, the square represents the laboratory indicators, and the diamond represents metabolites. OTUs operational taxonomy units; WBC, white blood cells; RBC, red blood cells; PLT, platelet; ALB, albumin; UA, uric acid; TBIL, total bilirubin; B, bacteria; PI, phenotype index; Meta, metabolite; rho, correlation index.
Characterization of the serum metabolites in EPRs and HCs
We followed previously enrolled EPs and eventually collected tongue swabs and serum samples from 22 recovered patients (EPRs). The serum of 22 EPRs and 44 HCs was analyzed by untargeted metabolomics. Under the condition that the analysis system is stable (Fig. 4A) and the fitting effect of the OPLS-DA model is good (Fig. 4C), OPLS-DA can well distinguish between EPRs and HCs (Fig. 4B). 229 metabolites with intergroup differences were selected from 1095 identified and quantified metabolites (VIP > 1, P < 0.05) (Supplementary Table 5). We used a volcano plot (Fig. 4E) to illustrate metabolites with upregulated and downregulated expression.

Metabolomic characteristics of EPRs (n = 22) are different from those of HCs (n = 44). (A) PCA score diagram results showed that the QC polymerization degree was high, and the analysis system was stable. (B) OPLS-DA showed a significant dispersion of metabolites between the two groups. (C) The permutation test indicates that the model had a good fitting effect. (D) The expression trend line chart showed the changes in the expression levels of metabolites in each group in each sample. The blue line represents the average expression levels of all metabolites in the subcluster. (E) The volcano plot shows downregulated and upregulated metabolites. (F) A KEGG topology bubble diagram shows the relative influence of different metabolites on metabolic pathways between the two groups. (G) Enrichment analysis histogram shows the enriched metabolic pathways of differential metabolites. *, p < 0.05, **, p < 0.01, ***, p < 0.001. PCA, principal component analysis; QC, quality control; OPLS-DA, Orthogonal Partial Least Squares Discrimination Analysis; KEGG, Kyoto Encyclopedia of Genes and Genomes; HCs, healthy controls; EPRs, patients whose seizures are under control.
The heatmap shows the relative expression levels of key metabolites in each sample. Compared with HCs, relative expression levels of 91 metabolites (e.g., malathion monocarboxylic acid, 7-Methyl-3-oxo-6-octenoyl-CoA, and aspartyl-threonine) in EPRs increased, and relative expression levels of 138 metabolites decreased. Notably, many of these differential metabolites are consistent with the differential metabolites found in EPs and HCs comparisons. The variation trends of EPRs and HCs metabolites in the two kinds of metabolite clusters were not consistent (Fig. 4D). In the 86 pathways of KEGG enrichment analysis (Fig. 4G, Supplementary Table 6), 18 metabolic pathways had statistical significance. Among them, nicotine addiction, leishmaniasis, caffeine metabolism, sphingolipid metabolism, and other metabolic pathways were significantly different. According to metabolic pathway analysis, D-glutamine and D-glutamate metabolism and sphingolipid metabolism were most affected between the two groups (Fig. 4F).
Three metabolite molecules including Serylisoleucine, 1-Methyladenosine and Heptaethylene glycol were identified by random forest analysis and fivefold cross-validation (Supplementary Table 7). Based on the optimal tag set, the POD index of EPRs was significantly higher than that of HCs, with an AUC reaching 100% (95% CI 100% to 100%, P < 0.0001).
Correlation analysis of the oral microecology, metabolic spectrum, and clinical indicators in EPRs and HCs
We performed an association analysis of metabolomics, oral microecology, and laboratory indicators for 22 EPRs and 44 HCs. As shown in Supplementary Fig. 3D, 10 OTUs, 10 metabolites, and TBIL showed a strong correlation. Heptathylene glycol correlated negatively with 5 OTUs (e.g., OTU30 (Lautropia mirabilis), OTU34 (Rothia aeria), OTU78 (Actinomyces HMT 169)). OTU30 (Lautropia mirabilis), OTU34 (Rothia aeria), OTU78 (Actinomyces HMT 169), and OTU133 (Porphyromonas catoniae) correlated positively with 9 metabolites but not heptethylene. We further depict the correlation between any two of the metabolites, microbial species, and laboratory indicators in Supplementary Fig. 3A, B, and C.
Characterization of serum metabolites before and after seizure control
To determine changes in metabolites and oral microflora before and after seizure control, we characterized the oral microecology and metabolomics of 19 EPs and 19 EPRs. LC–MS was performed on 19 EPs and serum samples. The results of the PCA score chart (Supplementary Fig. 4A) showed that the degree of polymerization of QC was high and that the data quality was reliable. In the case of the good fitting effect of the OPLS-DA model (Fig. 5B), EPs and EPRs were discrete (Fig. 5A). Furthermore, 18 intergroup differential metabolites were screened out from 1095 identified and quantified metabolite molecules (VIP > 1, P < 0.05) (Supplementary Table 8).

Metabolomics differences before and after seizure control. (A) The OPLS-DA score showed obvious metabolite dispersion of EPs (n = 19) and EPRs (n = 19). (B) The permutation test indicates that the model had a good fitting effect. (C) The volcano plot shows downregulated and upregulated metabolites. (D) A KEGG topology bubble diagram shows the relative influence of different metabolites on metabolic pathways between the two groups. (E) The heatmap shows the relative expression of each metabolite in all samples. (F) 12 metabolites were selected as the best potential biomarkers. (G) In the discovery cohort, the POD value of EPRs was significantly higher than that of EPs. (H) In the discovery cohort, the AUC was 1. OPLS-DA, Orthogonal Partial Least Squares Discrimination Analysis; KEGG, Kyoto Encyclopedia of Genes and Genomes; POD, probability of disease; AUC, area under the ROC curve; EPs, patients diagnosed with epilepsy; EPRs, patients whose seizures were under control. Centerline, median; box limits, upper and lower quartiles; circle or square symbol, mean; error bars, 95% CI.
A volcano plot (Fig. 5C) shows multiples of differences in metabolites between the two groups. According to the heatmap (Fig. 5E), the levels of 15 metabolic molecules in EPRs increased compared with EPs, whereas the levels of 3 metabolic molecules decreased. We observed that the average levels of metabolites in EPs and EPRs were different (Supplementary Fig. 4B). Among the 42 KEGG pathways to which different metabolites were enriched, 13 had statistical significance (Supplementary Fig. 4C, Supplementary Table 9). There were significant differences in metabolic pathways such as sphingolipid metabolism, and choline metabolism in cancer and necroptosis. Furthermore, MetaboAnalystR (Fig. 5D) suggested that different metabolites had the most significant effect on sphingolipid metabolism, followed by vitamin B6 metabolism, tyrosine metabolism, toluene degradation, and glycerophospholipid metabolism.
Finally, we performed a quintupled cross-validation analysis of differential metabolites to obtain a marker composed of 12 metabolites (Fig. 5F and Supplementary Fig. 4D, Supplementary Table 10). Based on the metabolite marker set, the POD index of EPRs in this cohort was significantly higher than that of EPs (Fig. 5G), and the AUC reached 100% (95% CI 100% to 100%, P < 0.0001) (Fig. 5H).
Correlation analysis of the oral microecology, metabolic spectrum, and clinical indicators before and after seizure control
We performed an association analysis of the microecology, metabolic spectrum, and clinical indicators in 19 EPRs and 19 EPs (Supplementary Fig. 5). In correlation analysis of 10 OTUs and 2 metabolic molecules (Supplementary Fig. 4F), (3-(2,5-dihydroxyphenyl)-3-oxo-1-phenylpropoxy) sulfonic acid correlated negatively with 6 OTUs (e.g., OTU17 (Veillonella atypica), OTU20 (Prevotella pallens)) but positively with OTU55 (Neisseria oralis). RBCs correlated positively with 10 OTUs (e.g., OTU51 (Neisseria elongata) and OTU8 (Neisseria perflava)) (Supplementary Fig. 4G). N-Acetyl-dl-tryptophan correlated negatively with Cr and ALB and BUN positively with (3-(2,5-dihydroxyphenyl)-3-oxo-1-phenylpropoxy) sulfonic acid (Supplementary Fig. 4E).
Discussion
The occurrence of epilepsy requires the participation of genetic, physiological, and environmental factors, and its repeated characteristics will not only bring physical and psychological obstacles to patients but also cause serious social and economic burdens. At present, the lack of effective early diagnostic tools makes the intervention treatment and prognosis recovery of epilepsy difficult10. The search for biomarkers that can be used in clinical diagnosis and tracking of disease-causing processes will help advance patient care, disease research, and the birth of new treatment strategies. Among the different biological samples, cerebrospinal fluid is the most accurate for diagnosis of epilepsy, but blood not only provides more comprehensive information, but the sampling is minimally invasive11. As a very ideal clinical diagnostic medium, its small molecule metabolites can cross the blood–brain barrier and may well represent the biological phenomena occurring in the central nervous system. Metabolomics identifies disease biomarkers in an efficient and high-throughput manner, elucidates complex biological mechanisms, monitors therapeutic effects, and is considered an effective tool in all aspects of disease diagnosis, classification, staging, treatment, and prognosis12,13. An in-depth understanding of metabolomic changes during the development of epilepsy can provide new insights into its pathophysiological mechanisms and provide new ideas for the development of effective anti-epilepsy strategies.
In this study, we compared the serum metabolites of a large sample of patients with epilepsy with those of healthy subjects and identified the serum metabolites in the former that differ from those in the latter. A total of 1090 metabolites were identified and quantified by LC–MS non-targeted metabolomics analysis, and 257 intergroup differential metabolites were found. Compared with HCs, Eps had 117 differently elevated metabolite levels, including malathion monocarboxylic acid, serylproline, aspartyl-threonine, and 7-Methyl-3-oxo-6-octenoyl-CoA, while 140 metabolites such as Asp—Phe, phenylalanylphenylalanine, artonol B, and 4—Hydroxy—alprenolol levels decreased. These elevated differential metabolites were mainly fatty acyls and carboxylic acids and derivatives. This is consistent with other studies, considering that it is caused by abnormal fatty acid metabolism during epilepsy14,15. Through further analysis of the KEGG pathway, the five most affected metabolic pathways were found, namely D-glutamine and D-glutamate metabolism, cutin, suberine and wax biosynthesis, caffeine metabolism, lysine biosynthesis, and alanine, aspartate and glutamate metabolism. By searching for differential metabolites and projecting them into the corresponding metabolic pathways, we found that adult patients with their first untriggered seizure had higher levels of caffeine metabolism, nicotine addiction, linoleic acid metabolism, choline metabolism in cancer, biosynthesis of amino acids, D-Glutamine and D-glutamate metabolism, purine metabolism, arginine biosynthesis, alanine, aspartate and glutamate metabolism and taste transduction compared with healthy volunteers, both at the time of illness and after 3–4 months of recovery. Brain imaging studies in humans have shown that nicotine can activate brain regions such as the prefrontal cortex, thalamus, and basal ganglia, and even had toxic effects at high doses that can cause nausea, confusion, seizures, and even death16. Nicotine addiction can enhance the ability of dopamine transmission, inducing long-term potentiation (LTP) of glutamatergic synapses in the ventral tegmental area (VTA) dopamine neurons17. As an excitatory neurotransmitter, glutamate had been shown to play an important role in epileptic seizures. In addition, the role played by purinergic signaling systems in mediating the excitability of neuronal networks was becoming increasingly clear18. It had also been confirmed that the top two enriched metabolic pathways involved in pediatric drug-resistant epilepsy are unsaturated fatty acid biosynthesis and linoleic acid metabolism19. Compared with EP, metabolites involved in sphingolipid metabolism in EPR were increased. Sphingolipid metabolism plays an important role in the homeostasis and function of the central nervous system and is closely related to Alzheimer's disease, Parkinson's disease, and multiple sclerosis20. Consistent with some previous findings, our study confirmed that serum metabolites were altered in adults with first unprovoked seizures and short-term control, and some metabolic pathways that enhance nervous system excitability or increase drug tolerance were increased. We believe that these are some key metabolic pathways in epileptogenesis and have certain significance for the diagnosis and treatment of epilepsy.
In addition, through the correlation analysis of oral microbiota, serum metabolomics, and laboratory indicators, we found that oral microbiota and serum metabolites were changed in patients with epilepsy and short-term control, and there was a correlation between oral microflora and serum metabolites, suggesting that oral microbiota and serum metabolomics may be linked in the regulation of epilepsy. The presence of metabolites and flora that remain at abnormal levels suggests that there may still be an inherent pathogenic biochemical basis for EPR. Metabolome-based markers have also been used in the diagnosis of diabetes21 and nonalcoholic steatohepatitis22. We used random forest analysis to construct relevant potential biomarkers based on serum metabolites and validated the diagnostic efficacy of the model with a randomized cohort. This diagnostic model will be helpful for early screening or diagnosis of epilepsy, especially when the diagnosis of epilepsy is not clear, and it may be an important auxiliary means.
In conclusion, based on serum metabolomics analysis, this study determined the changes in serum metabolites in EPs, EPRs, and HCs, and Spearman correlation analysis was performed to find the relationship between oral flora, serum metabolites, and clinical indicators during the onset of epilepsy. The diagnostic model based on potential biomarkers achieved excellent diagnostic performance in both discovery and validation queues. Specific serum metabolites reflect the changes and characteristics of epilepsy and seizure control in different states of the disease, providing a large sample of reliable evidence for the study of serum metabolism of epilepsy and new ideas for the development of effective anti-epilepsy strategies.
Methods
Research program
This study was approved by the Institutional Review Board of the First Affiliated Hospital of Zhengzhou University (No. 2021-KY-0574-002). All samples and clinical data involved in this study were collected with the informed consent of each participant. A total of 131 serum samples were prospectively collected from hospitalized adult patients with the first unprovoked seizure of epilepsy (epileptic patients, EPs). After 3–4 months of follow-up, seizures were controlled in 22 patients without using antiepileptic drugs (epileptic patients recovered, EPRs). The healthy controls (HCs) were 527 volunteers who underwent a physical examination in the First Affiliated Hospital of Zhengzhou University. Serum samples were subjected to untargeted metabolomics analysis. Study inclusion and exclusion criteria and the collection and testing of tongue coating specimens were in Supplementary Methods.
Serum sample preparation
The anterior elbow venous blood was collected into a common serum tube (BD, Oxford, UK), and the supernatant was collected immediately after centrifugation at 4 °C and 10,000 rpm for 10 min. Stored the supernatant at − 80 °C. For testing, the sample was thawed at 4 °C and precisely removed 100 µL into a 1.5 ml centrifuge tube. Then 400 µL methanol containing 0.02 mg/mL internal standard (L-2-chlorophenylalanine) was added and swirled for 30 s. Low-temperature ultrasonic extraction for 30 min (temperature 5 °C, power 40 kHz). Then we placed the sample at − 20 °C for 30 min, centrifuged it at 4 °C at a speed of 13,000 g for 15 min, and transferred the supernatant to a liquid chromatograph-mass spectrometer (LC–MS) injection vial for machine testing. In addition, each sample was mixed with 20 µL supernatant and used as the quality control (QC) sample. A tube of 200 µL serum was taken out to test liver function, renal function, and other indicators in the laboratory department of the First Affiliated Hospital of Zhengzhou University.
LC–MS experiment
The LC–MS analysis was performed using a tandem ultra-high performance liquid chromatography-Fourier transform mass spectrometry system (UHPLC-Q Exactive HF-X, Thermo Fisher Scientific, USA). The chromatographic column was ACQUITY UPLC HSS T3 (100 mm × 2.1 mm i.d., 1.8 µm; Waters, Milford, USA), the mobile phase A was 95% water + 5% acetonitrile (containing 0.1% formic acid), and the mobile phase B was 47.5% acetonitrile + 47.5% isopropyl alcohol + 5% water (containing 0.1% formic acid). The sample size was 2 μL, and the column temperature was 40 °C. The samples were ionized by electrospray, and the mass spectrum signals were collected by positive and negative ion scanning modes respectively. The quality control samples were prepared by mixing the extraction liquid of all samples in the same volume, and the volume of each QC was the same as that of the sample, which was processed and tested in the same way as the analytical sample. In the process of instrument analysis, a QC sample was inserted into every 5–15 analytical samples to investigate the stability of the entire detection process.
Chromatogram of total ions
The components of the sample separated by chromatography continuously entered the mass spectrum, and the mass spectrum was continuously scanned for data collection. A mass spectrum was obtained for each scan, and the total ion current intensity was obtained by adding all the ion intensities in each mass spectrum. The total ion chromatograms that time as the abscissa, ionic strength sum as ordinate. The detection effect was evaluated according to the total ion chromatograms of quality control samples in positive and negative ion modes.
Identification of metabolites
The raw data were imported into Progenesis QI (Waters Corporation, Milford, USA) for baseline filtering, peak identification, integration, retention time correction, peak alignment, etc. Finally, the data matrix containing retention time, mass-to-charge ratio, and peak intensity information was obtained. The MS and MS/MS mass spectrum information were matched with the metabolic database. MS mass error was set to less than 10 ppm, and metabolites were identified according to the secondary mass spectrum matching score. The main database for metabolites identification were some mainstream public databases and self-built databases including Human Metabolome Database (http://www.hmdb.ca/), MetaboAnalyst (https://www.metaboanalyst.ca/) and Metabolite Link (https://metlin.scripps.edu/).
Multivariate data analysis
After the data matrix was imported into the “ropls” package (v1.18.8) of R 4.1.1 (http://www.R-project.org/)23, the unsupervised principal component analysis (PCA) method was used to analyze the data, which could be used to find abnormal samples and evaluate QC repeatability. A supervised (orthogonal) partial least squares analysis (OPLS-DA) was then used to show the overall differences in metabolic profiles among groups and to look for the metabolites that differed between groups. In the OPLS-DA analysis, variables whose Variable Importance in Projection (VIP) was greater than 1 were important variables. The corrected T-test and multivariate analysis of OPLS-DA were used to screen out the metabolites with differences between groups (VIP > 1 and P value < 0.05). To prevent the model from overfitting, the fitting effect of the model was investigated by using 200 substitution tests.
Advanced analysis
For the differential metabolites obtained through multivariate statistical analysis, MetaboAnalystR 4.0 (https://github.com/xia-lab/MetaboAnalystR) was used for metabolite aggregation analysis24, and then we performed pathway enrichment analysis of the differential metabolites using Metabolic Pathway Analysis (MetPA) module (KEGG PATHWAY Database, http://www.genome.jp/kegg/pathway.html)25. To quantify pathway activity, the composite score for each pathway was standardized to 1, where the importance measure for each biomolecule was given a weighted score based on the relative importance of its position. Finally, the cumulative importance score of the current pathway was obtained by calculating the weighted score of corresponding metabolites. The higher the score, the greater the influence of the pathway. In addition, we used the“ggplot2” package (v3.3.5) in R 4.1.1 (http://www.R-project.org/) to perform a clustering analysis of the expression patterns of metabolites with statistical differences (Metabolite distance algorithm: Euclidean; Metabolite level clustering method: Complete)26.
Construction of diagnostic models
Serum samples from epilepsy patients and healthy controls were randomly divided into training cohort and test cohort at a ratio of 2:1. In the training cohort, we characterized the serum metabolites of 100 EPs and 200 HCs, constructed a classifier based on the random forest model (randomForest package (v4.6.14))27, and selected the best combination of biomarkers by five-fold cross validation. According to the determined optimal set of biomarkers, the probability of disease12 index was calculated for the training cohort and the test cohort. The POD index is the ratio of the number of samples predicted to be EPs to the number of samples predicted to be HC in a randomly generated decision tree. Then, the receiver operating characteristic (ROC) curve was constructed using the “pROC” package (v1.17.0.1) of R28, and the performance of the model was evaluated by the area under the curve. In addition, correlations among oral microbiota, lipid molecules and clinical indicators were elucidated based on Spearman correlation analysis.
Statistical analysis
The continuous variables with normal distribution were expressed as mean ± standard deviation and compared between the two groups using the t-test. Continuous variables that were not normally distributed were expressed as medians and interquartile ranges, and comparisons between groups were performed with the use of the Wilcoxon rank-sum test. The chi-square test or Fisher's exact test was used for categorical variables between the two groups. The statistical analysis was performed using SPSS V.26 for Windows (SPSS, Chicago, Illinois, USA). Statistical significance was defined by P < 0.05.
Limitations of the study
This study included adults who had their first seizure without an apparent trigger and who did not have another seizure after a short follow-up, and the reason for inclusion was partly related to clinical limitations. However, the clinical classification of epilepsy was very complex, and we only considered one of them. Adding more epilepsy classifications for subgroup analysis or long-term follow-up will provide more evidence for the diagnosis and treatment of epilepsy. In addition, we only found some metabolites and metabolic pathways related to seizures, and more specific mechanisms have not been detected. In-depth verification may bring more surprises.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board from the First Affiliated Hospital of Zhengzhou University (No. 2021-KY-0574-002). The study was performed in accordance with the Helsinki Declaration and Rules of Good Clinical Practice. All participants signed written informed consent after the study protocol was fully explained.
Patient and public involvement
Patients or the public were not involved in the design, conduct, reporting, or dissemination plans of our research.
Data availability
The raw Illumina read data for all tongue coating samples were deposited in the European Bioinformatics Institute European Nucleotide Archive database (Accession Number: PRJNA759716). The serum metabolomics data sets supporting the conclusions of this article are included within the article and its additional files. Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact: Zhigang Ren (fccrenzg@zzu.edu.cn).
References
Sheng, J., Liu, S., Qin, H., Li, B. & Zhang, X. Drug-resistant epilepsy and surgery. Curr. Neuropharmacol. 16, 17–28. https://doi.org/10.2174/1570159x15666170504123316 (2018).
Liu, X. & Chen, J. Research progress on ferroptosis and its role in epilepsy. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. https://doi.org/10.26402/jpp.2022.6.02 (2022).
Thijs, R. D., Surges, R., O’Brien, T. J. & Sander, J. W. Epilepsy in adults. Lancet (London, England) 393, 689–701. https://doi.org/10.1016/s0140-6736(18)32596-0 (2019).
Devinsky, O. et al. Epilepsy. Nat. Rev. Dis. Primers 4, 18024. https://doi.org/10.1038/nrdp.2018.24 (2018).
Goodacre, R., Vaidyanathan, S., Dunn, W. B., Harrigan, G. G. & Kell, D. B. Metabolomics by numbers: Acquiring and understanding global metabolite data. Trends Biotechnol. 22, 245–252. https://doi.org/10.1016/j.tibtech.2004.03.007 (2004).
Liu, P. et al. Discovery of metabolite biomarkers for acute ischemic stroke progression. J. Proteome Res. 16, 773–779. https://doi.org/10.1021/acs.jproteome.6b00779 (2017).
Abela, L. et al. N(8)-acetylspermidine as a potential plasma biomarker for Snyder-Robinson syndrome identified by clinical metabolomics. J. Inherit. Metab. Dis. 39, 131–137. https://doi.org/10.1007/s10545-015-9876-y (2016).
Beamer, E. et al. Elevated blood purine levels as a biomarker of seizures and epilepsy. Epilepsia 62, 817–828. https://doi.org/10.1111/epi.16839 (2021).
Engelke, U. F. et al. Untargeted metabolomics and infrared ion spectroscopy identify biomarkers for pyridoxine-dependent epilepsy. J. Clin. Investing. https://doi.org/10.1172/jci148272 (2021).
Niu, D., Sun, P., Zhang, F. & Song, F. Metabonomic analysis of cerebrospinal fluid in epilepsy. Ann. Transl. Med. 10, 449. https://doi.org/10.21037/atm-22-1219 (2022).
Donatti, A., Canto, A. M., Godoi, A. B., da Rosa, D. C. & Lopes-Cendes, I. Circulating metabolites as potential biomarkers for neurological disorders-metabolites in neurological disorders. Metabolites https://doi.org/10.3390/metabo10100389 (2020).
Murgia, F. et al. Metabolomics as a tool for the characterization of drug-resistant epilepsy. Front. Neurol. 8, 459. https://doi.org/10.3389/fneur.2017.00459 (2017).
Eid, T. Harnessing metabolomics to advance epilepsy research. Epilepsy Curr. 22, 123–129. https://doi.org/10.1177/15357597221074518 (2022).
Abuknesha, N. R. et al. Plasma fatty acid abnormality in Sudanese drug-resistant epileptic patients. Prostaglandins Leukot. Essent. Fatty Acids 167, 102271. https://doi.org/10.1016/j.plefa.2021.102271 (2021).
Taha, A. Y., Burnham, W. M. & Auvin, S. Polyunsaturated fatty acids and epilepsy. Epilepsia 51, 1348–1358. https://doi.org/10.1111/j.1528-1167.2010.02654.x (2010).
Melis, M. & Pistis, M. Targeting the interaction between fatty acid ethanolamides and nicotinic receptors: Therapeutic perspectives. Pharmacol. Res. 86, 42–49. https://doi.org/10.1016/j.phrs.2014.03.009 (2014).
Mansvelder, H. D. & McGehee, D. S. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 27, 349–357. https://doi.org/10.1016/s0896-6273(00)00042-8 (2000).
Beamer, E., Kuchukulla, M., Boison, D. & Engel, T. ATP and adenosine-Two players in the control of seizures and epilepsy development. Prog. Neurobiol. 204, 102105. https://doi.org/10.1016/j.pneurobio.2021.102105 (2021).
Guo, H. L. et al. Integrating metabolomics and lipidomics revealed a decrease in plasma fatty acids but an increase in triglycerides in children with drug-refractory epilepsy. Epilepsia open 8, 466–478. https://doi.org/10.1002/epi4.12712 (2023).
Alaamery, M. et al. Role of sphingolipid metabolism in neurodegeneration. J. Neurochem. 158, 25–35. https://doi.org/10.1111/jnc.15044 (2021).
Song, L. et al. Urine metabonomics reveals early biomarkers in diabetic cognitive dysfunction. J. Proteome Res. 16, 3180–3189. https://doi.org/10.1021/acs.jproteome.7b00168 (2017).
Qi, S. et al. Metabonomics screening of serum identifies pyroglutamate as a diagnostic biomarker for nonalcoholic steatohepatitis. Clin. Chim. Acta Int. J. Clin. CHEM. 473, 89–95. https://doi.org/10.1016/j.cca.2017.08.022 (2017).
Thévenot, E. A., Roux, A., Xu, Y., Ezan, E. & Junot, C. Analysis of the human adult urinary metabolome variations with age, body mass index, and gender by implementing a comprehensive workflow for univariate and OPLS statistical analyses. J. Proteome Res. 14, 3322–3335. https://doi.org/10.1021/acs.jproteome.5b00354 (2015).
Chong, J., Wishart, D. S. & Xia, J. Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinf. 68, 86. https://doi.org/10.1002/cpbi.86 (2019).
Lou, J. et al. Fecal microbiomes distinguish patients with autoimmune hepatitis from healthy individuals. Front. Cell. Infect. Microbial. 10, 342. https://doi.org/10.3389/fcimb.2020.00342 (2020).
Coleman, A., Bose, A. & Mitra, S. Metagenomics data visualization using R. Methods Mol. Boil. (Clifton, N.J.) 2649, 359–392. https://doi.org/10.1007/978-1-0716-3072-3_19 (2023).
Alderden, J. et al. Predicting pressure injury in critical care patients: A machine-learning model. Am. J. Crit. Care Off. Pub. Am. Assoc. Crit. Care Nurses 27, 461–468. https://doi.org/10.4037/ajcc2018525 (2018).
Robin, X. et al. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 12, 77. https://doi.org/10.1186/1471-2105-12-77 (2011).
Acknowledgements
We thank all the generous volunteer subjects who enrolled in the study. We are also very grateful to Hongyan Ren and Chao Liu (Shanghai Mobio Biomedical Technology Co., Ltd.) for their generous help in sequencing.
Funding
This study was sponsored by grants from Key Scientific Research Project of Henan Province University (24A320031), Key project of Henan Provincial Natural Science Foundation (HNSZRKXJJZDXM2023019), Central Plains Talent Program-Central Plains Youth Top Talents, Young and Middle-aged Academic Leaders of Henan Provincial Health Commission (HNSWJW-2022013), and Funding for Scientific Research and Innovation Team of The First Affiliated Hospital of Zhengzhou University (QNCXTD2023002).
Author information
Authors and Affiliations
Contributions
Z.R. and Y.J. designed the study. X.L, Y.J., Y.L., Z.L., L.J., T.W., Y.C., and S.L. collected clinical samples. X.L., L.J., T.W., and W.K. collected and analyzed the clinical data of the subjects. Z.L. and S.L extracted the bacterial DNA and analyzed the data. Z.R. and Y.J. performed MiSeq sequencing. Z.L. and S.L. wrote the manuscript, Z.R. revised the manuscript. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lian, X., Liu, Z., Liu, S. et al. Alterations in serum metabolomics during the first seizure and after effective control of epilepsy. Sci Rep 14, 19180 (2024). https://doi.org/10.1038/s41598-024-68966-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68966-8
- Springer Nature Limited