
Abstract
The effect of chemotherapy for anti-glioblastoma is limited due to insufficient drug delivery across the blood–brain–barrier. Poloxamer 188-coated nanoparticles can enhance the delivery of nanoparticles across the blood–brain–barrier. This study presents the design, preparation, and evaluation of a combination of PLGA nanoparticles (PLGA NPs) loaded with methotrexate (P-MTX NPs) and PLGA nanoparticles loaded with paclitaxel (P-PTX NPs), both of which were surface-modified with poloxamer188. Cranial tumors were induced by implanting C6 cells in a rat model and MRI demonstrated that the tumors were indistinguishable in the two rats with P-MTX NPs + P-PTX NPs treated groups. Brain PET scans exhibited a decreased brain-to-background ratio which could be attributed to the diminished metabolic tumor volume. The expression of Ki-67 as a poor prognosis factor, was significantly lower in P-MTX NPs + P-PTX NPs compared to the control. Furthermore, the biodistribution of PLGA NPs was determined by carbon quantum dots loaded into PLGA NPs (P-CQD NPs), and quantitative analysis of ex-vivo imaging of the dissected organs demonstrated that 17.2 ± 0.6% of the NPs were concentrated in the brain after 48 h. The findings highlight the efficacy of combination nanochemotherapy in glioblastoma treatment, indicating the need for further preclinical studies.
Similar content being viewed by others

Introduction
Despite advances in the treatment of cancers, glioblastoma multiforme (GBM), the most rampant malignant primary brain tumor1, is still considered to have a poor prognosis with a survival rate of a fewer than two years2. Common treatments for GBM have been the same for at least the past 20 years2. One of the reasons for this failure to cure and properly eliminate GBM is the existence of the blood–brain–barrier (BBB)3. BBB restricts the transport of materials to the brain4,5 as it lacks significant paracellular transport and selectively permits only specific substances4,6. Novel chemotherapeutic formulations have been introduced and studied to overcome such challenges associated with the BBB7. Transcellular and paracellular pathways are maintained as the two main routes for the delivery of substances across the BBB, however, the latter is restricted because of tight junctions. Two important transcellular approaches are endocytosis and receptor-mediated transcytosis8,9. It has been stated that PLGA nanoparticles (P NPs) have the ability to cross the BBB and their surface modification is a key tool to boost their targeted delivery10.
Polymeric nanoparticles (NPs) coated with targeting agents can be a solution to this age-old problem11. Poly (lactic-co-glycolic acid) (PLGA) is a reputable biodegradable and FDA-approved polymer for certain drug delivery applications12. Notably, PLGA exhibits the ability to partially cross the BBB13. Report has also indicated that NPs surface modification can enhance its delivery to the central nervous system (CNS)14. Tween is a gold standard for the surface coating of NPs in this field and poloxamers are potent as well15. Specifically, poloxamer 188 (P188) and polysurbate 80 are coatings which have been used on NPs for CNS targeted delivery16. P188 is an inexpensive and non-ionic surfactant17, which enhances the internalization of NPs into cells and allows NPs to transfer across the BBB18. It is proposed that P188-coated NPs can adsorb apolipoprotein E and interact with LDL receptors on endothelial cells18. In addition to targeted delivery, poloxamer can prevent P-glycoprotein (P-gp) efflux systems and P-gp pumps drugs from the interior space of the cells to the extracellular matrix (ECM), leading to drug resistance16,19,20,21.
Paclitaxel (PTX) is a widely utilized chemotherapeutic drug; however, its application is restricted because of the extreme toxicity of its solvent, cremophor EL22. Depolymerization of microtubules following cell death in late G2 phase or M phase happens in cancer cells treated with PTX23. While hydrophobic drugs with a molecular weight below 400 Da can diffuse across the lipid-based environment of the BBB24, PTX’s molecular weight is significantly higher, which restricts its passage across the BBB as well as simple diffusion25. PTX is a potent chemotherapeutic, therefore, studies have focused to facilitate its delivery with the aid of nanostructures as a carrier11. The effect of this drug on GBM has been studied by single or co-delivery systems, yielding promising results26,27. Albeit, the use of Cremophor EL remains a significant drawback in the application of PTX. This problem could be solved with the aid of NPs based chemotherapies28. Similarly, methotrexate (MTX) an analog of folic acid, inhibits folic acid synthesis by involving enzymatic activity29. MTX molecular weight is ~ 454 g/mol with a logP of ~ −1.8 which demonstrates its tendency to dissolve in water compared to hydrophobic solvents25; therefore, the efficient passage of MTX across the BBB is diminished30. The effectiveness of MTX in GBM treatment has been proven in the clinic11, but untargeted toxicity resulting from the systemic distribution of MTX is still an unsolved obstacle to its use31.
Using two or more drugs in drug delivery will increase anti-cancer performance due to the different mechanisms of action to kill cancer cells32. PTX kills cancer cells by sticking to microtubules and results in G2/M cell death whereas MTX is the analogue of folic acid which interferes in nucleotide synthesis33. This hypothesis was proven by different drugs (such as erlotinib and doxorubicin34,35, paclitaxel and etoposide36, as well as paclitaxel and naringenin37) in co-delivery or combination therapy using NPs for treating GBM.
In our previous work, we observed that MTX and PTX loaded into PLGA NPs coated with PVA/P188 possessed the increased cytotoxicity on U-87 MG (human GBM) and B65 (rat GBM) cell lines33. In this current study, we aimed to check the effectiveness of a combination therapy of P-MTX NPs and P-PTX NPs coated with PVA/P188 on GBM cells and tumors in an animal model. The rapid growth of a tumor, intensive vasculature, similarity to human GBM, a larger tumor size, and their independency to immunosuppressants, were the major reasons for using C6-induced GBM model38.
For the diagnosis of GBM in the clinic, magnetic resonance imaging (MRI) is the first modality39, and positron emission tomography (PET) scan is a gold standard procedure to reveal the metabolic states and neurobiochemistry in the body40. Fluorodeoxyglucose (18F-FDG) is one of the highly used radiotracers in clinical PET scans41. The tumor metabolic state (necrosis or active tumors) can be distinguished based on the severity of the radiotracer uptake4. MRI and PET scan are invaluable procedures during GBM treatment41. Consequently, our current study was performed to determine the efficacy of a combination therapy of MTX and PTX by PLGA-based NPs formulations using a rat GBM model. The biodistribution of NPs was studied using carbon quantum dots (CQD) loaded in PLGA NPs by fluorescent tracking NPs. To evaluate the anti-glioma efficacy of NPs, MRI and PET scan were applied. Our model assayed survival rate, neurological tests, and histopathology of major organs, showing much promise for PLGA-based NPs loaded with PTX and MTX to treat GBM.
Material and methods
Materials
PLGA (50:50 MW 30,000 g.mol−1), MTX (purity > 95%) and PTX (purity > 97%) were obtained from the Xi’an Xinlu Biotech Company (China). PVA (80% hydrolyzed, MW 30,000 g.mol−1) was purchased from Merck (Germany). Acetone (purity > 99.9%) was bought from Dr. Mojallali’s Industrial Chemical Complex Company (Iran). P188 (solid pellet) was bought from the Alfa Aesar Company. CQDs were obtained from the Shimi Sanat Roshd Sahand Company. Dimethyl sulfoxide (DMSO), fetal bovine serum (FBS), trypsin, (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid) (MTT) powder, penicillin–streptomycin, phosphate-buffered saline (PBS), goat serum, tris buffered saline (TBS), and H2O2 were purchased from Sigma-Aldrich (Germany). Proteinase K was purchased from the Roche Company. C6 cell line was obtained from the Cell Bank of Pasteur Institute (Iran). Male Wistar rats (220 ± 20 mg) were obtained from the Royan Institute (Iran). Hematoxylin and Eosin, Annexin V/Propidium iodide were purchased from the BioLegend Company. Oxytetracycline spray was purchased from Daru Darman Salafchegan.
Preparation of the NPs
P-MTX NPs and P-PTX NPs
NPs were prepared by precipitation method according to our previous research with some modifications33. Specifically, 50 mg of PLGA and 5 mg of MTX or PTX were dissolved in 5 mL acetone separately. The organic phase was added to the water-based phase (PVA, 90 mL, 1% W/V) and P188 (10 mL, 1% W/V) was added as the coating agent and stabilizer for the NPs. After evaporation of the organic phase during the night, the dispersant was centrifuged (Eppendorf5810) at 16,000 g for 15 min and washed two times with distilled water. Afterwards, the pellet was dispersed in sucrose (2% W/V) using a Vortex mixture (Thermo Scientific™). The dispersant was freeze-dried (−50 °C) (Telstar, lyoQuest) and stored at 4 °C for other characterization studies.
PLGA NPs loaded with CQD (P-CQD NPs)
P-CQD NPs were prepared by an emulsion solvent evaporation method. At first, the best solvent for CQD was visually determined. The particular amount of CQD was dissolved in acetone, distilled water, DMSO, and DCM. It was visually observed that the DMSO solution was the most transparent one (Fig. S9. B). For the preparation of P-CQDNPs, briefly, 25 mg of CQD was dissolved in 1 mL of DMSO, and 50 mg of PLGA was dissolved in acetone. These organic phases were added to the PVA/P188 solution at a ratio of 9:1 (100 mL, 1% W/V). Then, the suspension was sonicated at 200 W for 5 min (Topsonics, UHP-200). All other processes (including centrifugation, washing, and freeze drying) were done similarly to the preparation of P-MTX NPs and P-PTX NPs.
Characterization of the NPs
Particle size and zeta potential analysis
The mean diameter of the NPs was measured using dynamic light scattering (DLS) (ScatterScope1, K-One, S. Korea) before and after the centrifugation. To investigate the zeta potential of the NPs, a Zeta Potential Analyzer (Malvern, ZEN 3600) was applied.
Morphological analysis of the NPs
Scanning electron microscopy (SEM) was applied to assess the size and morphology of the NPs. Dried suspensions of NPs on aluminum foil were sputter-coated with gold and palladium, and the particle morphology was assessed using SEM under a voltage acceleration of 15 kV.
Stability of the NPs
The diameter of the NPs was measured over the time to assess stability. For this, the prepared NPs were stored at 4 °C, and their hydrodynamic diameters were measured for one, two, and three months after preparation.
Determination of drug loading (%), encapsulation efficiency (%) and in vitro drug release
Drug loading, encapsulation efficiency, and in vitro drug release of P-MTX NPs and P-PTX NPs were studied using UV–Vis spectroscopy according to our previous study33. Encapsulation efficiency and drug loading of P-CQD NPs were obtained by UV–Vis spectroscopy at 430 nm. In vitro release studies were done by the dispersion of 20 mg of P-CQD NPs in 5 mL of a PBS solution at pH 7.3. The dispersion was poured into a previously prepared dialysis tube (MWCO 6–8 KDa). The prepared dialysis tube was put into falcon tubes containing a PBS (pH 7.3) medium, caped, and placed in a shaking incubator (labTech) at 500 g at 37 °C. At intervals of 1, 6, 24, 48, and 72 h, 10 mL of the medium was collected, and its absorption was read at 430 nm. In order to maintain the sink conditions, 10 mL of the new medium was added to the falcon tube. This process was repeated three times, independently.
GC-FID technique
Residual acetone was evaluated using the GC-FID (gas chromatography with downstream flame ionization detector) technique. P-MTX NPs and P-PTX NPs samples (1 g) were equilibrated at room temperature for 30 min and put into the head space vial based on USP (United States Pharmacopeia) standard protocol, OVI (Organic Volatile Impurities) monograph. Analysis was performed using an Agilent 7890B gas chromatograph (USA). An Agilent DB-624 MS capillary column (30 m × 0.250 mm × 1.4 µm) was used for the separation of the analytes. The injector temperature was set at 140 °C. The initial temperature of the column oven program was 40 °C which was held for 20 min. The temperature increased to 250 °C at a heating rate of 10 °C/min. The carrier gas was N2 at a constant flow of 1.5 mL/min and a split ratio of 1:2 was applied. The area under the curve of the standard solution of acetone and samples was used to determine the concentration of acetone in samples.
Hemolysis assay
A blood sample from a healthy volunteer donor was collected and mixed with a sodium citrate solution (3.8 wt. %) at a ratio of 9:1 (blood to sodium citrate). Dispersions of P NPs, MTX, PTX, MTX + PTX, P-MTX NPs, P-PTX NPs and P-MTX NPs + P-PTX NPs were prepared at several concentrations (0.065 mg/mL, 0.125 mg/mL, 0.250 mg/mL, 0.5 mg/mL, 1mg/mL, and 2 mg/mL), incubated with diluted blood and placed in a shaking incubator for 3 h. Cremophor EL was applied for PTX solubilization, the combination of Cremophor EL and normal saline (1:1) was used to dissolve PTX + MTX. Normal saline was used for the other formulations. Then, the samples were centrifuged for 10 min at 500 g. The supernatant optical density (OD) was read at 540 nm by a microplate reader (BioTek). Triton-X100 and normal saline serum were applied as a positive and negative control, respectively. All protocols were clarified and approved by the Ethics Committee of Tehran University of Medical Sciences (IR.TUMS.MEDICINE.REC.1399.614) and all methods were performed in accordance with the relevant guidelines and regulations. Informed consent was obtained from all subjects and/or their legal guardian(s).
Cellular assays
Cell culture
Rat glial cells (C6) were obtained from the Pasteur Institute (Iran). Cells were placed in a 95% CO2 humidified incubator at 37 °C. DMEM-F12 medium containing 1% V/V penicillin/streptomycin and 10% V/V fetal bovine serum (FBS) was used as the culture media for each passage, which was done every 48–72 h.
Cell viability assays
C6 cells were seeded in a 96-well plate at a concentration of 3 × 105 cells per well42 and incubated for 24 h. Afterward, cells were treated with P-MTX NPs, P-PTX NPs, PTX, MTX, P NPs, a mixture of PTX and MTX, and a mixture of P-PTX NPs and P-MTX NPs, at several concentrations for 24, 48 and 72 h. Cells were washed with a PBS solution at the specified time points. 100 µL of an MTT solution (0.5 mg/mL) was poured into each well. Then, the plate was covered with aluminum foil to avoid light and incubated for 4 h. 200 µL of DMSO was added to each well, and after 2 min of shaking, the absorbance was read at 570 nm by a microplate reader (BioTek).
Flow cytometry
C6 cells (106 cells) were treated with PTX + MTX and P-MTX NPs + P-PTXNPs at their IC50 concentrations for 48 h. After centrifugation at 300 g for 5 min, the pellet was washed three times. 100 µL of Annexin V-FITC and propidium iodide was added to 500 µL of the cells. After 30 min of incubation in a dark room, the cells were washed by centrifugation. Each sample was placed in a flow cytometer, and the percent of necrosis, early apoptosis, late apoptosis, and healthy cells was measured.
Animal studies
Male wistar rats (about 2 months of age) were used for in vivo studies. Animals were purchased from the Royan Institute (Tehran, Iran) and kept in 12 h of darkness, 12 h of lightness, and 22 °C with enough food and water (3 rats in each cage). Before any measurement, at least one week was considered for adaptation43. All protocols were clarified and approved by the Ethics Committee of Tehran University of Medical Sciences (IR.TUMS.MEDICINE.REC.1399.614) and all methods were performed in accordance with the relevant guidelines and regulations. The study is reported in accordance with ARRIVE guidelines (https://arriveguidelines.org).
Orthotopic GBM model
Rats were divided into 10 groups and each group contained 6 rats after tumor implantation. Rats were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) / xylazine (13 mg/kg)44. After ensuring deep anesthesia in rats, they were placed in a stereotaxic apparatus on a controlled heat pad. Artificial tear gel and wet gauze were applied to protect the eyes. First, the hair at the site was completely shaved, and after washing and sterilization, a small incision was made 1 cm long. The tumor area was implanted in the frontal lobe according to other studies with brief modification45,46. A tiny hole was then drilled (1 mm diameter) in the frontal lobe of the skull and 10 µL of C6 cells (8 × 105)36 were injected via a 25 µL Hamilton syringe (26s gauge) slowly (1µL/min), (stereotaxic navigation: anteroposterior (AP) 2, medio-lateral (ML) + 1.5, dorso-ventral (DV) 3). After the injection was finished, the syringe was kept in place for 2 min to prevent the cells from pouring out of the hole. Then, the syringe was removed. The hole was sealed with bone wax (Animalab, Co.) and three sutures were used with silk thread to seal the injection site with oxytetracycline sprayed on the skin. The entire procedure lasted 15–20 min for each animal, and a heat pad was used during recovery.
Magnetic resonance imaging (MRI)
To confirm tumor formation and follow the change in tumor size, a small animal 3T MRI (Siemens Healthcare, Erlangen, Germany; slew rate, 200 mT/m/ms; maximum amplitude, 45 mT/m), FOV read = 80 mm, FOV phase 100.0%, Section thickness = 5.0 mm, Voxel size = 0.4 × 0.4 × 5.0, TR = 7.8 ms and TE = 3.69 ms, was applied 14, 28, 42 and 56 DPI in the National Brain Mapping Laboratory (NBML). Anesthetized animals were put into the animal holder. Slices of T2-weighted MR images were captured. The tumor area was measured manually via AxioVision Rel 4.8 software. To calculate the tumor volume, the tumor area was multiplied by slice thickness.
Positron emission tomography (PET)
A small animal PET scan (micro-PET, PNP Co, Tehran, Iran) was used to check the variation in metabolism in the tumor before and after treatment on days 15, 29 and 43 after tumor implantation. The rats were injected with 1 mCi of 18F-FDG via tail vein, and the anesthetized animals were placed in an animal holder, and three-dimensional regions of interest (ROI) were drawn around the brain manually.
The ROIs were converted to a brain-to-background ratio (BBR) as: (ROI counts per voxel)/ (background counts per voxel). The back of the neck region was used as the background. 3D images were captured. Then, analysis of tumor volume and radionuclide brain uptake, and metabolic tumor volume were completed.
Chemotherapy regimen
Tumor-bearing rats were divided into 10 groups randomly (n = 6) including: 1) physiological saline, 2) P NPs, 3) MTX, 4) PTX, 5) MTX + PTX, 6) ½ (MTX + PTX), 7) P-MTX NPs, 8) ½ (P-MTX NPs + P-PTX NPs), 9) P-PTX NPs, and 10) P-MTX NPs + P-PTX NPs were injected via tail vein. Moreover, a sham group was considered. Administration of the chemotherapeutics MTX at 1 mg/kg47 and PTX at 3 mg/kg36 was continued every week, and the total volume of injection was 250–500 µL (depending on the animal weight). To disperse NPs, injectable normal saline was applied and to solubilize free drugs, medical DMSO and normal saline (1:99) were used.
Survival rate and body weight change
To study the effect of chemotherapeutics on animal growth or general body condition, animal weight was recorded every week, and a graph of weight change was constructed. Every animal death was registered (6 out of 6 in each group), and euthanasia was considered in the case of weight loss of more than 15%, hemiplegia, or starvation over more than 48 h. A Kaplan–Meyer diagram was constructed, and the mean survival rate was also measured.
Movement assays
An open field device was used to determine the animal posture and movement during the study at 9 am43. Before any experiments, the chamber was cleaned with 70% ethanol. Each animal was placed in a test area for 10 min and crosswalk, grooming, and rearing number of rats was recorded.
Quantitative histopathology analysis
Slides of the brain, heart, liver, spleen, lung, right kidney, and right testis of all groups were deparaffinized and stained with hematoxylin followed by eosin staining. Slides were sectioned at 0.5 µm thickness, and pictures were captured by optical microscopy (Jenamed2, Camera uEye). Quantitative analysis was done on the liver, spleen, kidney, and testis tissues. Hepatocyte nuclei diameter, hepatocyte diameter, number of ruined cells, and number of Kupffer cells were counted manually according to other studies48,49. The Bowman capsule diameter, proximal epithelium thickness, proximal lumen diameter, distal epithelium thickness, and distal lumen diameter were measured on the kidney slides50. The diameter of the white pulp was also measured in spleen samples. The number of leyding51,52,53, Sertoli54,55, spermatid56, spermatocyte56, spermatogonia57, spermatozoid58, epithelium thickness59,60 and seminiferous tubular diameter55,61 were assessed. Statistical analysis was performed only among the P-PTX NPs, P-MTX NPS + P-PTX NPs, and the sham groups.
In-vivo apoptosis assays
During our study, as will be presented, it was concluded that P-PTX NPs and P-MTX NPs + P-PTX NPs regimens exhibited greater potency than other treatments. Consequently, it was decided to identify which of these regimens is of paramount importance. The tumor-bearing rats from P-PTX NPs and from P-MTX NPs + P-PTX NPs were chosen to check tumor markers in case the tumor was not eliminated. Slides of the brain tissues from P-PTX NPs, P-MTX NPs + P-PTX NPs, and the control (tumor bearing rats) were deparaffinized as follows: The slides were placed in xylene for 5 min, twice, then in 100% ethanol for 3 min, and in 95% ethanol for 3 min. After that, they were rinsed with PBS/Tween 20 solution for 2 min, twice. Then, H2O2: methanol (1:9) was used to cover the slides for 10 min. After washing with PBS three times, proteinase K was applied for 30 min. Slides were washed, and triton X 3% was poured on them. Samples were washed again and treated with terminal deoxynucleotidyl transferase (TdT) for 2 h at 37 °C, followed by PBS washing and DAPI staining. Pictures were acquired by a fluorescent microscope (Olympus BX 43).
Immunohistochemistry assays
Slides of P-PTX NPs, P-MTX NPs + P-PTX NPs, and controls (tumor-bearing rats) were dipped in a TBS solution for 20 min, washed with PBS, coated with triton 3% for 20 min, and washed again. Goat serum was used to block secondary antibodies (40 min). Slides were treated with diluted primary antibodies (anti-NF-ĸB) in PBS (1:100) at 4 °C for 24 h. Slides were washed with PBS for 4 times (each time 5 min), and a diluted secondary antibody in PBS (1:150) was applied at 37 °C for 90 min in dark conditions. After washing with PBS, DAPI was used to stain cell nuclei. Pictures were obtained by a fluorescent microscope (Olympus BX 43). A Ki-67 antibody, MMP-2, VEGF and p53 were also used to determine cell propagation62, prognosis63, angiogenesis64, and apoptosis65, respectively. For the quantitative analysis of samples, ImageJ software was applied.
Blood assays
To investigate the safety of NPs and organ toxicity, a blood assay was performed. According to animal care standard, blood assays were only done on groups with the best anti-tumor effect (P-PTX NPs, P-MTX NPs + P-PTX NPs and healthy rats). Three rats in each group were anesthetized, and blood samples were acquired via cardiac puncture. Samples were sent to the lab quickly for investigation. Blood urea, creatinine, alanine transaminase (ALT), aspartate transaminase (AST), and alkaline phosphatase (ALP) were measured and analyzed.
Biodistribution tests
Three tumor-baring rats were anesthetized with ketamine (100 mg/kg) / xylazine (13 mg/kg). Then, the full body was shaved, and 10 mg of P-CQD NPs (dispersed in 0.3–0.5 mL physiological saline) were slowly injected via the tail vein. 1 h after injection, NPs tracking was performed via the KODAK imaging system within 2 min of exposure time. Excitation and emission filters were set at 430 nm and 535 nm, respectively. After the last time point of imaging, animals were sacrificed, and the brain, heart, lung, spleen, liver, and kidneys were extracted for ex-vivo imaging. Quantitative analysis of NPs distribution was also calculated.
Statistical analysis
All data are reported as the mean ± SD. The Shapiro–Wilk test was applied to assess the normality of the data. Student t-tests and one-way analysis of variance (ANOVA), were used to analyze data using GraphPad Prism 9 software (P < 0.05). If data did not exhibit normal distribution, results were determined via Kruskal Wallis test.
Ethics approval and consent to participate
All protocols were clarified and approved by the Animal Ethics Committee of Tehran University of Medical Sciences (IR.TUMS.MEDICINE.REC.1399.614).
Results and discussion
Preparation and physiochemical characterization of P-MTX NPs and P-PTX NPs
The NPs were prepared by precipitation method according to our previous study33. PVA and P188 were applied as coatings. According to the SEM images, the P-MTX NPs had a semi-spherical morphology, and the P-PTX NPs had a spherical morphology (Fig. 1A–D). Also, a smooth coating was evident around the PLGA core (Fig. 1B). In the SEM images (mean size distribution based on a sample of at least 50 NPs) of the P-MTX NPs, most of the NPs were 50–100 nm (mean = 73.7 ± 9 nm) (Fig. 1E); however, the size range of the P-PTX NPs was about 150–250 nm (mean = 199.2 ± 38 nm) (Fig. 1F). The mean hydrodynamic diameter of the NPs was analyzed using DLS before and after centrifugation (Figure S1). The hydrodynamic diameters of the P-MTX NPs and the P-PTX NPs were 117 ± 20 nm and 215 ± 41 nm, respectively, which increased to 133 ± 18 nm and 221 ± 36 nm after centrifugation (Fig. S1). It was observed that there was a partial increase in the hydrodynamic mean diameter of the NPs after centrifugation which could be attributed to NP agglomeration.

Physiochemical characterization of NPs: (A, C) SEM images of P-MTX NPs (Scale bars = 100 nm and 200 nm, respectively), (B, D) SEM images of P-PTX NPs (Scale bars = 200 nm and 1 µm, respectively), (E) size distribution of P-MTX NPs according to SEM images, (F) size distribution of P-PTX NPs according to SEM images, (G) in vitro release profile, (H) zeta potential of P-MTX NPs and P-PTX NPs, and (I) hydrodynamic size change of P-MTX NPs and P-PTX NPs before and after centrifugation, one, two and three months after preparation (n = 3).
In this study, the mean hydrodynamic diameter of the P-MTX NPs was 146 ± 15 nm, 166 ± 19 nm, and 174 ± 16 nm after one, two, and three months of storage, respectively. The size of the P-PTX NPs was 253 ± 14 nm, 276 ± 23 nm, and 315 ± 66 nm after one, two, and three months of storage, respectively (Fig. 1I and Fig. S1). Changes in sample size and polydispersity are common phenomena when using the present lyophilization storage method, as observed in another study66. The mean zeta potential of the P-MTX NPs and P-PTX NPs was −29.3 ± 1 mV and −18.4 ± 1 mV, respectively (Fig. 1H). According to Jang et al PLGA NPs containing MTX had a range of zeta potentials from −17 to −20 mV depending on the preparation method. It was also observed that the addition of poloxamer to the preparation of the NPs resulted in a more negative zeta potential67. In another study, the zeta potential of PLGA NPs containing PTX with various types and concentrations of poloxamers and 0.05% Sodium Lauryl Sulfate (SLS) coatings was from −19 to −40 mV68.
In the nanoprecipitation process, drugs were physically entrapped alongside block copolymers (PLGA) and the encapsulation efficiency of P-MTX NPs and P-PTX NPs was 70.66 ± 7.57% and 89.33 ± 3.78%, respectively. The drug loading of P-MTX NPs was 10.1 ± 1.15% and it was 7.66 ± 0.577% for the P-PTX NPs, which was comparable to our previous study33.
In vitro drug release profiles express essential information for determining the therapeutic pharmacodynamics in the body, which was assayed in PBS media at pH 7.3. The P-MTX NPs showed a minor burst release during the first 10 h and a slow prolonged release until 120 h; however, PTX was sustainably released from the P-PTX NPs initially until 120 h, and at least 20% of PTX remained in the NPs at that time (Fig. 1G). To identify the residual acetone in samples GC-FID was applied. According to the analysis and calculations, the residual acetone was 3.6 ppm and 3.54 ppm in P-PTX NPs and P-PTX NPs, respectively (Fig. S2).
To check the safety of NPs on RBCs, the hemolytic activity of NPs and free drugs on RBCs during in vitro conditions was performed. It was obvious that the hemolysis for the P-MTX NPs + P-PTX NPs was notably lower compared to MTX + PTX. The concentration-dependent hemolysis effect for the MTX + PTX group was completely observable; however, it was not significantly noticeable for the P-MTX NPs + P-PTX NPs (Fig. S3A). The difference between these groups was more distinguishable, especially at the higher concentrations of 1 mg/mL and 2 mg/mL, which was still about 5% for the P-MTX NPs + P-PTX NPs; although, it reached 7% for the MTX + PTX (Fig. S3B and C). It is assumed that this little amount of hemolysis may be due to the presence of P188 on the surface of the NPs. In our previous study, the percent hemolysis from NPs coated with just PVA was lower than the NPs coated with PVA/P18833. According to Fig. S3B, the rate of hemolysis in the P NPs was 4.1 ± 0.1% and 5.57 ± 0.23%, which was slightly lower than the MTX + PTX; 4.83 ± 0.3% and 5.92 ± 0.32% at the lowest (0.065 mg/mL) and the highest (2 mg/mL) concentrations, respectively.
Cellular assessments
To determine NPs toxicity against glioma cells, MTT assays were performed on the C6 rat glioma cell line. It was observed that the cytotoxicity of NPs and free drugs was concentration- and time-dependent. It was further observed that free drugs, PTX or MTX + PTX was more potent against C6 cells during the first 24 h than even after 48 h (Fig. 2A, B). However, the outcome changed after 72 h of treatment as the NPs groups, especially the P-MTX NPs + P-PTX NPs formulation, was the most cytotoxic group among the others. These results could be attributed to the sustained release of drugs from NPs, which resulted in a higher effectiveness after 72 h (Fig. S4A–D). This outcome is similar to our previous work on B65 and U-87 MG cell lines33.

(A, B) MTT assay on the C6 cell line with P NPs, MTX, PTX, P-MTX NPs, P-PTX NPs, MTX + PTX, and P-MTX NPs + P-PTX NPs after 48 h of treatment, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, flow cytometry results of C6 cells (C) control (without treatment) (D) treated with P-PTX NPs and (E) P-MX NP + P-PTX NPs, Q1: necrosis, Q2: late apoptosis, Q3: early apoptosis, and Q4: live cells.
Apoptosis and necrosis of C6 cells when exposed to P-PTX NPs and P-MTX NPs + P-PTX NPs were studied by a fluorescence-activated cell sorter (FACS). 94.6% of cells were alive in the control (tumor bearing rats) group (Fig. 2C). It was observed that 8.99%, 22.5%, and 14.4% of cells were in early apoptosis, late apoptosis, and necrosis, for the P-MTX NPs + P-PTX NPs treated cells (Fig. 2E). In P-PTX NPs treated cells, the percentage of early apoptosis, late apoptosis and necrosis was 6.24%, 12.6% and 21.5%, respectively (Fig. 2D). Therefore, it can be concluded that the combination of P-MTX NPs with P-PTX NPs resulted in a lower necrosis percentage and a lower apoptosis.
Animal studies
Tumor imaging, body weight and survival analysis
In order to investigate anti-GBM efficacy in vivo, rat tumor induction was completed using the cancerous C6 cell line. Fig. S5 is a schematic view of the tumor implantation procedure and imaging processes. MRI showed tumor formation on the 14 DPI, and animals were treated with formulations weekly. Images of the brain tumors and also the health conditions of the animals were assessed according to the timetable (Fig. 3).

Schematic view of the timetable of the animal studies.
According to the MRI images of the horizontal and coronal views, all animals in all groups had an increase in tumor size except the P-PTX NPs and P-MTX NPs + P-PTX NsP group (Fig. 4A, B and Fig. S6B). Body weight changes for all groups, except the sham and P-PTX NPs, showed a decrease in tumor growth. Although the body weight of the P-PTX NPs and P-MTX NPs + P-PTX NPs increased, it was lower than the normal rat growth (the sham), which can be attributed to tumor existence and chemotherapy regimens (Fig. 4C). For the control group (tumor-bearing rats), P NPs, PTX, MTX, PTX + MTX and ½ (MTX + PTX) groups, rats did not survive until the third MRI (which was done on 42 DPI). P-MTX NPs and ½ (P-MTX NPs + P-PTX NPs) were more effective than the mentioned groups, however, the rats died in these two groups. The mean survival values for the P-MTX NPs and ½ (P-MTX NPs + P-PTX NPs) groups were 38 and 39 DPI, respectively. P-PTX NPs and P-MTX NPs + P-PTX NPs were 54 and 59 DPI, respectively. Two rats of each group ultimately survived until 70 DPI, and the tumor completely vanished under MRI. Although the other four rats in the P-PTX NPs and P-MTX NPs + P-PTX NPs groups died, the tumor growth rate was still slower than in the other groups (Fig. 4D, E).

(A) Horizontal view of MRI images on 14, 28, 42, and 56 DPI, tumors were specified with a yellow circle. (B) Tumor volume diagram according to MRI pictures, data = mean ± SD (analysis was done only among P-PTX NPs + P-MTX NPs vs. P-PTX NPs and it was ns.) (C) Body weight changes of the animals during the study (analysis was done only among P-PTX NPs + P-MTX NPs, P-PTX NPs and sham, only sham vs. P-MTX NPs + P-PTX NPs was significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). (D) Kaplan-Meyer diagram of the survival fraction of rats (n = 6). (E) Median survival of each treatment group and control (tumor-bearing rats) (n = 6).
In addition to tumor size, the metabolic status of the tumor is principal in determining prognosis, although rarely studied69. For this purpose, PET scanning is the best way to determine the cellular function in the tumor milieu40. PET scanning from animal brains was performed on 15, 29, and 43 DPI only on the control (Fig. 5A and supplementary information), P-PTX NPs (Fig. 5B and supplementary information), and P-MTX NPs + P-PTX NPs (Fig. 5C and supplementary information). It should be noticed that the last two groups were selected for their better impression of the tumor. Results showed that radionuclide absorption (BBR: Brain to Background Ratio) increased in one of control (tumor-bearing rats) groups. In another one, BBR slightly decreased, and the third sample died before the second scanning (Fig. 5D). Enhancement of radionuclide absorption is due to tumor growth, but its decrement could be due to fewer movement of animal (that is, unfavorable physical conditions because of tumor growth). The BBR of P-PTX NPs samples generally decreased, possibly because of tumor necrosis or diminution of tumor size (Fig. 5E). Rats treated with P-MTX NPs + P-PTX NPs showed a general reduction in BBR (Fig. 5F). In one case, the absorption of radionuclide in brain decreased harshly, classified as green. However, the tumor remained visible in the MRI images, which can be attributed to the necrosis of the tumor without any inflammation (Fig. F C). Metabolic tumor volumes obtain from PET scan (Fig. 5G).

PET scan results of the: (A) control (tumor-bearing rats), (B) P-PTX NPs and (C) P-MTX NPs + P-PTX NPs, BBR (brain to background ratio) of (D) control (tumor-bearing rats), (E) P-PTX NP, (F) P-MTX NPs + P-PTX NPs and (G) metabolic tumor volume in the control (tumor-bearing rats), P-PTX NPs, and P-MTX NPs + P-PTX NPs groups (i : indistinguishable).
Movement analysis
To check the general health condition of rats, movement, grooming, and rearing counting was done using an open-field assay. As shown in Fig. 6A, B, the number of crosswalks during 10 min decreased in the control (tumor-bearing rats), P NPs, PTX, MTX, MTX + PTX, ½ (MTX + PTX), P-MTX NPs, and ½ (P-MTX + P-PTX) NPs groups continuously over time; meanwhile there wasn’t any impressive change in the number of crosswalks among the sham, P-PTX NPs, and P-MTX NPs + P-PTX NPs groups over time. The trend of grooming diagram is a bit more complicated; there was no meaningful difference over time for the MTX group. However, the amount of grooming decreased in the control, P NPs, PTX, MTX + PTX, ½ (MTX + PTX) treated rats from 13 to 27 DPI (Fig. 6E, F). In the P-PTX NPs, P-MTX NPs + P-PTX NPs, and sham groups, we observed successive fluctuations in grooming behavior. However, by 55 days post-implantation (DPI), the grooming number in these groups had reached to a relatively equal level (Fig. 6E, F). In the control, P NPs, PTX, MTX + PTX, ½ (MTX + PTX), ½ (P-MTX + P-PTX), and P-MTX NPs treated rats, we observed a continuous decrease in the number of rearing behaviors. Notably, Notably, the P-MTX NPs + P-PTX NPs group exhibited a subtractive graph with a slight slope. However, in the sham and P-PTX NPs groups, an increasing trend was observed (55 DPI) after a reduction in rearing number (27 DPI), reaching a higher level by 55 DPI (Fig. 6C, D).

(A, B) The number of crossing analysis only was done among the sham, P-PTX NPs and P-MTX NPs + P-PTX NPs and none of them was significant, (C, D) the number of rearing, analysis only was done among the sham, P-PTX NPs and P-MTX NPs + P-PTX NPs and none of them was significant, (E, F) the number of rearing of animals in the treatment groups and also control (tumor-bearing rats without treatments) and sham groups, analysis only was performed among the sham, P-PTX NPs and P-MTX NPs + P-PTX NPs and none of them was significant, and the blood assays of (G) Blood Urea, (H) Creatinine, (I) ALT, (J) AST, (K) ALP of P-PTX NPs and P-MTX NPs + P-PTX NPs compared to healthy rats, all were non-significant.
Blood assays
To evaluate the blood toxicity of the formulation, NPs were injected into rats at 7 doses, blood samples were taken 24 h later, and liver and kidney factors were measured. Blood urea and creatinine are factors that estimate renal function70. As shown in Fig. 6G–K, no significant change was observed in the treatment groups compared to the untreated groups. Alkaline phosphatase (ALP), alanine transaminase (ALT), and aspartate transaminase (AST) are considered as enzymatic signs of liver function. Elevated levels of these enzymes are associated with liver injuries71.
Immunofluorescence and immunohistochemistry
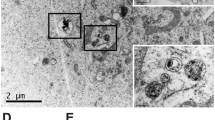
This study further determined apoptosis percentage in tumor and near-tumor environments among these two groups and the control group (tumor-bearing rats). Apoptosis and necrosis are two types of cell death. Apoptosis is programmed and cells will die without damaging adjacent cells and their environment72. To determine apoptosis rate, the TUNEL assay is considered as one of the best methods72. Immunofluorescence images showed that apoptosis was significantly more in P-MTX NPs + P-PTX NPs compared to control (Fig. 7A, C).

(A) Apoptosis rate (TUNEL assay) of the control (tumor-bearing rats), P-PTX NPs, and P-MTX NPs + P-PTX NPs, (scale bar = 20 µm, The scale bars are the same in all pictures), (B) NF-ĸB expression of the control P-PTX NPs and P-MTX NPs + P-PTX NPs treated rat brain (scale bar = 20 µm, The scale bars are the same in all pictures), (C) quantitative analysis of TUNEL, and (D) quantitative analysis of NF-ĸB, (Data = mean ± SD, *P < 0.05, ** P < 0.01, ***P < 0.001, ****P < 0.0001).
According to the immunofluorescence assay, NF-ĸB was expressed in P-MTX NPs + P-PTX NPs to a lower extent than the control (Fig. 7B). These differences in NF-ĸB expression were significant on P-MTX NPs + P-PTX NPs compared to the control which could be due to minor inflammation in the brain and better prognosis in the treated group (Fig. 7D).
To better distinguish the efficacy of P-PTX NPs and P-MTX NPs + P-PTX NPs, Ki-67, VEGF, p53 and MMP-2 expression was assayed in the tumor area by immunohistochemistry (Fig. 8A). It was observed that p53 was expressed to a higher extent in P-MTX NPs + P-PTX NPs compared to the control and P-PTX NPs. The expression of Ki-67 was significantly lower in P-MTX NPs + P-PTX NPs treatments (Fig. 8C, E).

(A) H&E and immunohistochemistry results of p53, Ki-67, VEGF and MMP-2 factors on the control (tumor-bearing rats), P-PTX NPs, P-MTX NPs + P-PTX NPs groups (scale bar = 100 µm, the scale bars are the same in all pictures), (B) quantitative analysis of p53, Ki-67, VEGF and MMP-2 expression, (Data = mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001) (C) MRI results, picture of the excised brain, and scanned image of H&E of brain slide of the rats applied in the immunohistochemistry and immunofluorescence studies.
For immunohistochemistry results, only the change in the expression of Ki-67 of the P-MTX NPs + P-PTX NPs than control was significant (Fig. 8B). The brains of the rats that were applied in immunohistochemistry and immunofluorescence are shown in Fig. 8C. MRI images in Fig. 8C certify the existence of GBM even after treatment. The picture of the excised brain, and scanned image of H&E of brain slide of the rats applied in the immunohistochemistry and immunofluorescence studies is seen in Fig. 8C.
Histopathology analysis
To assess the health condition of the primary organs, samples were prepared from each organ. The quantitative tissue analysis provides valuable insights into the condition of each tissues. Consequently, this study conducted quantitative analyses of tissues. In the H&E image of the liver, healthy hepatocytes were observed in a healthy arranged array with a normal Rimac column in the control (tumor-bearing rats) liver in Fig. 9 (yellow arrow). The hepatocyte nuclei were globular in the center of the cell (green arrow). Mean hepatocyte nuclei and mean hepatocyte diameters were about 8 µm and 18 µm, respectively (Fig. S7A, B). The number of Kupfer and ruined cells were counted at 10–15 and 30–40, respectively (Fig. S7C, D). Spleen sections were typical, and the white pulp diameter was about 200–220 µm (Fig. S7E). A longitudinal cut from the heart showed cells were located beside each other together and ordered. Also, lung, kidney, and testis slides showed regular morphology. The Bowman’s capsule diameter, proximal epithelium thickness, proximal lumen diameter, distal epithelium thickness, and distal lumen diameter were measured in the kidneys (Fig. S7F, G, H, I, J). Cell counts were done for the testis samples containing leyding, Sertoli, spermatid, spermatocyte, spermatogonium, and spermatozoid. Epithelium thickness and seminiferous lumen diameter were measured as well (Fig. S7K, L, M, N, O, P, Q, R). The qualitative investigation of the P NPs treated slides were normal; however, the proximal and distal epithelium thicknesses were reduced, and, on the other hand, the proximal and distal lumen diameters increased. In the testis samples, the number of Sertoli increased; however, the number of spermatogonium decreased.

H&E images of the liver, spleen, heart, lung, kidney, and testis for all groups of the study (scale bar = 100 µm, the scale bars are the same in all pictures of each column).
In the liver slides of ½ (MTX + PTX) and MTX + PTX rats, hepatocytes seem disarrayed (green arrow); moreover, partial vacuolization was observed in higher doses (yellow arrow). Hepatocyte nuclei diameter increased for the MTX + PTX treated group (Figure S7). The spleen seemed healthy in both groups, but the white pulp diameter was diminished for the ½ (MTX + PTX) treatment. The distal epithelium thickened and the distal lumen enlarged for both groups. Additionally, The number of Leyding cells decreased, and spermatogonium cells were reduced particularly at a half concentration dose (½ (MTX + PTX)).
For the MTX and PTX liver samples, pathological changes were distinguished by vacuolization and bleeding for the MTX and PTX groups, respectively (Fig. 9 yellow arrow and Fig. S7A, B). There were complex changes in hepatocytes and their nuclei diameters. For the MTX-treated rats, the white pulp diameter was less, and also the epithelium thickness increased to some extent in the spleen and lung, respectively. There was no drastic qualitative and quantitative pathological change in the heart and kidney samples. It seems that the testis was inflamed during organ excision for the PTX treated group. Partial disorderliness was seen for the H&E samples (Fig. S7C, D), and in quantitative analysis, epithelium thickness and the number of spermatozoids were reduced (Fig. 9). Qualitative changes were seen in this tissue with some areas showing cell destruction, making it challenging to distinguish between them (Fig. S7). In the ½ (P-MTX + P-PTX) NPs group, hepatocyte nuclei diameter was reduced, and in the P-MTX NPs slides, the Rimac space was enlarged, but the cells were arrayed. Heart, lung spleen, and testis seemed unchanged for the ½ (P-MTX + P-PTX) NPs and P-MTX NPs groups. The Bowman’s capsule and proximal and distal lumen diameter increased for both of them compared to the control.
The P-PTX NPs group was full of amorphous and vacuolized hepatocytes (Fig. S7F) and Rimac was hardly detectable. P-MTX NPs + P-PTX NPs induced vacuolization too; however, the Rimac space was unchanged. Hepatocyte diameter and the number of ruined cells increased for both groups. However, the number of Kupffer cells remained relatively stable. Slides of the spleen, heart, and lung seem unchanged. The Bowman’s capsule diameter increased sharply for the P-MTX NPs + P-PTX NPs group, and for the testis tissue, some variations containing enlarged seminiferous tubules and thicker epithelium occurred (Fig. S8). The testis tissue from the P-PTX NPs treated rat changed partially. The space between the base membrane and cells partially increased (Fig. S8E). In this study, the condition of the tissues was briefly examined, but more intense histological studies are still needed to fully assess the influence of these NPs on tissue health.
P-CQD NPs and biodistribution
To investigate bio-distribution, CQDs were applied. TEM images of CQDs indicated that the CQDs were semi-spherical with a size range below 10 nm (mean = 3.57 ± 1.9 nm) (Fig. 10A). Emission spectra of the CQD were around 400–500 nm (Fig. 10D), and the crystalline structure of the CQDs was assayed via XRD. Interlayer spaces were confirmed by the existence of a wide peak at 22 degrees at 3.39 Å (Fig. S9C)73,74. FTIR results showed bands at 1120, 1390, and 1720 cm-1 which are from the stretching vibration of C–O75, O–H74, and C = O76 functional groups and also peaks at 3100 and 3400 cm-1 which are due to the stretching vibration of –NH77 and –OH78 groups. These functional groups are pursuant to the elements in the CQDs structure (Fig. S9A)74.

(A) TEM image of CQD (Scale bar = 10 nm), (B) SEM image of P-CQD NPs (Scale bar = 100 nm), (C) in vitro release profile of CQD from P-CQD NPs data = mean ± SD, n = 5, (D) emission spectra of CQD, (E) schematic image of P-CQD NPs, (F) fluorescent bio tracking of P-CQD NPs 1, 6, 24, and 48 h after IV injection, (G) fluorescent intensity of dissected organs after 48h, (H) fluorescent intensity of injected sample (before injection), (I) average signal count emitted from each organ, (Data = mean ± SD) and (J) percent of signal count in organs, (Data = mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
After making sure the CQDs were suitable, P-CQD NPs were prepared. Since these NPs can stimulate the biodistribution of P-MTX NPs and P-PTX NPs in the body, it was necessary to have similar physiochemical properties. The P-CQD NPs hydrodynamic size was 150 ± 23 nm with a zeta potential of −27.3 ± 0.6 mV, which was relatively similar to P-MTX NPs and P-PTX NPs. SEM images showed semi-spherical NPs with a rough surface that may be because of the adherence of the CQDs on the surface of the NPs (Fig. 10B). The in vitro release of CQDs from P-CQD NPs was also studied to ensure that these NPs are suitable for biodistribution studies lasting up to 48 h. In vitro release results showed that only 30% of the loaded CQDs were released from the NPs during the first 48 h (Fig. 10C). Therefore, we can conclude that most of the adsorption intensity in the biodistribution study was related to the distribution of the NPs, not free CQDs. Figure 10 E is a schematic view of P-CQD NPs applied for the biodistribution studies.
First of all, NP emission was checked to ensure the fluorescent emission of the samples (Fig. 10H). Here, the aim was to check for the biodistribution in GBM-bearing rats and after confirming tumor formation via MRI (Fig. S10), rats were anesthetized 1, 6, 24, and 48 h after injection to track the P-CQD NPs. It was observed that the NPs were distributed all over the body (Fig. 10F). After 48 h, animals were sacrificed, and significant organs were scanned to detect P-CQD NPs. As seen (Fig. 10F, G), the NPs exhibited remarkable brain accumulation, but also concentrated in large quantities in the lungs (Fig. 10G). According to Fig. 10I, J, the lowest and highest signals were from the spleen and lungs, respectively. The highest NPs concentration was seen in the kidneys (two kidneys were considered as one organ) compared to other organs. A notable finding in this study was the modest accumulation of particles in the liver. In the current study, only 17.2 ± 0.6% of NPs (among major organs) were able to reach the brain after 48 h of injection, but this formulation effectively controlled GBM in rats, suggesting that it could serve as a straightforward and effective approach for enhancing the targeted delivery of these nanoparticles to the brain.
Discussion
In this study, we prepared P-MTX NPs and P-PTX NPs to evaluate their combination effect in the GBM model. According to the SEM images the mean diameter of P-MTX NPs and P-PTX NPs were 73.7 ± 9 nm and 199.2 ± 38 nm, respectively. It is stated that polymeric NPs with a size of below 200 nm are able to cross the BBB. In animal studies, the optimal size range of NPs lies between 70 and 200 nm15,79. Moreover, during clathrin-mediated transcytosis, which is one of the routs for NPs uptake, the size of vesicles exceed 200 nm79. Studies on rats indicated that the pore size of brain tumors ranges from 100 to 380 nm80. In addition, P188 was used to deliver NPs via endogenous active targeting via anchoring apoE to the surface of P188-coated NPs and their entry via LDL receptors of the BBB33,81. In the current research, it seems logical that P-MTX NPs and P-PTX NPs can cross BBB to some extent.
The size of both P-MTX NPs and P-PTX NPs exhibited a partial increase during the 3-month storage period in lyophilized form. That is a common phenomenon during a lyophilized storing process, as observed in another study, too66,82,83.
In our study, P-PTX NPs had spherical morphology; however, in P-MTX NPs they were non-spherical particles. The shape will affect the protein corona of NPs84 and result in changes in the pharmacokinetic, anti-tumor efficacy and bio-fate of NPs85,86. Besides, there is extensive research on the effect of shape on biodistribution and cell internalization of inorganic NPs, nano-disc, and nanorods87,88,89,90,91,92,93, and there is literature on linking PLGA NPs shape and biodistribution94.
The release of MTX from P-MTX NPs lasted up to 5 days whereas the release of PTX from P-PTX NPs was not as prolonged. A sustained release of PTX was also seen in other studies95,96, But it is claimed that the burst release of MTX is a common phenomenon in the preparation of NPs, because MTX is usually located in the outer layers of the NPs97,98.
After preparing P-MTX NPs and P-PTX NPs, we verified their suitability for further studies by measuring the residual acetone concentration using GC-FID. Since residual solvents can cause toxicity and side effects, their detection and elimination in NP samples are crucial. GC-FID is highly accurate for identifying residual solvents like acetone in formulations99,100,101. In this study, acetone was employed as the solvent in the preparation of P-MTX NPs and P-PTX NPs. Animal studies have shown that acetone injections can cause neurological and hematological side effects, with high intraperitoneal doses proving fatal in rats102.
The hemolytic activity of the combination form of P-MTX NPs + P-PTX NPs was compared to MTX + PTX. It was observed that the amount of hemolysis was much less in the combination form of NPs than free drugs. According to the MTT assay the cytotoxicity of free drugs in combination form was more notable than combination NPs, during the first 24 h. However, this trend is expected to reverse over time. The cytotoxicity of P-MTX NPs + P-PTX NPs is expected to increase significantly after 72 h of treatment. We hypothesize that this effect can be attributed to extended drug release. Our previous research33 and a study performed by Maleki et al demonstrated a considerable cytotoxic effect of free drugs compared to the nanoformulation during the initial 24 h36. Flow cytometry results presented that the co-administration of P-MTX NPs and P-PTX NPs induced a higher amount of apoptosis and less necrosis than the co-administration of MTX and PTX.
In our study, the C6 cell line was applied for the insertion of a cranial brain tumor due to factors such as integrin, ICAM, CD44, osteopontin, laminin, fibronectin, phosphacan, nexin, angiotensinogen, MMP, and collagen type IV which are well expressed in the C6-derived GBM model103. In addition to the aforementioned characteristics, the lack of necessity for immunocompromised rodents suggests that C6 cells are an optimal choice for tumor implantation38. In order to assess the in vivo anti-GBM efficacy of the formulations, C6 tumors were implanted and then subjected to regular imaging.
The standard imaging approach for brain tumors always contains at least two planes of T2 MRI104 and intracranial GBM (as a hyperintense) can be distinguished significantly from normal brain tissue via both T1 and T2 MRI105. According to the MRI and PET scan images, it was concluded that two formulations, P-PTX NPs and P-MTX NPs + P-PTX NPs were successful in the treatment of GBM. Remarkably, two rats in each group exhibited survival until 70 DPI, and exhibited complete tumor regression. However, it is noteworthy that although other rats in the P-PTX NPs and P-MTX NPs + P-PTX NPs groups experienced mortality, the tumor enlargement rate in these two groups remained relatively slower compared to the other groups, including those treated with free drugs. The difference in response to treatment can be attributed to variations in stem cells, cellular responses, and the structural composition of tumor tissues106.
The well-being of rats was assessed by quantifying their movement, grooming, and rearing using an open-field experiment. Based on the findings, the number of crosswalks, grooming and rearing of P-PTX NPs, P-MTX NPs + P-PTX NPs were partially equal to the sham group. Studies have shown conflicting results regarding the relationship between disease severity and movement status in rats107,108,109. In order to assess the impact of P-PTX NPs and P-MTX NPs + P-PTX NP formulations on the health of rats, a series of injections consisting of seven repeating doses of NPs were administered. There was no significant difference detected between the treatment groups and the untreated groups. The enzymatic activity of the liver is represented via ALP, ALT, and AST, which exhibited the increased levels in the case of liver injury110. Our investigation did not find any significant variation in the levels of ALP, ALT, and AST. The safety of PTX in NPs formulation has also been validated in another study111. According to the TUNEL test it was observed that the rats treated with P-PTX NPs and P-MTX NPs + P-PTX NPs had a higher percentage of apoptosis compared to the control group (tumor-bearing rats). Although MTX acts as an anti-inflammatory substance at low doses (5–25 mg/week), it causes apoptosis by ROS generation112, and changes in methylenetetrahydrofolatereductase expression113. NF-ĸB is known as an anti-inflammatory factor; however, it was observed that in stressful circumstances (such as hypoxia or under UV-C irradiation), it would regulate apoptosis114. On the other hand, NF-ĸB is also over-expressed in cancer, trauma, or inflammation115, and its activation is associated with a poor prognosis indicating tumor growth and exacerbated tumor conditions116. Notably, NF-ĸB is highly activated, particularly in GBM117. In this study, treatment with P-MTX NPs and P-PTX NPs led to a reduction in NF-ĸB concentration, indicating a promising therapeutic response. To distinguish the best response to the treatment between P-MTX NPs + P-PTX NPs and P-PTX NPs, the expressed Ki-67, VEGF, p53 and MMP-2 was quantified. Ki-67 is a marker to estimate proliferation and metastasis in tumors118. VEGF plays a role in angiogenesis119 using the amount of expression as a factor to estimate the severity of GBM64. Matrix metalloproteinases (MMP) play a role in normal physiological conditions, such as the development of the fetus and tissue regeneration and remodeling processes. Infiltration and invasiveness of cancer cells are related to MMP activity120. It is claimed that a greater amount of MMP-9 is related to an exacerbated condition in cancer120. P53 is one of the most critical proteins in apoptosis and tumor suppression121.
The hypothesis of this assay was that even if a tumor was resistant to treatment according to MRI images, there would be the difference in cancer cell apoptosis, angiogenesis, and inflammation among the control and P-PTX NPs and P-MTX NPs + P-PTX NPs treated rats. The expression of Ki-67 was remarkably reduced in the P-MTX NPs + P-PTX NPs and it was observed that P-PMTX NPs + P-PTX NPs were superior to the P-PTX NPs treatment. In the current study, the efficacy of the NPs formulations against GBM tumors was proven; however, we were uncertain about their side effect. Therefore, we aimed to check the organ's histopathology quantitatively and qualitatively. As we can conclude, the toxic effect of free drugs was harsher than the NPs formulations. Only, in P-MTX NPs + P-PTX NPs treated rats, the number of dead cells was elevated in the liver sample. Furthermore, the epithelium thickness and seminiferous tubule diameter in the testicular tissue increased significantly. The safety of PTX in the PLGA NPs on the liver, kidney and heart tissues was confirmed in a study121. In another investigation, it was revealed that coating the PLGA NPs with P188 will reduce the side effects of the drug122. The biodistribution of the P-CQD NPs coated with P188/PVA was also checked in this study. According to the in vivo fluorescent tracking of the NPs, a noticeable percent of P-CQD NPs reached the brain after 48 h of i.v. administration. However, we found that a noticeable concentration of NPs was found in the lungs. In the other study, it was revealed that PLGA NPs coated with P188 were mostly found in organs like the lung, kidney, and spleen which are part of the reticuloendothelial system123. Additionally, it was mentioned that non-ionic and P188-coated NPs are good choices for lung drug delivery, as well124.
It is stated that surface coating of NPs with surfactants such as P188, poloxamer 407, tween 80 and cholic acid can boost a NPs uptake by brain cells and surface coverage with a surfactant could be considered for better brain targeting16. One mechanism for this better uptake is anchoring apoE to the surface of NPs and their entry via LDL receptors9. As there are promising results of P188 modified PLGA NPs for treating GBM in our studies and other studies125,126, it seems that the application of P188 should be highly considered for GBM targeted delivery. Fig. S11 is a schematic view of NPs preparation and the hypothesized delivery to the brain and Fig. S12 is the graphical view of the aim of this study.
Conclusion
In this study P-MTX NPs and P-PTX NPs were prepared. The surface of NPs was coated with PVA and P188. Efficacy of different formulations containing MTX, PTX, MTX + PTX, ½ (MTX + PTX), P-MTX NPs, P-PTX NPs, ½ (P-MTX NPs + P-PTX NPs) and P-MTX NPs + P-PTX NPs were assayed using a C6 GBM model on rats. The results of this study, even within one treatment group, were diverse, but indicated that P-MTX NPs in combination with P-PTX NPs (P-MTX NPs + P-PTX NPs) could be an improved chemotherapy regimen for the treatment of GBM. However, the survival and MRI of P-PTX NPs treated rats were similar to P-MTX NPs + P-PTX NPs treated rats, partially. PET scans and immunohistochemistry results of Ki-67, VEGF, p53, and MMP-2 factors of the brain showed prominence for the P-MTX NPs + P-PTX NP regimen. Further, the apoptosis rate in the brain by the TUNEL assay in P-MTX NPs + P-PTX NPs was higher than P-PTX NPs treated rats. Histopathology analysis of all treatment regimens were also performed on the liver, spleen, heart, kidneys, lungs and testis. It was observed that under the P-MTX NPs + P-PTX NP treatment regimen, the spleen, heart, and lung were healthy and any change was not seen in the histopathology slides of these tissues, but some changes were seen in the liver, kidney and testis. Fluorescence imaging demonstrated successful brain delivery of the NPs. However, enhanced targeting efficacy may require additional modifications such as incorporation of alternative targeting agents alongside P188. Furthermore, integrating antioxidants such as vitamins, beta-carotene, and minerals into the NP formulation could mitigate organ toxicity to a degree.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Pan, Y. et al. Localized NIR-II laser mediated chemodynamic therapy of glioblastoma. Nano Today 43, 101435 (2022).
Di Mascolo, D. et al. Conformable hierarchically engineered polymeric micromeshes enabling combinatorial therapies in brain tumours. Nat Nanotechnol 16(7), 820–829 (2021).
Zhang, F. et al. Effect of axitinib regulating the pathological blood–brain barrier functional recovery for glioblastoma therapeutics. CNS Neurosci Ther 28(3), 411–421 (2022).
Li, Z. et al. Activatable luminescent probes for imaging brain diseases. Nano Today 39, 101239 (2021).
Huang, J. et al. Application of blood brain barrier models in pre-clinical assessment of glioblastoma-targeting CAR-T based immunotherapies. Fluids Barriers CNS 19(1), 1–15 (2022).
Stater, E. P. et al. The ancillary effects of nanoparticles and their implications for nanomedicine. Nat Nanotechnol 16(11), 1180–1194 (2021).
Mathew, E. N. et al. Delivering therapeutics to glioblastoma: overcoming biological constraints. Int J Mol Sci 23(3), 1711 (2022).
Stamatovic, S. M., Keep, R. F. & Andjelkovic, A. V. Brain endothelial cell-cell junctions: how to “open” the blood brain barrier. Curr Neuropharmacol 6(3), 179–192 (2008).
Hartl, N., Adams, F. & Merkel, O. M. From adsorption to covalent bonding: Apolipoprotein E functionalization of polymeric nanoparticles for drug delivery across the blood–brain barrier. Adv ther 4(1), 2000092 (2021).
Del Amo, L. et al. Surface functionalization of PLGA nanoparticles to increase transport across the BBB for Alzheimer’s disease. Appl. Sci. 11(9), 4305 (2021).
Madani, F. et al. Polymeric nanoparticles for drug delivery in glioblastoma: State of the art and future perspectives. J Control Release 349, 649–661 (2022).
Paul, P. S. et al. Unconjugated PLGA nanoparticles attenuate temperature-dependent beta-amyloid aggregation and protect neurons against toxicity: Implications for Alzheimer’s disease pathology. J Nanobiotechnology 20(1), 67 (2022).
Latronico, T. et al. Luminescent PLGA nanoparticles for delivery of darunavir to the brain and inhibition of matrix metalloproteinase-9, a relevant therapeutic target of HIV-associated neurological disorders. ACS Chem Neurosci 12(22), 4286–4301 (2021).
Seko, I. et al. Development of curcumin and docetaxel co-loaded actively targeted PLGA nanoparticles to overcome blood brain barrier. J. Drug Deliv. Sci. Technol. 66, 102867 (2021).
Cai, Q. et al. Systemic delivery to central nervous system by engineered PLGA nanoparticles. Am J Transl Res 8(2), 749 (2016).
Joseph, A. et al. Surfactants influence polymer nanoparticle fate within the brain. Biomaterials 277, 121086 (2021).
Ossama, M. et al. Augmented cytotoxicity using the physical adsorption of Poloxamer 188 on allicin-loaded gelatin nanoparticles. J Pharm Pharmacol 73(5), 664–672 (2021).
Hartl, N., Adams, F. & Merkel, O. M. From adsorption to covalent bonding: Apolipoprotein E functionalization of polymeric nanoparticles for drug delivery across the blood–brain barrier. Adv. Ther. 4(1), 2000092 (2021).
Saxena, V., Hussain, M. D. Poloxamer 407/TPGS mixed micelles for delivery of gambogic acid to breast and multidrug-resistant cancer. Int. J. Nanomed. 2012; p. 713–721.
Song, C. K. et al. Enhanced in vitro cellular uptake of P-gp substrate by poloxamer-modified liposomes (PMLs) in MDR cancer cells. J. Microencapsulation 28(6), 575–581 (2011).
Attia, M. S. et al. Pharmaceutical polymers and P-glycoprotein: Current trends and possible outcomes in drug delivery. Mater. Today Commun. 34, 105318 (2023).
Huang, S.-T. et al. Liposomal paclitaxel induces fewer hematopoietic and cardiovascular complications than bioequivalent doses of Taxol. Int. J. Oncol. 53(3), 1105–1117 (2018).
Astarita, E. M. et al. Adenomatous polyposis coli loss controls cell cycle regulators and response to paclitaxel in MDA-MB-157 metaplastic breast cancer cells. PloS one 16(8), e0255738 (2021).
Pardridge, W. M. Drug transport across the blood–brain barrier. J Cereb Blood Flow Metab 32(11), 1959–1972 (2012).
Zhang, D. Y. et al. Ultrasound-mediated delivery of paclitaxel for glioma: A comparative study of distribution, toxicity, and efficacy of albumin-bound versus cremophor formulations US-delivered ABX extends survival in GBM PDX mouse model. Clin Cancer Res 26(2), 477–486 (2020).
Zhao, M. et al. Codelivery of paclitaxel and temozolomide through a photopolymerizable hydrogel prevents glioblastoma recurrence after surgical resection. J. Controlled Release 309, 72–81 (2019).
Wang, L. et al. Paclitaxel and naringenin-loaded solid lipid nanoparticles surface modified with cyclic peptides with improved tumor targeting ability in glioblastoma multiforme. Biomed Pharmacother 138, 111461 (2021).
Danhier, F. et al. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: In vitro and in vivo evaluation. J Control Release 133(1), 11–17 (2009).
Deda, D. K. et al. Cytotoxicity of methotrexate conjugated to glycerol phosphate modified superparamagnetic iron oxide nanoparticles. J Nanosci Nanotechnol 21(3), 1451–1461 (2021).
Corley, C. & Allen, A. R. A bibliometric analysis of cyclophosphamide, methotrexate, and fluorouracil breast cancer treatments: Implication for the role of inflammation in cognitive dysfunction. Front Mol Biosci 8, 683389 (2021).
Di Filippo, L. D. et al. Drug delivery nanosystems in glioblastoma multiforme treatment: Current state of the art. Curr Neuropharmacol 19(6), 787–812 (2021).
Zhao, M. et al. Nanocarrier-based drug combination therapy for glioblastoma. Theranostics 10(3), 1355 (2020).
Madani, F. et al. Paclitaxel/methotrexate co-loaded PLGA nanoparticles in glioblastoma treatment: Formulation development and in vitro antitumor activity evaluation. Life Sci 256, 117943 (2020).
Lakkadwala, S. et al. Dual functionalized liposomes for efficient co-delivery of anti-cancer chemotherapeutics for the treatment of glioblastoma. J Control Release 307, 247–260 (2019).
Lakkadwala, S. & Singh, J. Co-delivery of doxorubicin and erlotinib through liposomal nanoparticles for glioblastoma tumor regression using an in vitro brain tumor model. Colloids Surf B 173, 27–35 (2019).
Maleki, H. et al. Effect of paclitaxel/etoposide co-loaded polymeric nanoparticles on tumor size and survival rate in a rat model of glioblastoma. Int. J. Pharm. 604, 120722 (2021).
Wang, L. et al. Paclitaxel and naringenin-loaded solid lipid nanoparticles surface modified with cyclic peptides with improved tumor targeting ability in glioblastoma multiforme. Biomed. Pharmacother. 138, 111461 (2021).
Giakoumettis, D., Kritis, A. & Foroglou, N. C6 cell line: the gold standard in glioma research. Hippokratia 22(3), 105–112 (2018).
Xia, W. et al. Deep learning for automatic differential diagnosis of primary central nervous system lymphoma and glioblastoma: multi-parametric magnetic resonance imaging based convolutional neural network model. J Magn Reson Imaging 54(3), 880–887 (2021).
Miranda, A. et al. PET imaging of freely moving interacting rats. Neuroimage 191, 560–567 (2019).
Bolcaen, J. et al. Assessment of the effect of therapy in a rat model of glioblastoma using [18F] FDG and [18F] FCho PET compared to contrast-enhanced MRI. Plos one 16(3), e0248193 (2021).
Huang, C. et al. Shikonin kills glioma cells through necroptosis mediated by RIP-1. PloS one 8(6), e66326 (2013).
Yang, J. et al. Nanoparticle-based co-delivery of siRNA and paclitaxel for dual-targeting of glioblastoma. Nanomedicine 15(14), 1391–1409 (2020).
Bianco, J. et al. Novel model of orthotopic U-87 MG glioblastoma resection in athymic nude mice. J Neurosci Methods 284, 96–102 (2017).
San-Galli, F. et al. Assessment of the experimental model of transplanted C6 glioblastoma in Wistar rats. J Neuro-Oncol 7(3), 299–304 (1989).
Namatame, H. et al. Photodynamic therapy of C6-implanted glioma cells in the rat brain employing second-generation photosensitizer talaporfin sodium. Photodiagn Photodyn Ther 5(3), 198–209 (2008).
Battaglia, L. et al. Solid lipid nanoparticles by coacervation loaded with a methotrexate prodrug: Preliminary study for glioma treatment. Nanomedicine 12(6), 639–656 (2017).
Morovvati, H., Najafzadeh, H. & Azizian, H. Evaluation of effect of curcumin on changes of liver in adrenalectomised rats. J Babol Univ Medical Sci 15(3), 59–64 (2013).
Morovvati, H. et al. The effects of vitamin E on liver and kidney damage induced by dianabol in small laboratory mice. J Babol Univ Medical Sci. 20(9), 36–47 (2018).
Momeni, H. R. & Eskandari, N. Effect of curcumin on kidney histopathological changes, lipid peroxidation and total antioxidant capacity of serum in sodium arsenite-treated mice. Exp Toxicol Pathol 69(2), 93–97 (2017).
Haider, S. G. Cell biology of Leydig cells in the testis. Int Rev Cytol 233(4), 181–241 (2004).
Yang, J.-M. et al. Cadmium-induced damage to primary cultures of rat Leydig cells. Reprod Toxicol 17(5), 553–560 (2003).
Lv, Y. et al. Dicyclohexyl phthalate blocks Leydig cell regeneration in adult rat testis. Toxicology 411, 60–70 (2019).
Scott, H. M. et al. Relationship between androgen action in the “male programming window”, fetal sertoli cell number, and adult testis size in the rat. Endocrinology 149(10), 5280–5287 (2008).
Sukmawati, Y. et al. Vitamin E ameliorates testicular histological features and androgen binding protein levels in testicle of rats induced by allethrin. J Adv Vet Anim Res 6(4), 486 (2019).
Gupta, R. et al. Antifertility studies of the root extract of the Barleria prionitis Linn in male albino rats with special reference to testicular cell population dynamics. J Ethnopharmacol 70(2), 111–117 (2000).
Tremblay, A. R. & Delbes, G. In vitro study of doxorubicin-induced oxidative stress in spermatogonia and immature Sertoli cells. Toxicol Appl Pharmacol 348, 32–42 (2018).
El-Desoky, G. E. et al. Improvement of mercuric chloride-induced testis injuries and sperm quality deteriorations by Spirulina platensis in rats. PloS one 8(3), e59177 (2013).
Kooti, W. et al. Protective effects of celery (Apium Graveolens) on testis and cauda epididymal spermatozoa in rat. Int J Reprod Biomed 12(5), 365–366 (2014).
Kaplanoglu, G. T. et al. Effects of green tea and vitamin E in the testicular tissue of streptozotocin-induced diabetic rats. Saudi Med J 34(7), 734–743 (2013).
Monsefi, M. et al. Mesenchymal stem cells repair germinal cells of seminiferous tubules of sterile rats. Iran J Reprod Med 11(7), 537 (2013).
Kumar, S. et al. Development of a novel animal model to differentiate radiation necrosis from tumor recurrence. J Neuro-Oncol 108(3), 411–420 (2012).
Zhang, H. et al. MMP-2 expression and correlation with pathology and MRI of glioma. Oncol Lett 17(2), 1826–1832 (2019).
Chen, B., et al., Radiogenomic analysis of vascular endothelial growth factor in patients with glioblastoma. 2022.
Gousias, K., Theocharous, T. & Simon, M. Mechanisms of cell cycle arrest and apoptosis in glioblastoma. Biomedicines 10(3), 564 (2022).
Holzer, M. et al. Physico-chemical characterisation of PLGA nanoparticles after freeze-drying and storage. Eur J Pharm Biopharm 72(2), 428–437 (2009).
Madani, F. et al. Preparation of methotrexate loaded PLGA nanoparticles coated with PVA and Poloxamer188. Nanomed Res J 3(1), 19–24 (2018).
Sakhi, M., et al. Design and characterization of paclitaxel-loaded polymeric nanoparticles decorated with trastuzumab for the effective treatment of breast cancer. Front Pharmacol, 2022. 13.
Belloli, S. et al. Characterization of biological features of a rat F98 GBM model: A PET-MRI study with [18F] FAZA and [18F] FDG. Nucl Med Biol 40(6), 831–840 (2013).
Pandya, D., Nagrajappa, A. K. & Ravi, K. Assessment and correlation of urea and creatinine levels in saliva and serum of patients with chronic kidney disease, diabetes and hypertension: A research study. J Clin Diagn Res 10(10), 58 (2016).
Vagvala, S. H. & O’Connor, S. D. Imaging of abnormal liver function tests. Clin Liver Dis 11(5), 128 (2018).
King, T., Cell injury, cellular responses to injury, and cell death. Elsevier’s Integrated Pathology, 2007: p. 1–20.
Peng, J. et al. Fabrication of graphene quantum dots and hexagonal boron nitride nanocomposites for fluorescent cell imaging. J Biomed Nanotechnol 9(10), 1679–1685 (2013).
Gharepapagh, E. et al. Preparation, biodistribution and dosimetry study of Tc-99m labeled N-doped graphene quantum dot nanoparticles as a multimodular radiolabeling agent. New J Chem 45(8), 3909–3919 (2021).
Parveen, A., S. Agrawal, and A. Azam. Thermal properties of transition metals doped (A: Co, Ni and Cu) BiFe0. 9A0. 1O3. in AIP Conference Proceedings. 2016. AIP Publishing LLC.
Yao, S.-F., Chen, X.-T. & Ye, H.-M. Investigation of structure and crystallization behavior of Poly (butylene succinate) by fourier transform infrared spectroscopy. J Phys Chem B 121(40), 9476–9485 (2017).
Kato, H. et al. Fourier transform infrared spectroscopy study of conducting polymer polypyrrole: Higher order structure of electrochemically-synthesized film. J Phys Chem 95(15), 6014–6016 (1991).
Chen, C., Gao, J. & Yan, Y. Observation of the type of hydrogen bonds in coal by FTIR. Energy Fuels 12(3), 446–449 (1998).
Ceña, V., Játiva, P. Nanoparticle crossing of blood–brain barrier: A road to new therapeutic approaches to central nervous system diseases. 2018, Future Medicine. p. 1513–1516.
Gaumet, M. et al. Nanoparticles for drug delivery: The need for precision in reporting particle size parameters. Eur J Pharm Biopharm 69(1), 1–9 (2008).
Hartl, N., Adams, F. & Merkel, O. M. From adsorption to covalent bonding: Apolipoprotein E functionalization of polymeric nanoparticles for drug delivery across the blood–brain barrier. Adv. Therap. 4(1), 2000092 (2021).
Holzer, M. et al. Physico-chemical characterisation of PLGA nanoparticles after freeze-drying and storage. Eur. J. Pharm. Biopharm. 72(2), 428–437 (2009).
Ribeiro, A. F. et al. Characterization and in vitro antitumor activity of polymeric nanoparticles loaded with Uncaria tomentosa extract. Anais da Acad. Brasileira de Ciências 92(01), e20190336 (2020).
Hadji, H. & Bouchemal, K. Effect of micro-and nanoparticle shape on biological processes. J. Controlled Release 342, 93–110 (2022).
Jasinski, D. L., Li, H. & Guo, P. The effect of size and shape of RNA nanoparticles on biodistribution. Mol. Ther. 26(3), 784–792 (2018).
Toy, R. et al. Shaping cancer nanomedicine: The effect of particle shape on the in vivo journey of nanoparticles. Nanomedicine 9(1), 121–134 (2014).
Huang, L. H. et al. Shape-dependent adhesion and endocytosis of hydroxyapatite nanoparticles on A7R5 aortic smooth muscle cells. J. Cell. Physiol. 235(1), 465–479 (2020).
Herd, H. et al. Nanoparticle geometry and surface orientation influence mode of cellular uptake. ACS Nano 7(3), 1961–1973 (2013).
Xie, X. et al. The effect of shape on cellular uptake of gold nanoparticles in the forms of stars, rods, and triangles. Sci Rep 7, 3827 (2017).
Li, L. et al. Biodistribution, excretion, and toxicity of mesoporous silica nanoparticles after oral administration depend on their shape. Nanomed.: Nanotechnol. Biol. Med. 11(8), 1915–1924 (2015).
Talamini, L. et al. Influence of size and shape on the anatomical distribution of endotoxin-free gold nanoparticles. ACS Nano 11(6), 5519–5529 (2017).
Sangabathuni, S. et al. Mapping the glyco-gold nanoparticles of different shapes toxicity, biodistribution and sequestration in adult zebrafish. Sci. Rep. 7(1), 4239 (2017).
Wang, J. et al. In vivo pharmacokinetic features and biodistribution of star and rod shaped gold nanoparticles by multispectral optoacoustic tomography. RSC Adv. 5(10), 7529–7538 (2015).
Kaplan, M. et al. Effects of particle geometry for PLGA-based nanoparticles: Preparation and in vitro/in vivo evaluation. Pharmaceutics 15(1), 175 (2023).
Khuroo, T. et al. Simultaneous delivery of paclitaxel and erlotinib from dual drug loaded PLGA nanoparticles: Formulation development, thorough optimization and in vitro release. J. Mol. Liq. 257, 52–68 (2018).
Bacanlı, M. et al. Evaluation of cytotoxic and genotoxic effects of paclitaxel-loaded PLGA nanoparticles in neuroblastoma cells. Food Chem. Toxicol. 154, 112323 (2021).
Vakilinezhad, M. A., Alipour, S. & Montaseri, H. Fabrication and in vitro evaluation of magnetic PLGA nanoparticles as a potential Methotrexate delivery system for breast cancer. J. Drug Deliv. Sci. Technol. 44, 467–474 (2018).
Mujokoro, B. et al. Combination and co-delivery of methotrexate and curcumin: preparation and in vitro cytotoxic investigation on glioma cells. J. Pharmaceut. Innov. 15, 617–626 (2020).
Ramos, F. et al. In-situ forming PLGA implants: Towards less toxic solvents. Int. J. Pharmaceut. 657, 124121 (2024).
Tankiewicz, M., Namieśnik, J. & Sawicki, W. Analytical procedures for quality control of pharmaceuticals in terms of residual solvents content: Challenges and recent developments. TrAC Trends Anal. Chem. 80, 328–344 (2016).
Espada, R. et al. Effect of aggregation state on the toxicity of different amphotericin B preparations. Int. J. Pharm. 361(1–2), 64–69 (2008).
Espinoza, S. M., Casanas Pimentel, R. G., San Martin Martinez, E. UV–Vis spectroscopic quantification of residual acetone during the development of nanoparticulate drug delivery systems. Pharm. Dev. Technol., 2019. 24(6): 751–760.
Grobben, B., De Deyn, P. & Slegers, H. Rat C6 glioma as experimental model system for the study of glioblastoma growth and invasion. Cell Tissue Res. 310, 257–270 (2002).
Martucci, M. et al. Magnetic resonance imaging of primary adult brain tumors: state of the art and future perspectives. Biomedicines 11(2), 364 (2023).
Arias-Ramos, N. et al. Integrative magnetic resonance imaging and metabolomic characterization of a glioblastoma rat model. Brain Sci. 14(5), 409 (2024).
Di Mascolo, D. et al. Conformable hierarchically engineered polymeric micromeshes enabling combinatorial therapies in brain tumours. Nature Nanotechnol. 16(7), 820–829 (2021).
Souza, T. K. F. et al. Image and motor behavior for monitoring tumor growth in C6 glioma model. PLoS One 13(7), e0201453 (2018).
Wang, Y.-Y. et al. Impaired hippocampal synaptic plasticity in C6 glioma-bearing rats. J. Neuro-oncol. 103, 469–477 (2011).
de Oliveira, É. A. et al. Evaluation of GX1 and RGD-GX1 peptides as new radiotracers for angiogenesis evaluation in experimental glioma models. Amino Acids 48, 821–831 (2016).
Vagvala, S. H. & O’Connor, S. D. Imaging of abnormal liver function tests. Clin. Liver Dis. 11(5), 128 (2018).
Wang, B. et al. Improved anti-glioblastoma efficacy by IL-13Rα2 mediated copolymer nanoparticles loaded with paclitaxel. Sci. Rep. 5(1), 16589 (2015).
Herman, S., Zurgil, N. & Deutsch, M. Low dose methotrexate induces apoptosis with reactive oxygen species involvement in T lymphocytic cell lines to a greater extent than in monocytic lines. Inflammation Res 54(7), 273–280 (2005).
Celtikci, B. et al. Methotrexate-induced apoptosis is enhanced by altered expression of methylenetetrahydrofolate reductase. Anti-Cancer Drugs 20(9), 787–793 (2009).
Khandelwal, N. et al. Nucleolar NF-κB/RelA mediates apoptosis by causing cytoplasmic relocalization of nucleophosmin. Cell Death Differ 18(12), 1889–1903 (2011).
Singh, S. & Singh, T. G. Role of nuclear factor kappa B (NF-κB) signalling in neurodegenerative diseases: an mechanistic approach. Curr Neuropharmacol 18(10), 918–935 (2020).
Park, M. H. & Hong, J. T. Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells 5(2), 15 (2016).
Uddin, M., et al., Natural small molecules targeting NF-κB signaling in glioblastoma. Front Pharmacol, 2021: p. 2174.
Gutiérrez-Almeida, C. E. et al. Proliferation and apoptosis regulation by G protein-coupled estrogen receptor in glioblastoma C6 cells. Oncol Lett 24(1), 1–9 (2022).
Zhang, X. et al. Artificial apoptotic cells/VEGF-loaded injectable hydrogel united with immunomodification and revascularization functions to reduce cardiac remodeling after myocardial infarction. Nano Today 39, 101227 (2021).
Yan, W. et al. Identification of MMP-9 specific microRNA expression profile as potential targets of anti-invasion therapy in glioblastoma multiforme. Brain Res 1411, 108–115 (2011).
Mujokoro, B. et al. Combination and co-delivery of methotrexate and curcumin: preparation and in vitro cytotoxic investigation on glioma cells. J Pharm Innov 15(4), 617–626 (2020).
Pereverzeva, E. et al. Toxicological study of doxorubicin-loaded PLGA nanoparticles for the treatment of glioblastoma. Int. J. Pharm. 554, 161–178 (2019).
Acharya, S. R. & Reddy, P. R. Brain targeted delivery of paclitaxel using endogenous ligand. Asian J. Pharm. Sci. 11(3), 427–438 (2016).
Cortés, H. et al. Non-ionic surfactants for stabilization of polymeric nanoparticles for biomedical uses. Materials 14(12), 3197 (2021).
Gelperina, S. et al. Drug delivery to the brain using surfactant-coated poly (lactide-co-glycolide) nanoparticles: Influence of the formulation parameters. Eur. J. Pharm. Biopharm. 74(2), 157–163 (2010).
Wohlfart, S. et al. Efficient chemotherapy of rat glioblastoma using doxorubicin-loaded PLGA nanoparticles with different stabilizers. PloS one 6(5), e19121 (2011).
Acknowledgements
This work was supported by Tehran University of Medical Sciences, grant No. 9711148003, Iran. The authors would like to acknowledge Preclinical Lab, Core Facility, Tehran University of Medical Sciences, Tehran, Iran and Iranian National Brain Mapping Laboratory (NBML) for providing the in vivo imaging and image processing services for this research.
Funding
This work was supported by Tehran University of Medical Science, grant No. 99-1-148-47998.
Author information
Authors and Affiliations
Contributions
F.M. : Investigation, Methodology, Writing—original draft. H.M. : Validation. T.J.W : Writing—review & editing. S.N.A.: Methodology. S.M.R: Validation. M.R.H: Methodology. M.K: Conceptualization, Supervision, Project administration . M.A: Supervision, Project administration, Writing—review & editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Madani, F., Morovvati, H., Webster, T.J. et al. Combination chemotherapy via poloxamer 188 surface-modified PLGA nanoparticles that traverse the blood–brain–barrier in a glioblastoma model. Sci Rep 14, 19516 (2024). https://doi.org/10.1038/s41598-024-69888-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-69888-1
- Springer Nature Limited