
Abstract
Osteosarcoma (OS) is the most common primary malignant tumour of the bone with high mortality. Here, we comprehensively analysed the hypoxia signalling in OS and further constructed novel hypoxia-related gene signatures for OS prediction and prognosis. This study employed Gene Set Enrichment Analysis (GSEA), Weighted correlation network analysis (WGCNA) and Least absolute shrinkage and selection operator (LASSO) analyses to identify Stanniocalcin 2 (STC2) and Transmembrane Protein 45A (TMEM45A) as the diagnostic biomarkers, which further assessed by Receiver Operating Characteristic (ROC), decision curve analysis (DCA), and calibration curves in training and test dataset. Univariate and multivariate Cox regression analyses were used to construct the prognostic model. STC2 and metastasis were devised to forge the OS risk model. The nomogram, risk score, Kaplan Meier plot, ROC, DCA, and calibration curves results certified the excellent performance of the prognostic model. The expression level of STC2 and TMEM45A was validated in external datasets and cell lines. In immune cell infiltration analysis, cancer-associated fibroblasts (CAFs) were significantly higher in the low-risk group. And the immune infiltration of CAFs was negatively associated with the expression of STC2 (P < 0.05). Pan-cancer analysis revealed that the expression level of STC2 was significantly higher in Esophageal carcinoma (ESCA), Head and Neck squamous cell carcinoma (HNSC), Kidney renal clear cell carcinoma (KIRC), Lung squamous cell carcinoma (LUSC), and Stomach adenocarcinoma (STAD). Additionally, the higher expression of STC2 was associated with the poor outcome in those cancers. In summary, this study identified STC2 and TMEM45A as novel markers for the diagnosis and prognosis of osteosarcoma, and STC2 was shown to correlate with immune infiltration of CAFs negatively.
Similar content being viewed by others
Introduction
Osteosarcoma (OS) is the most common primary malignant tumour of the bone, usually occurring at the knee and proximal humerus. OS incidence has a bimodal age distribution with peaks in the 10–19 and 60–79 age groups1. Although rare, the incidence of OS is reported to be 2–4.2 cases per million population per year, the clinical outcome of OS has always been poor, and a high degree of OS has high mortality2. Furthermore, the underlying mechanism and potential therapeutic targets remain to be fully elucidated. Hence, developing new predictive and prognostic biomarkers for OS is essential.
It has been known for many years that hypoxia condition is the hallmark of most solid tumors3. Under hypoxia, hypoxia-inducible factors (HIF) become activated and further bind to hypoxia-response elements (HRE) in target genes4. Thus, hypoxia regulates tumour biological processes, including angiogenesis, cell survival, metabolism and metastasis5. TMEM45A is a member of the transmembrane protein family, with 275 amino acids, which is highly expressed in the skin and participates in epidermal keratinization6,7. TMEM45A has been reported to be upregulated by the hypoxia-inducible factor (HIF-1A)8,9. A recent study has indicated that TMEM45A is highly expressed in ovarian cancer and may regulate tumour cell proliferation via the TGF-β signalling pathway10. Furthermore, elevated TMEM45A expression levels were linked to reduced overall survival in breast cancer patients8. STC2 is a 302-amino acid protein upregulated in the breast, muscle, heart, testis and pancreas11. STC2 has been reported to be regulated by HIF-1A and associated with cell proliferation12. The expression of STC2 was significantly elevated in breast cancer13, colorectal cancer14, gastric cancer15, and prostate cancer16.
However, the interaction between hypoxia and OS has not been well investigated. Here, we comprehensively analysed the hypoxia signalling in OS and further constructed novel hypoxia-related gene signatures for OS prediction and prognosis. The expression of the hypoxia-related gene has been validated in datasets and molecular experiments. Furthermore, the correlation between hypoxia-related genes and immune cell infiltration was assessed through immune infiltration analyses. Our present research provides novel OS diagnosis and prognosis tools, which may contribute to clinical decision-making and individual treatment.
Materials and methods
Data source
All datasets in this study were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), and the GDC TARGET-OS cohort was acquired from the UCSC database (https://xenabrowser.net/). GSE99671 contained 18 normal individuals and 18 osteosarcoma patients. In GSE16088, there were 4 normal individuals and 16 osteosarcoma patients. GSE225588 was performed on 6 normal individuals and 6 osteosarcoma patients. GSE21257 analysed 53 osteosarcoma patients. Detailed information on datasets is described in Table 1, and the workflow of this study is shown in Fig. 1.

The workflow of the study. OS Osteosarcoma, DEGs Differentially expressed genes, WGCNA Weighted correlation network analysis, GSEA Gene Set Enrichment Analysis, LASSO The least absolute shrinkage and selection operator, KM Kaplan Meier, DCA Decision curve analysis.
Identification and visualization of differentially expressed genes (DEGs)
We evaluated the distribution of gene expression levels in different samples in GSE99671. Principal component analysis (PCA) and sample hierarchical clustering tree were applied to explore the intergroup difference and intragroup sample duplications. Abnormal samples were removed from subsequent analysis. The "DESeq2" package17 was used for screening the DEGs with the cut-off criteria of P < 0.05 and fold change (FC) > 1, which was further visualised by "pheatmap" and "ggplot2" packages in the heat map and volcano map.
WGCNA
The GSE99671 dataset containing the expression profile and clinical traits of 33 samples was fitted into the construction of weight gene co-expression networks with the "WGCNA" package18. The top 50% of genes with maximal differences among the samples were selected as the input matrix for subsequent WGCNA. Pearson correlation tests were performed to construct a matrix to evaluate the module-trait relationships between genes and disease status according to the soft-threshold value. The modules with P < 0.05 were considered significantly correlated with disease status, and the modules with the highest correlation coefficient were selected as hub modules for further analysis.
Identification of the hypoxia-related genes
To identify the importance of hypoxia status in osteosarcoma. We performed Gene Set Enrichment Analysis (GSEA) between osteosarcoma and normal samples in the GSE99671 dataset based on DEGs and 200 hypoxia hallmark genes from the Molecular Signatures database (MSigDB). Enrichment score and P value were calculated to predict the hypoxia status. As a result, the hypoxia pathway was activated in the osteosarcoma group compared with the normal group. Intersecting genes of hypoxia hallmark genes and hub modules were screened as the hypoxia-related genes, which played critical roles in osteosarcoma.
Construction and evaluation of a diagnostic model for OS
Firstly, the GSE99671 dataset was randomly divided into a training set (70%) and a test set (30%). Secondly, we performed the LASSO (The Least Absolute Shrinkage and Selection Operator) algorithm to select suitable variables for constructing the diagnostic model using the "glmnet" package. Then, the "rms" package was used to build the logistic regression model in the training set. We applied ROC (the receiver operating characteristic) and area under the curve (AUC) to evaluate the diagnostic effectiveness in the training set, test set and external dataset (GSE16088). Decision curves were plotted utilising the "dcurves" package. Calibration curves were drawn by "rms" packages in the training set and the whole GSE99671 dataset to assess the divergence between the predicted and actual diagnostic probabilities. A nomogram was built to evaluate the diagnostic probability of osteosarcoma quantitatively.
Construction and evaluation of a prognostic model for OS
The TARGET cohort was randomly divided into a training set (70%) and a test set (30%). Before establishing the model, the LASSO Cox regression algorithm tested the hypoxia-related genes to select the hypoxia-related signature in the training set. Furthermore, clinical information, including age, sex and metastasis combined with the gene signature, were tested by univariate and multivariate Cox regression analysis (P < 0.05). Finally, we calculated the risk score using the formula as follows:
coefi indicated the correlation coefficient of each variable, and Xi indicated the gene expression level. Based on the risk score, we computed the best cut-off value to divide patients into high- and low-risk groups. The Kaplan–Meier (K–M) method was used to draw survival curves, and the log-rank test was applied to calculate differences in overall survival between low- and high-risk groups. Time-dependent ROC and AUC values were performed by the "timeROC" package. A nomogram was established to quantitatively evaluate the survival probabilities of osteosarcoma patients based on the multivariate Cox regression analysis result. Calibration curves of 1, 3 and 5 years were drawn by the "rms" package to estimate the divergence between the predicted probability and the actual survival probability. Decision curves were performed by the "ggDCA" package to determine the clinical applicability of our established nomogram. Time-ROC, risk-score and K–M plot were also conducted in the validation dataset (GSE21257).
Cell culture and reagents
Human OS cells HOS, 143B, Saos-2, U-2OS, and human osteoblast cell hFOB1.19 were supplied by the America Type Culture Collection (ATCC, USA). hFOB1.19 and 143B cells were kept in Dulbecco's modified Eagle's medium (DMEM) (Gibco, USA) supplied with 10% fetal bovine serum(FBS, Pricella, China) and 1% penicillin/streptomycin at 37 °C. HOS were cultured at 37 °C in MEM (Pricella, China) enriched with 10% FBS and 1% penicillin/streptomycin. U-2OS and Saos-2 were natured in McCoy's 5A medium with 10% FBS and 1% penicillin/streptomycin in an incubator humidified with 5% CO2 and 37 °C.
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
RNA isolation from cells was performed with Trizol reagent (Invitrogen), and cDNA was synthesised using the ReverTra Ace qPCR RT Master Mix (Toyobo) according to the manufacturer's instructions. qRT-PCR was conducted using the SYBER Select Master Mix (Thermo Scientific) on the QuantStudio 4 Flex Real-Time PCR system (Thermo Scientific) with the primers listed in Table 2. ACTB was used as internal control, and the 2−ΔΔCt method was applied to calculate the relative expression of STC2 and TMEM45A. All experiments were conducted in triplicate.
Immune cell infiltration analysis between high- and low-risk groups of osteosarcoma patients
OS patients in the TARGET cohort were divided into two groups according to the risk scores: high-risk and low-risk. The immune cell infiltrations were conducted by EPIC, MCPCOUNTER and XCELL methods. The correlation between STC2 and immune cells was analysed by the "ggpubr" package.
Pan-cancer analysis of STC2 expression and prognosis
Pan-cancer analyses were performed on the online tools, including GEPIA (http://gepia.cancer-pku.cn/detail.php)19 for expression analysis and Kaplan–Meier Plotter (https://kmplot.com/analysis/index.php?p=service)20 for prognosis analysis.
Statistical analysis
The Wilcoxon test was employed to assess the discrepancy between two samples, whereas the Kruskal–Wallis test was utilised to evaluate the divergence between multiple samples. A p-value of less than 0.05 was regarded as statistically significant. The symbols “*”, “**”, and “***” were used to indicate p-values of less than 0.05, less than 0.01, and less than 0.001, respectively.
Results
Identification of DEGs in OS dataset
The GSE99671 datasets contained 18 normal and 18 OS samples. After PCA and hierarchical clustering analysis, 3 samples were removed from the subsequent analysis (Fig. 2a,b). With the cut-off criteria (P < 0.05 & |FC|> 1), 218 upregulated DEGs and 541 downregulated DEGs were identified. DEGs were visualized in a volcano plot and heatmap (Fig. 2c,d).

Identification of DEGs in GSE99671. (a) PCA analysis. (b) Hierarchical clustering analysis. (c) Volcano map. (d) Heat map.
Identification of modules associated with OS traits by WGCNA analysis
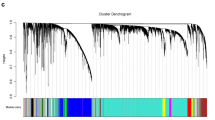
The WGCNA package was applied to construct the gene co-expression networks of the GSE99671 dataset. The soft thresholding power was set to 7 based on scale independence and mean connectivity (Fig. 3a,b). Twenty-six co-expression modules were identified by dynamic tree cut package (Fig. 3c). P < 0.05 was considered as significant module-trait relationship between module genes and clinical traits (Fig. 3d). With the same correlation coefficient, brown and pink modules were selected for further analysis. The correlation between module genes and traits was analysed and visualized in Fig. 3e,f.

Identification of hub module by the weight gene co-expression network analysis (WGCNA) and assessment of module-trait relationships. (a,b) Soft threshold selection. (c) The cluster dendrogram of 26 modules. (d) The relationship of gene modules and clinical trait status. (e,f) Scatterplots of gene significance vs. module membership in brown and pink modules.
Identification of the hypoxia-related genes
GSEA analysis revealed that OS samples exhibited a significant hypoxia enrichment signature compared to normal samples in GSE99671 (P < 0.001) (Fig. 4a). With the cut-off criteria (MM > 0.5 and GS > 0.2), pink and brown modules containing 752 genes were selected as hub modules in the OS dataset. The intersection of hub genes and hypoxia hallmark genes was conducted using the "VennDiagram" package. Twenty-one genes were screened as candidate markers for OS diagnosis (Fig. 4b).

Identification of candidate genes for OS. (a) Gene Set Enrichment Analysis of hypoxia pathway in GSE99671 (P < 0.01). (b) Intersection of hub module genes and hypoxia hallmark genes.
Construction and evaluation of a diagnostic model for OS
The LASSO regression was performed using the "glmnet" package on the 21 candidate genes in the training set. The result of the LASSO regression identified 2 genes (STC2 and TMEM45A) as characteristic genes of OS (Fig. 5a,b). We built a logistic model for OS diagnosis using the core genes, further quantified in a nomogram (Fig. 5c). The results showed the excellent diagnostic performance of the prediction model based on the 2 genes, with an area under the ROC curve of 0.995, 0.958 and 1 in the training set, test set and external validation dataset (GSE16088), respectively (Fig. 5d-f). DCA analysis was performed to assess the clinical usefulness of this novel prediction model. As shown in Fig. 5g,h, the model showed an excellent net benefit at a 0.0 to 1.0 threshold in both the training and test sets. Consistently, the calibration curve of the model for OS diagnosis showed consistently accurate prediction between the training set and the whole set (Fig. 5i,j).

Construction and validation of the diagnostic model. (a,b) LASSO regression for variable selection. (c) The construction and nomogram establishment of the diagnostic model with the STC2 and TMEM45A. (d-f) ROC analysis of the diagnostic model in training, test, and external datasets, respectively. (g,h) DCA curve analysis in training set and test set. (i,j) Calibration curve in the training set and the whole dataset.
Construction and verification of a prognostic model
Univariate and multivariate Cox regressions were conducted to analyse the relationship between hub gene expression, clinical features, and the overall survival of OS patients. Cox analysis indicated that the expression of STC2 and metastasis significantly correlated with patients' overall survival in the training set (Fig. 6a,b). Based on the multivariate Cox regression analysis, a nomogram was constructed using the expression of STC2 and metastasis status (Fig. 6c). Each patient was assigned a total points value by calculating the points for each prognostic parameter. Higher points indicated worse clinical outcomes. ROC curve was performed to evaluate the sensitivity and specificity of the prognostic signature. The AUC values for 1-, 2-, and 3-year survival rates were 0.96, 0.82, and 0.80, respectively (Fig. 6d). Using the gene expression and the corresponding coefficient derived from the Cox proportional hazards regression model, risk score = 0.54*STC2 expression. The distribution of the risk score in the TARGET datasets is shown in Fig. 6e. With the best cut-off value of the risk score, patients were divided into high-risk (n = 45) and low-risk (n = 45) groups. K–M survival analysis was applied to compare patients' overall survival in the high-risk and low-risk groups. As shown in Fig. 6f, patients in the high-risk group were significantly lower than those in the low-risk group. To verify the stability and reliability of the prognostic model, we used the GSE21257 dataset for external verification. The AUC values for 1-, 2-, and 3-year survival rates were 0.88, 0.91, and 0.88, respectively, in GSE21257 (Fig. 7a). The distribution of the risk score in GSE21257 is shown in Fig. 7b. K–M plot indicated that the overall survival of patients in the high-risk group was significantly lower than in the low-risk group (log-rank test, P < 0.05) (Fig. 7c). DCA curve was performed to assess the clinical usefulness of this prognostic model. As shown in Fig. 7d, the prognostic model possessed a higher net benefit at the corresponding threshold. We also employed the calibration curves in 1, 2, and 3 years for the TARGET cohort. As shown in Fig. 7e–g the prediction lines of the nomogram for 1, 2, and 3-year survival probability in the TARGET were extremely close to the ideal performance, indicating a high accuracy of the nomogram. Therefore, combining the hypoxia-related gene and the clinical feature, this nomogram might be a novel and accurate prognostic model for OS in clinical practice.

Establishment and assessment of prognostic model. (a) Univariate Cox regression analysis of candidate genes and clinical features in TARGET OS cohort. (b) Multivariate Cox regression analysis of STC2 and metastasis. (c) Nomogram of prognostic model. (d) ROC analysis of 1-, 3-, 5-year survival probability. (e) High- and low-risk group risk score rank, survival status and expression of STC2. (f) Survival analysis between high-low-risk group using KM method in TARGET OS cohort (P < 0.05).

Validation and a calibration curve of a prognostic model. (a) ROC analysis of 1-, 3-, 5-year survival probability in GSE21257. (b) High- and low-risk group risk score rank, survival status and expression of STC2 in GSE21257. (c) Survival analysis between high-low-risk group using KM method in GSE21257 (P < 0.05). (d) DCA curve of 1-, 3-,5-year survival probability in TARGET OS dataset. (e-g) The calibration curve of the nomogram predicted 1-, 3-,5-year survival probability.
Verification of the expression of STC2 and TMEM45A in the dataset and OS cell lines
The expression levels of STC2 and TMEM45A were significantly higher in the OS group than in the normal group in both GSE99671 and GSE225588 datasets (Fig. 8a-d). To further verify the expression of hub genes in normal and OS tissues, we cultured the osteoblast cell line hFOB1.19 and OS cell lines (HOS, 143B, Saos-2 and U-2OS) and detected the expression of STC2 and TMEM45A by RT-qPCR, consistently, the results showed compared with osteoblast, OS cell lines had higher expression of STC2 and TMEM45A (Fig. 8e,f).

Validation of gene levels in datasets and in vitro. (a,b) Expression of STC2 and TMEM45A in GSE99671. (c,d) Expression of STC2 and TMEM45A in GSE225588. (e,f) Expression of STC2 and TMEM45A in vitro.
Immune cell infiltration analysis between high- and low-risk groups of osteosarcoma patients
Three methods, including EPIC, MCPCOUNTER and XCELL, were used to estimate the difference in immune cells between the high-risk and low-risk group patients in the TARGET dataset (Fig. 9a-c). Interestingly, the cancer-associated fibroblast cell was identified as significantly downregulated among the three methods. The expression of STC2 was correlated considerably with cancer-associated fibroblast cell infiltration (Pearson test, P < 0.05) (Fig. 9d). Thus, our prognostic model might be highly associated with an immunosuppressive microenvironment.

Immune cell infiltration analysis between high- low-risk groups. (a) Comparison of immune cell abundance between high- and low-risk group in TARGET OS dataset by EPIC method. (b) Comparison of immune cell abundance between high- and low-risk group in TARGET OS dataset by MCPCOUNTER method. (c) Comparison of immune cell abundance between high- and low-risk group in TARGET OS dataset by XCELL method. (d) Correlation between expression of STC2 and infiltration score of cancer-associated fibroblast in EPIC, MCPCOUNTER, and XCELL analysis (Pearson test, P < 0.05).
Pan-cancer analysis of STC2 expression and prognosis
To further elucidate the role of STC2 in cancer, we performed a pan-cancer analysis of STC2 expression and prognosis. As shown in Fig. 10a, the gene level of STC2 was highly expressed in ESCA, HNSC, KIRC, LUSC, and STAD. Consistently, the high expression level of STC2 was associated with poor prognosis (Fig. 10b–f).

Pan-cancer analysis of STC2 expression and prognosis. (a) Expression of STC2 in cancers. (b) Survival analysis between high and low levels of STC2 in ESCA. (c) Survival analysis between high and low levels of STC2 in HNSC. (d) Survival analysis between high and low levels of STC2 in KIRC. (e) Survival analysis between high and low levels of STC2 in LUSC. (f) Survival analysis between high and low levels of STC2 in STAD.
Discussion
Although rare as OS is, an estimated 800–900 cases are diagnosed annually in the United States. The 5-year survival rate is approximately 60% among osteosarcoma patients without metastases and only 20% in patients with metastases or recurrence21. Advances have been made in the treatment of OS, but the prognosis of OS is still unsatisfactory. The advancement of bioinformatics technology has led to the development of an increasing number of disease markers, which are now being utilized in the diagnosis and prognosis of various diseases. Notably, studies have shown that hypoxia enhances gastric cancer's metastatic ability, and upregulation of HIF-1α is a poor prognostic indicator in gastric cancer patients22,23. In addition, hypoxia promotes breast cancer metastasis via directly regulating variable HIF target genes24,25. Hypoxia-related gene signatures have been developed and verified to be a promising tool for risk stratification in breast and gastric cancer26,27. As for OS, necroptosis-associated lncRNA signature, ferroptosis-related lncRNA signature and pyroptosis-related gene signature have been developed28,29,30. However, this study developed a novel diagnostic and prognostic model for osteosarcoma from a unique hypoxic perspective.
In our study, we applied bioinformatic methods to screen out TMEM45A and STC as biomarkers for OS diagnosis and prognosis, which are regulated by hypoxia. In the diagnostic model, AUC values in the training set, test set, and external dataset (GSE16088) were 0.955, 0.958, and 1, respectively. In the prognostic model, AUC values of 1-, 3-, and 5-year survival probability were 0.96, 0.82, and 0.80, respectively.
With the cut-off values of risk scores, patients in the low-risk group had higher survival times than those in the high-risk group. The expression level of STC2 was significantly negatively associated with the infiltration of cancer-associated fibroblasts (CAFs). Our results indicated that both STC2 and TMEM45A were ideal biomarkers for OS.
TMEM45A is a transmembrane protein with a length of 275 amino acids, which is upregulated in many cancers: breast cancer8, liver cancer8, ovarian cancer10, Clear cell renal cell carcinoma31, and glioma cancer32. Highly expressed TMEM45A is correlated with poor prognosis10,33. Enca et al. reported that TMEM45A could be upregulated by hypoxia9. Another research also elucidated that TMEM45A was upregulated in hypoxia conditions and further mediated chemotherapy resistance in breast cancer cells8. In conclusion, TMEM45A may be an oncogenic gene regulated by hypoxia and further involved in proliferation, migration, and metastasis.
Human STC2 protein is a glycosylated peptide hormone with a length of 302 amino acids, broadly expressed in various tissues, including the pancreas, spleen, lung, placenta, and skeletal muscles. Biologically, STC2 plays a critical role in calcium regulation, ion transport, glucose regulation, and angiogenesis34. Recent studies have proved STC2 as a universal biomarker in solid tumors. Upregulated STC2 have been reported in breast cancer13, colorectal cancer14, gastric cancer15, prostate cancer16, hepatocellular carcinoma33, ovarian cancer35, lung cancer36, and so on. A pan-cancer analysis has consistently revealed that STC2 is upregulated in 20 cancers37. STC2 has been reported to be modulated by Hypoxia-inducible factor-1(HIF-1) on the transcription level and further modulate the process of cell proliferation, cell survival, and tumour metastasis via diverse pathways38. Thus, STC2 overexpression is associated with poor prognosis in various cancers37,38.
In tumour immunity, Johanna et al. 39 have reviewed that dendritic cells tend to induce T cell activation in the tumour microenvironment and are associated with improved prognosis of OS. Tumour-associated macrophages (TAMs) have been proven to promote tumour growth by suppressing T lymphocytes in the OS39. High infiltration of natural killer T cells (NKT) in cancer patients is correlated with good clinical outcomes40,41. Evidence also indicates that the expression of STC2 is highly associated with antitumor immunity37,42. In our study, we have identified that the upregulated STC2 is associated with poor prognosis of OS and negatively associated with the infiltration of the CAFs. Consistently, the infiltration of CAFs is significantly higher in the low-risk group of OS43. Initially, cancer-associated fibroblasts (CAFs) were presumed to promote tumorigenesis, while they have been reported to have a dual role in tumorigenesis owing to their heterogeneity44. CAFs participate in tumour cell growth and immunity by secreting extracellular matrix and many other cytokines, such as CC-chemokine 2 (CCL2), colony-stimulating factor 1 (CSF1), insulin-like growth factor 1 (IGF1) and vascular endothelial growth factor (VEGF)45. CAFs regulate angiogenesis and subsequently promote tumour progression through the secretion of VEGFA and CXCL1246,47. Furthermore, CAFs provide metabolic support to tumour cells by releasing alanine, deoxycytidine, proline, and other substances48,49,50,51. Conversely, CAFs have also been demonstrated to exert a tumor-restraining effect. The depletion of αSAM+ CAFs has been shown to accelerate the progression of pancreatic cancer in a mouse model, resulting in a reduction in the survival time52. Consistently, a higher level of αSAM+ CAFs is associated with superior overall survival outcomes in patients with pancreatic cancer. The results of this study demonstrate that CAFs play a tumour-restraining role in osteosarcoma. However, further investigation is necessary to identify the specific mechanism. Future studies should investigate the efficacy of targeted therapy for CAFs to regulate different subpopulations of CAFs with greater precision.
This study still has some limitations. First, all datasets concluded in this study were downloaded from the online database. More prospective studies are needed to confirm the diagnostic and prognostic model based on the hypoxia-related genes. Second, gene levels of the hypoxia-related genes were validated in datasets and in vitro. At the same time, more experiments should be conducted to investigate the function and potential target of STC2 and TMEM45A.
Conclusion
In summary, we constructed novel hypoxia-related gene signatures for OS diagnosis and prognosis, with favourable prediction performance and promising clinical practice in OS. Furthermore, STC2 and TMEM45A can be candidate targets for OS treatment.
Data availability
Our data can be found in the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/ GSE99671, GSE16088, GSE21257 and GSE225588) database and TCGA (TARGET-OS cohort, https://portal.gdc.cancer.gov/) database.
References
Beird, H. C. et al. Osteosarcoma. Nat. Rev. Dis. Primers 8, 77. https://doi.org/10.1038/s41572-022-00409-y (2022).
Rojas, G. A., Hubbard, A. K., Diessner, B. J., Ribeiro, K. B. & Spector, L. G. International trends in incidence of osteosarcoma (1988–2012). Int. J. Cancer 149, 1044–1053. https://doi.org/10.1002/ijc.33673 (2021).
Brahimi-Horn, M. C., Chiche, J. & Pouyssegur, J. Hypoxia and cancer. J. Mol. Med. (Berlin) 85, 1301–1307. https://doi.org/10.1007/s00109-007-0281-3 (2007).
Yang, G., Shi, R. & Zhang, Q. Hypoxia and oxygen-sensing signaling in gene regulation and cancer progression. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21218162 (2020).
Shi, R., Liao, C. & Zhang, Q. Hypoxia-driven effects in cancer: Characterization, mechanisms, and therapeutic implications. Cells https://doi.org/10.3390/cells10030678 (2021).
Hayez, A. et al. High TMEM45A expression is correlated to epidermal keratinization. Exp. Dermatol. 23, 339–344. https://doi.org/10.1111/exd.12403 (2014).
Kim, A. L. et al. TMEM45A is dispensable for epidermal morphogenesis, keratinization and barrier formation. PLoS ONE https://doi.org/10.1371/journal.pone.0147069 (2016).
Flamant, L. et al. TMEM45A is essential for hypoxia-induced chemoresistance in breast and liver cancer cells. BMC Cancer 12, 391. https://doi.org/10.1186/1471-2407-12-391 (2012).
Martin-Rendon, E. et al. Transcriptional profiling of human cord blood CD133+ and cultured bone marrow mesenchymal stem cells in response to hypoxia. Stem Cells 25, 1003–1012. https://doi.org/10.1634/stemcells.2006-0398 (2007).
Guo, J. et al. Inhibition of TMEM45A suppresses proliferation, induces cell cycle arrest and reduces cell invasion in human ovarian cancer cells. Oncol. Rep. 33, 3124–3130. https://doi.org/10.3892/or.2015.3902 (2015).
Moore, E. E. et al. Stanniocalcin 2: Characterization of the protein and its localization to human pancreatic alpha cells. Horm. Metab. Res. 31, 406–414. https://doi.org/10.1055/s-2007-978764 (1999).
Law, A. Y. S. & Wong, C. K. C. Stanniocalcin-2 is a HIF-1 target gene that promotes cell proliferation in hypoxia. Exp. Cell Res. 316, 466–476. https://doi.org/10.1016/j.yexcr.2009.09.018 (2010).
Bouras, T. et al. Stanniocalcin 2 is an estrogen-responsive gene coexpressed with the estrogen receptor in human breast cancer. Cancer Res. 62, 1289–1295 (2002).
Ieta, K. et al. Clinicopathological significance of stanniocalcin 2 gene expression in colorectal cancer. Int. J. Cancer 125, 926–931. https://doi.org/10.1002/ijc.24453 (2009).
Yan, G. R. et al. Characterization of phosphoproteins in gastric cancer secretome. OMICS 15, 83–90. https://doi.org/10.1089/omi.2010.0056 (2011).
Tamura, K. et al. Stanniocalcin 2 overexpression in castration-resistant prostate cancer and aggressive prostate cancer. Cancer Sci. 100, 914–919. https://doi.org/10.1111/j.1349-7006.2009.01117.x (2009).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Tang, Z. et al. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–W102. https://doi.org/10.1093/nar/gkx247 (2017).
Nagy, A., Munkacsy, G. & Gyorffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 11, 6047. https://doi.org/10.1038/s41598-021-84787-5 (2021).
Meltzer, P. S. & Helman, L. J. New horizons in the treatment of osteosarcoma. N. Engl. J. Med. 385, 2066–2076. https://doi.org/10.1056/NEJMra2103423 (2021).
Isobe, T. et al. Clinicopathological significance of hypoxia-inducible factor-1 alpha (HIF-1alpha) expression in gastric cancer. Int. J. Clin. Oncol. 18, 293–304. https://doi.org/10.1007/s10147-012-0378-8 (2013).
Li, M. et al. HIF in gastric cancer: Regulation and therapeutic target. Molecules https://doi.org/10.3390/molecules27154893 (2022).
Chang, P. H. et al. Interplay between desmoglein2 and hypoxia controls metastasis in breast cancer. Proc. Natl. Acad. Sci. USA https://doi.org/10.1073/pnas.2014408118 (2021).
Wang, R. et al. Hypoxia-inducible factor-dependent ADAM12 expression mediates breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. USA https://doi.org/10.1073/pnas.2020490118 (2021).
Liu, Y. et al. Development and validation of a hypoxia-immune-based microenvironment gene signature for risk stratification in gastric cancer. J. Transl. Med. 18, 201. https://doi.org/10.1186/s12967-020-02366-0 (2020).
Yang, X. et al. A combined hypoxia and immune gene signature for predicting survival and risk stratification in triple-negative breast cancer. Aging (Albany NY) 13, 19486–19509. https://doi.org/10.18632/aging.203360 (2021).
Wang, G., Zhang, X., Feng, W. & Wang, J. Prediction of prognosis and immunotherapy of osteosarcoma based on necroptosis-related lncRNAs. Front. Genet. 13, 917935. https://doi.org/10.3389/fgene.2022.917935 (2022).
Zhang, Y. et al. A novel pyroptosis-related signature for predicting prognosis and indicating immune microenvironment features in osteosarcoma. Front. Genet. 12, 780780. https://doi.org/10.3389/fgene.2021.780780 (2021).
Zhang, Y. et al. Comprehensive analysis of a ferroptosis-related lncRNA signature for predicting prognosis and immune landscape in osteosarcoma. Front. Oncol. 12, 880459. https://doi.org/10.3389/fonc.2022.880459 (2022).
Jiang, H., Chen, H., Wan, P., Liang, M. & Chen, N. Upregulation of TMEM45A promoted the progression of clear cell renal cell carcinoma in vitro. J. Inflamm. Res. 14, 6421–6430. https://doi.org/10.2147/JIR.S341596 (2021).
Zhang, L., Wu, F. & Zhao, J. Transmembrane protein 45A regulates the proliferation, migration, and invasion of glioma cells through nuclear factor kappa-B. Anticancer Drugs 31, 900–907. https://doi.org/10.1097/CAD.0000000000000890 (2020).
Tang, Y., Guo, C., Chen, C. & Zhang, Y. Characterization of cellular senescence patterns predicts the prognosis and therapeutic response of hepatocellular carcinoma. Front. Mol. Biosci. 9, 1100285. https://doi.org/10.3389/fmolb.2022.1100285 (2022).
Joshi, A. D. New insights into physiological and pathophysiological functions of stanniocalcin 2. Front. Endocrinol. 11, 172. https://doi.org/10.3389/fendo.2020.00172 (2020).
Buckanovich, R. J. et al. Tumor vascular proteins as biomarkers in ovarian cancer. J. Clin. Oncol. 25, 852–861. https://doi.org/10.1200/JCO.2006.08.8583 (2007).
Liu, Y. N. et al. Acquired resistance to EGFR tyrosine kinase inhibitors is mediated by the reactivation of STC2/JUN/AXL signaling in lung cancer. Int. J. Cancer 145, 1609–1624. https://doi.org/10.1002/ijc.32487 (2019).
Jiang, Z. H. et al. A pan-cancer analysis reveals the prognostic and immunotherapeutic value of stanniocalcin-2 (STC2). Front. Genet. 13, 927046. https://doi.org/10.3389/fgene.2022.927046 (2022).
Qie, S. & Sang, N. Stanniocalcin 2 (STC2): A universal tumour biomarker and a potential therapeutical target. J. Exp. Clin. Cancer Res. 41, 161. https://doi.org/10.1186/s13046-022-02370-w (2022).
Wang, Z. et al. Innate immune cells: A potential and promising cell population for treating osteosarcoma. Front. Immunol. 10, 1114. https://doi.org/10.3389/fimmu.2019.01114 (2019).
Hishiki, T. et al. Invariant natural killer T infiltration in neuroblastoma with favorable outcome. Pediatr. Surg. Int. 34, 195–201. https://doi.org/10.1007/s00383-017-4189-x (2018).
Tachibana, T. et al. Increased intratumor Valpha24-positive natural killer T cells: A prognostic factor for primary colorectal carcinomas. Clin. Cancer Res. 11, 7322–7327. https://doi.org/10.1158/1078-0432.CCR-05-0877 (2005).
Shen, B., Zhang, G., Liu, Y., Wang, J. & Jiang, J. Identification and analysis of immune-related gene signature in hepatocellular carcinoma. Genes https://doi.org/10.3390/genes13101834 (2022).
Wang, X., Xie, C. & Lin, L. Development and validation of a cuproptosis-related lncRNA model correlated to the cancer-associated fibroblasts enable the prediction prognosis of patients with osteosarcoma. J. Bone Oncol. https://doi.org/10.1016/j.jbo.2022.100463 (2023).
Gieniec, K. A., Butler, L. M., Worthley, D. L. & Woods, S. L. Cancer-associated fibroblasts-heroes or villains?. Br. J. Cancer 121, 293–302. https://doi.org/10.1038/s41416-019-0509-3 (2019).
Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598. https://doi.org/10.1038/nrc.2016.73 (2016).
O’Connell, J. T. et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc. Natl. Acad. Sci. USA 108, 16002–16007. https://doi.org/10.1073/pnas.1109493108 (2011).
Orimo, A. et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121, 335–348. https://doi.org/10.1016/j.cell.2005.02.034 (2005).
Dalin, S. et al. Deoxycytidine release from pancreatic stellate cells promotes gemcitabine resistance. Cancer Res. 79, 5723–5733. https://doi.org/10.1158/0008-5472.CAN-19-0960 (2019).
Olivares, O. et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 8, 16031. https://doi.org/10.1038/ncomms16031 (2017).
Parker, S. J. et al. Selective alanine transporter utilization creates a targetable metabolic niche in pancreatic cancer. Cancer Discov. 10, 1018–1037. https://doi.org/10.1158/2159-8290.CD-19-0959 (2020).
Sousa, C. M. et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. https://doi.org/10.1038/nature19084 (2016).
Ohlund, D. et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 214, 579–596. https://doi.org/10.1084/jem.20162024 (2017).
Acknowledgements
We thank the researchers who made their datasets publicly available.
Author information
Authors and Affiliations
Contributions
B.W. and J.C. are responsible for the design of research methods, experimental design, data collection, processing and analysis, as well as writing the initial draft of the paper and participating in discussions and revisions. Other authors assist with data collection, processing, and analysis. X.Q. and X.C. are responsible for the overall planning, design, and implementation of the research. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wen, B., Chen, J., Ding, T. et al. Development and experimental validation of hypoxia-related gene signatures for osteosarcoma diagnosis and prognosis based on WGCNA and machine learning. Sci Rep 14, 18734 (2024). https://doi.org/10.1038/s41598-024-69638-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-69638-3
- Springer Nature Limited