
Abstract
Cadmium, a common metal pollutant, has been demonstrated to induce type 2 diabetes by disrupting pancreatic β cells function. In this study, transcriptome microarray was utilized to identify differential gene expression in oxidative damage to pancreatic β cells following cadmium exposure. The results indicated that a series of mRNAs, LncRNAs, and miRNAs were altered. Of the differentially expressed miRNAs, miR-29a-3p exhibited the most pronounced alteration, with an 11.62-fold increase relative to the control group. Following this, the target gene of miR-29a-3p was identified as Col3a1 through three databases (miRDB, miRTarbase and Tarbase), which demonstrated a decrease across the transcriptome microarray. The upstream target gene of miR-29a-3p was identified as NONMMUT036805, with decreased expression observed in the microarray. Finally, the expression trend of NONMMUT036805/miR-29a-3p/Col3a1 was reversed following NAC pretreatment. This was accompanied by a reduction in oxidative damage indicators, MDA/ROS/GSH-Px appeared to be negatively affected to varying degrees. In conclusion, this study has demonstrated that multiple RNAs are altered during cadmium exposure-induced oxidative damage in pancreatic β cells. The NONMMUT036805/miR-29a-3p/Col3a1 axis has been shown to be involved in this process, which provides a foundation for the identification of potential targets for cadmium toxicity intervention.
Similar content being viewed by others
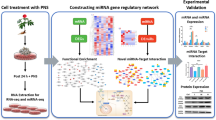


Introduction
As a highly toxic endocrine disruptor, cadmium is a common productive environmental heavy metal pollutant1. A number of studies have indicated that cadmium can enter the body through the digestive and respiratory tracts and accumulate in multiple tissues and organs over an extended period. Most of previous researches have concentrated on the acute toxicity and carcinogenicity. In fact, epidemiological study has showed that cadmium exposure concentrations were positively associated with the risk of gestational diabetes in pregnant women2. As same in the general population, elevated urinary cadmium levels were also found to be positively associated with the development of diabetes mellitus3. It is of greater significance that a recent a dose–response meta-analysis has indicated that even a very low dose of cadmium exposure can promote the development of type 2 diabetes, and this promoting effect is dose-dependent4. Furthermore, experimental studies have shown that cadmium can disrupt glucose homeostasis and result in hyperglycemia in experimental animals5. Mechanism exploring results indicated that cadmium accumulated in pancreatic tissue and preferentially in islets, of which induce apoptotic cell death and interfere insulin synthesis and secretion6,7,8. These adverse effects are attributed to a number of mechanisms, including cadmium-induced oxidative stress, programmed cell death, and inflammatory responses9,10,11. Nevertheless, the precise mechanisms remain to be identified in full.
Cadmium has been identified as an inducer of oxidative stress in various tissues and cells through two main pathways. On the one hand, cadmium facilitates the release of intracellular free iron, which participates in the catalytic decomposition of hydrogen peroxide and leads to the formation of hydroxyl radicals, thus inducing lipid peroxidation12. On the other hand, cadmium can consume glutathione, which results in the inhibition of cadmium clearance. This leads to a disruption of the cellular redox state. Cadmium-induced lipid peroxidation also inhibits the activity of antioxidant defense system enzymes12. Due to the low levels of antioxidant enzymes present in pancreatic β cells, they are particularly vulnerable to the damaging effects of reactive oxygen species (ROS)13,14. Our preceding study demonstrated that exposure to cadmium results in a reduction in glutathione (GSH) levels and an increase in ROS accumulation in islet cells. Furthermore, cadmium exposure induces aberrant expression of non-coding RNAs that are involved in the regulation of cellular events such as oxidative damage and cell death15. More importantly, long non-coding RNAs (LncRNAs) and mircoRNAs (miRNAs), the two most extensively studied non-coding RNAs, have been confirmed to play a role in the oxidative damage caused by cadmium. However, there are multitude of LncRNAs and miRNAs, which presents challenge when identifying specific targets for intervention. Fortunately, LncRNAs can act as sponge to adsorb specific miRNAs, which has been defined as a competitive endogenous RNA (ceRNA) regulatory axis. The ceRNA regulatory axis has been demonstrated to play a pivotal role in the reversal of oxidative damage. Does such a ceRNA regulatory axis exist during oxidative damage induced by cadmium? It is currently unclear.
In this study, genome-wide expression profiling microarray analysis was performed in mouse insulinoma cells (NIT-1) following the induction of oxidative damage by cadmium sulfate (CdSO4). Subsequently, the results of the screening were validated and analyzed by means of bioinformatics, the findings of which were utilized to construct the ceRNA regulatory axis. Finally, N-acetyl-L-cysteine (NAC), a widely used antioxidant, was employed to ascertain whether the ceRNA regulatory axis is implicated in the cadmium-induced oxidative damage process.
Materials and methods
Cell culture
The pancreatic β cell line NIT-1 cells (Ginio Biotechnology Co., Ltd., Guangzhou, China) were cultured in an incubator with saturated humidity, a temperature of 37 °C, and a CO2 concentration of 5%. The culture medium consisted of 1640 medium (Life Technologies/Gibco, Grand Island, NY, USA) with 10% fetal bovine serum (Life Technologies/Gibco, Grand Island, NY, USA). Additionally, 100 Unit/mL penicillin and 100 μg/mL streptomycin (New Cell & Molecular Biotech Co., Ltd, Suzhou, China) were added to the culture medium on a routine basis.
Cell models
Cadmium-induced damage model15: CdSO4 was used as the test material in this study. It is the most abundant in the environment, is highly soluble in water and poses a serious pollution risk. NIT-1 cells were treated with varying concentrations (0 μmol/L, 1 μmol/L, 2 μmol/L, 4 μmol/L, 8 μmol/L, 16 μmol/L, 32 μmol/L and 64 μmol/L) of CdSO4 for 24 h. Cell viability was detected by CCK-8 assay. Three concentrations exhibiting cell viability above 70% were selected for the induction of oxidative damage. Finally, the CdSO4 concentrations were determined to be 0 μmol/L, 1 μmol/L, 2 μmol/L and 4 μmol/L.
Antioxidant model: NIT-1 cells were treated with N-Acetyl-L-cysteine (NAC, Shanghai yuanye Bio-Technology Co., Ltd., Shanghai, China) at concentrations of 0 mmol/L, 0.625 mmol/L, 1.25 mmol/L, 2.5 mmol/L, 5 mmol/L, 10 mmol/L, and 20 mmol/L for 24 h. The concentration of NAC with the highest cell viability was selected as the pretreatment concentration. The experiments were divided into four groups: a control group, a NAC group, a CdSO4 group, and a NAC + CdSO4 group. In the NAC group and NAC + CdSO4 group, 5 mmol/L NAC was added to the cells for 2 h pretreatment. Following the removal of the liquid, 4 μmol/L CdSO4 was added to the CdSO4 group and the NAC + CdSO4 group and placed in an incubator for 24 h. At this point, a series of measurements were taken.
Differentially expressed gene screening
Total RNA was prepared from the control group (0 μmol/L CdSO4) and the treatment group (4 μmol/L CdSO4) with qualified quality. The transcriptome expression profile was detected on the Clariom D mouse microarray from Affymetrix GeneChip by Shanghai Baygene Biotechnology Co., Ltd. The procedure was followed: total RNA preparation, hybridization, image scanning, and raw data acquisition. Subsequently, the raw data underwent RMA correction, with probe information being annotated in the process. Finally, the differentially expressed genes (DEGs) were analyzed by intergroup moderated T-test based on sample groups. The fold change (FC) of RNA expression in different groups was compared from the above DEGs, and the genes with upregulated or downregulated were identified for further analysis. Filter conditions: For up-regulation, the criteria were a FC of at least 2 and a false discovery rate (FDR) of less than 0.05. For down-regulation, the criteria were a FC of at least -2 and an FDR of less than 0.05. All gene expression data have been uploaded to the GEO database with the accession number GSE253072.
RT-qPCR
Total RNA was extracted by using RNA simple total RNA kit (Tiangen Biotech Beijing Co., Ltd.) for mRNA detection. Total RNA with a length greater than 200 nt and less than 200 nt was extracted by using miRcute miRNA isolation kit (Tiangen Biotech Beijing Co., Ltd.). Less than 200 nt of total RNA was employed for miRNA detection and greater than 200 nt for LncRNA detection. Complementary DNA (cDNA) was synthesized following the FastKing gDNA dispelling RT superMix (Tiangen Biotech Beijing Co., Ltd.). cDNA was synthesized following the miRcute plus miRNA first-strand cDNA kit (Tiangen Biotech Beijing Co., Ltd.). RT-qPCR was performed on QuantStudio 3 (Thermo Fisher Scientific, Waltham, MA, USA). The mRNA and lncRNA expression levels were examined in accordance with the two-step method outlined in the instructions for the SuperReal premix plus (SYBR Green) kit (Tiangen Biotech Beijing Co., Ltd.). The miRNA levels were quantified using the miRcute plus miRNA qPCR Kit (Tiangen Biotech Beijing Co., Ltd.). The quality of the PCR process was evaluated based on the amplification and melting curves. And Gapdh was applied as internal control for mRNA and LncRNA, Rnu6 was applied as internal control for miRNA. The relative expression levels of the target genes were calculated by 2-△△Ct method. Primer sequences were listed in Table 1.
Western blot
NIT-1 cells were inoculated in 6-well plates and allowed to adhere to the wall. The cells were then treated with CdSO4 and/or NAC for 24 h. At the conclusion of this period, 180 μL of a ready-made cell lysate was added. Subsequently, the reaction was conducted at 4 °C for 10 min after thorough agitation, and the supernatant was harvested by high-speed centrifugation at low temperature (4 °C, 13,000 rpm, 10 min) to obtain the protein samples. The protein concentration was determined by the BCA method (Beyotime Biotech. Inc., Shanghai, China).
A total of 50 μg of protein samples were successively loaded onto SDS-PAGE gels at concentrations of 5% and 12% for separation purposes. Following this, the samples were transferred to polyvinylidene difluoride (PVDF) membranes. Following a two-hour incubation period at room temperature with 5% skimmed milk, the primary antibody to COL3A1 (Beijing Solarbio Science & Technology Co., Ltd.) was applied. On the subsequent day, the membranes were incubated with a secondary antibody conjugated to horseradish peroxidase (Beijing Zhongsui Jinqiao Biotechnology Co., 1:6000) for one hour at room temperature. Subsequently, the immunoreactive bands were visualised with an enhanced chemiluminescence reagent (Millipore, Temecula, CA) and imaged with a JS-M6 chemiluminescence imaging system (Peiqing Technology Co., Ltd., Shanghai, China). The intensity of the protein bands was quantified using the Image J software (National Institutes of Health, USA). GAPDH was employed as an internal control, and the fold change in protein expression was calculated based on the protein levels observed in the control group.
CCK-8 assay
The inoculation of 96-well plates should be conducted in accordance with the density of 1 × 104 cells per well. CdSO4 or NAC should be added overnight following 24 h of wall attachment. The culture medium should be configured with 10% CCK-8, and the old medium discarded. The fresh medium should be added rapidly and the cells incubated for 3 h. Absorbance at 450 nm was measured by Multifunctional Fluorescent Plate Reader (Flash Biotechnology Co., Ltd., Shanghai, China), and the cell viability = A450 mean of the treated group / A450 mean of the control group × 100%.
Oxidative Stress Detection
Detection of ROS
According to the manual of the reactive oxygen species assay kit (Beyotime Biotech. Inc., Shanghai, China), cells were incubated (37 ℃, 20 min) after diluting the DCFH-DA probe (final concentration of 10 μmol/L). Cells were observed and photographed with a fluorescence microscope (excitation wavelength 488 nm, NIB610-FL, Ningbo Yongxin Optics Co., Ltd., Ningbo, China). The fluorescence intensity was quantified by Image Pro Plus, and the average fluorescence intensity of ROS = the sum of fluorescence intensity/ region area.
Detection of MDA
The MDA content was measured by lipid peroxidation MDA assay kit (Beyotime Biotech. Inc., Shanghai, China). Firstly, a standard curve was prepared for the standards. Then, the reaction system was formulated in a 1.5 mL centrifuge tube following the guideline, mixing and heating at 100 ℃ for 15 min. Finally, the samples were cooled to room temperature and centrifuged (1000 g, 10 min), and 200 μL of the upper solution was used to measure the absorbance at 532 nm. A standard curve regression was established with the concentration of the standards as an independent variable (x), and the absorbance as a dependent variable (y). Absorbance of the samples were substituted into the regression equation and the MDA concentration of the samples were calculated.
Detection of SOD
The reaction system was prepared in a 96-well plate as per the instructions of the total superoxide dismutase assay kit with WST-8 (Beyotime Biotech. Inc., Shanghai, China). Then, the absorbance at 450 nm was measured after incubation at 37 ℃ for 30 min. The absorbance was used to calculate the percentage inhibition, which was (A blank control 1 – A sample)/(A blank control 1 – A blank control 2) × 100%. The SOD enzyme activity units in the sample to be tested = SOD enzyme activity units in the assay system = percentage inhibition/(1 – percentage inhibition).
Detection of GSH-Px
NIT-1 cells were inoculated into a 6-well plate, and protein was extracted after treatment. GSH-Px was detected by following the protocol of the cellular glutathione peroxidase assay kit with NADPH (Beyotime Biotech. Inc., Shanghai, China). The reaction system was prepared referring to the instructions and incubated at room temperature for 15 min. Lastly, each well was added with 10 μL of peroxide reagent solution and the absorbance at 340 nm was measured immediately by a Multifunctional Fluorescent Plate Reader, at which time the 0 min value was recorded. Six values were recorded at 2 min intervals. Glutathione peroxidase activity in the assay system = [(sample△A340 – blank△A340)/min]/(0.00622 × 0.276).
Statistical analysis
All statistical analysis of data from the experiments was performed in SPSS 26.0. One-way ANOVA was performed for comparisons between multiple groups, and the LSD method was used for comparisons between groups. Kruskal–Wallis rank sum test was used when the variance was not equal, and Bonferroni method was used for two-way comparisons between groups. The significant level was set to 0.05.
Results
Screening results of differentially expressed genes
As illustrated in Fig. 1A, the violin profiles exhibited a comparable shape across all samples. Each violin was of uniform width, and the three horizontal lines indicating the 25th, 50th and 75th quartiles were in alignment, indicating that there were no chips with abnormal quality after RMA correction, and no significant batch effects were present. Consequently, the data could be used for further analysis. The principal component analysis demonstrated a clear separation of the two samples in the three principal components, indicating a difference between the control and experimental groups (Fig. 1B). We found 348 DEGs with 296 exhibiting increased expression and 52 exhibiting decreased expression (Fig. 1C). In detail, there are 75 differentially expressed mRNAs, of which 42 were up-regulated and the top three were Hsph1, Chordc1 and Hmox1, while 33 were down-regulated and the three most dramatically decreased were Stc1, Pdgfc and Ppap2b (Fig. 1D). The levels of 20 LncRNAs were substantially up-regulated, with the top three changes ranked as NONMMUT029382, ENSMUST00000162103, ENSMUST00000117235. In contrast, the three LncRNAs, NONMMUT036805, NONMMUT065427 and NONMMUT014565, were down-regulated (Fig. 1E). In addition, Fig. 1F illustrated the differential expression of 16 miRNAs, with 4 miRNAs exhibiting upregulation and 12 miRNAs exhibiting downregulation. Among them, miR-29a exhibited the highest fold change, at 11.62-fold that observed in the control group.

Differentially expressed genes following 4 μmol/L CdSO4 induced oxidative damage to NIT-1 cells. (A) Illustration of violin in control group and CdSO4 group. (B) Principal component analysis scatter matrix. (C) Volcano diagram. (D) Differential expression of mRNAs in CdSO4 group compared with control group. (E) Differential expression of LncRNAs in CdSO4 group compared with control group. (F) Differential expression of miRNAs in CdSO4 group compared with control group. Figure 1A-C were generated by the R software (version 3.6.3, https://www.r-project.org/), while Figure 1D-F were produced by TBtools(v1.098745, https://github.com/CJ-Chen/TBtools-II).
Construction and validation of ceRNA regulatory axis
Given that miR-29a showed the greatest fold change with 11.62-fold, we initially sought to validate the levels of miR-29a-3p in CdSO4-treated NIT-1 cells. The data are presented in Fig. 2A. In detail, the changes in miR-29a-3p were 0.98-fold, 14.86-fold and 19.77-fold in the treatment of 1, 2 and 4 μmol/L CdSO4. Levels of miR-29a-3p exhibited a notable alteration in the 2 and 4 μmol/L CdSO4 groups when compared with the control group (F = 82.61, P < 0.05), thereby corroborating the veracity of the microarray outcomes. Subsequently, the target genes of miR-29a-3p were predicted synergistically with miRDB, miRTarbase and Tarbase. This process yielded a relatively accurate target gene for miR-29a-3p. These genes were then intersected with the 75 differentially expressed mRNAs, resulting a common gene, namely Col3a1 (Fig. 2B). This gene exhibited a decrease in protein expression and mRNA level with the elevation of CdSO4 treatment concentrations. Following the RNAhybrid database prediction, it was demonstrated that the 3′ untranslated region of Col3a1 was capable of binding to the sequence of the seed region of miR-29a-3p (Fig. 2E). After 24 h of treatment with 1 μmol/L, 2 μmol/L, and 4 μmol/L CdSO4, the relative expression of Col3a1 mRNA was 1.09-fold, 0.75-fold (P < 0.05), and 0.26-fold (P < 0.05) (Fig. 2C). The protein expression of COL3A1 in the 1, 2 and 4 μmol/L groups was 0.89-fold, 0.79-fold and 0.63-fold higher than that in the 0 μmol/L group, respectively. The observed differences were statistically significant when comparing the 2 and 4 μmol/L groups with the 0 μmol/L group (F = 13.26, P = 0.00) (Fig. 2D). Moreover, the levels of three LncRNAs were negatively correlated with miR-29a expression, as evidenced by the microarray screening results. The full-length sequence of NONMMUT036805 was predicted to bind to the seed region sequence of miR-29a-3p by the RNAhybrid database (Fig. 2F). In parallel, the levels of NONMMUT036805 exhibited a 1.12-fold increase under 1 μmol/L CdSO4 treatment, 0.25-fold (P < 0.05) decrease under 2 μmol/L CdSO4 treatment and a 0.28-fold (P < 0.05) reduction under 4 μmol/L CdSO4 treatment (Fig. 2G). It is proposed that NONMMUT036805 may function as a ceRNA for miR-29a-3p in the context of CdSO4 treatment.

Construction and validation of ceRNA regulatory axis. The target genes of miR-29a-3p were predicted, and then cross-referenced with the DEGs identified by microarray. As the same, changes in miR-29a-3p, NONMMUT036805 and Col3a1 were observed in NIT-1 cells following treatment with 0 μmol/L, 1 μmol/L, 2 μmol/L and 4 μmol/L of CdSO4.The results were derived from three independent experiments,and the quantitative results were expressed as the mean ± standard deviation (Supplementary table). The symbol "a" indicates a significant difference compared with the control (P < 0.05). (A) The relative expression level of miR-29a-3p. (B) Intersection of the genes tested in this study with those predicted to be targeted by miR-29a-3p. (C) The relative expression level of Col3a1 mRNA. (D) Flod change in COL3A1 protein expression. (E) Prediction of 3′ untranslated region of Col3a1 mRNA bound to miR-29a-3p seed region sequence. (F) The full-length sequence of NONMMUT036805 bound to the seed region sequence of miR-29a-3p. (G) The relative expression level of NONMMUT036805.
Effects of NAC on oxidative stress induced by cadmium
In order to mitigate the oxidative damage effects of CdSO4 on NIT-1 cells, NIT-1 cells were pretreated with 5 mmol/L NAC for 2 h, after which 4 μmol/L CdSO4 was added and the cells were incubated for a further 24 h. In Fig. 3A, cell viability of the NAC + CdSO4 group (84.89%) was greater than that of the CdSO4 group (75.15%), a difference which was statistically significant (F = 58.07, P < 0.05). As illustrated in Fig. 3B, the apoptotic rates of NAC group, CdSO4 group, and NAC + CdSO4 group were 9.17%, 37.17%, and 21.00%, respectively. The statistical inference results further indicated that the apoptosis rates in CdSO4 group and NAC + CdSO4 group were both higher than that in control group (F = 50.32, P < 0.05), while the rate in NAC + CdSO4 group was lower than that in CdSO4 group (P < 0.05). Figure 3C demonstrated that the mean fluorescence intensity of ROS in control, NAC, CdSO4 and NAC + CdSO4 groups were 0.03, 0.04, 0.10 and 0.08, respectively. Further analysis indicated that ROS level in CdSO4 group was higher than that in control and NAC + CdSO4 group (P < 0.05). As shown in Fig. 3D, the concentration of MDA in CdSO4-treated cells was 18.43 μmol/L, which was higher than that observed in control and NAC groups. NAC + CdSO4 group (4.26 μmol/L) exhibited a lower MDA concentration than that observed in CdSO4 group, yet a higher concentration than that observed in control and NAC groups. This suggested that NAC exerted an antagonistic effect on the elevation of the MDA concentration induced by CdSO4. The SOD enzyme activity units in control, NAC, CdSO4 and NAC + CdSO4 groups were 2.05 U, 1.80 U, 1.15 U and 1.40 U. This result indicated that CdSO4 caused the greatest decrease in SOD activity (P < 0.05) while there was an increase after the NAC intervention when compared to the CdSO4 group (P < 0.05, Fig. 3E). In the final figure (Fig. 3F), the GSH-Px enzyme activity in CdSO4 group was 9.91 mU/mL, which was higher than that in NAC + CdSO4 group (14.56 mU/mL, P < 0.05).

The effect of NAC on oxidative damage caused by cadmium. NIT-1 cells were treated with 5 mmol/L NAC, 4 μmol/L CdSO4, and a combination of the two. Cell viability and apoptosis were quantified via CCK-8 assay and hoechst staining assay. The average fluorescence intensity of ROS was determined by a fluorescent probe (DCFH-DA). The MDA content was quantified using the thiobarbituric acid assay. The activity of SOD enzyme was quantified by colorimetric analysis of the WST-8. The activity of GSH-Px enzyme was determined by the nicotinamide NADPH method. The results were derived from three independent experiments, and the quantitative results were expressed as the mean ± standard deviation. The symbol "a" indicates a significant difference compared with the control. The symbol "b" indicates a significant difference compared with NAC group. The symbol "c" indicates a significant difference compared with CdSO4 group. (A) Cell viability of NIT-1 cells following NAC pretreatment. (B) Apoptosis rates of NIT-1 cells following NAC pretreatment. (C) Average fluorescence intensity of ROS in NIT-1 cells following NAC pretreatment. (D) MDA content of NIT-1 cells following NAC pretreatment. (E) Units of SOD enzyme activity in NIT-1 cells following NAC pretreatment. (F) GSH-Px enzyme activity of NIT-1 cells after NAC pretreatment.
Effects of NAC on cadmium-induced changes of ceRNA regulatory axis
To elucidate the impact of cadmium treatment on the NONMMUT036805/miR-29a-3p/Col3a1 regulatory axis and its subsequent effect on pancreatic β cells functionality, the levels of NONMMUT036805, miR-29a-3p and Col3a1 were quantified in NIT-1 cells after treatment with CdSO4 and/or NAC. The fold change of NONMMUT036805 in NAC, CdSO4 and NAC + CdSO4 groups was 2.20-fold, 0.19-fold and 0.61-fold of that in control group. The downward trend observed in CdSO4-treated group was reversed by NAC pretreatment (P < 0.05, Fig. 4A). The changing trends of miR-29a-3p in NAC, CdSO4 and NAC + CdSO4 groups were 1.01-fold, 31.64-fold and 14.45-fold of that in control group. The upward trend observed in CdSO4-treated group was reversed by NAC pretreatment (P < 0.05, Fig. 4B). As illustrated in Fig. 4C, the level of Col3a1 mRNA in CdSO4 treatment was found to be 0.16-fold that of control group. This reduction was, however, reversed by NAC pre-treatment, with a level of 0.39-fold change observed. A similar trend was observed in the expression of COL3A1 protein. In detail, the pretreatment with NAC significantly reversed the reduction in COL3A1 protein expression induced by CdSO4 treatment. The protein expression levels were 0.53 and 0.87-fold change in comparison to that in control group (P < 0.05) (Fig. 4D).

The effects of NAC on the NONMMUT036805/miR-29a-3p/Col3a1 regulatory axis in the context of cadmium exposure. Pancreatic β cells were pretreated with 5 mmol/L NAC for 2 h, following by the treatment of CdSO4 for another 24 h. Thereafter, the levels of NONMMUT036805, miR-29a-3p, and Col3a1 in different groups were quantified. The results were derived from three independent experiments, and the quantitative results were expressed as the mean ± standard deviation. The symbol "a" indicates a significant difference compared with the control. The symbol "b" indicates a significant difference compared with NAC group. The symbol "c" indicates a significant difference compared with CdSO4 group. (A) The relative expression level of NONMMUT036805 in NIT-1 cells after NAC pretreatment. (B) The relative expression level of miR-29a-3p in NIT-1 cells after NAC pretreatment. (C) The relative expression level of Col3a1 mRNA in NIT-1 cells after NAC pretreatment. (D) Flod change in COL3A1 protein expression of NIT-1 cells after NAC pretreatment.
Discussion
Cadmium exposure can cause cellular damage through a variety of mechanisms, among which oxidative damage has been extensively studied and widely recognized. Our previously published data also showed elevated levels of oxidative damage level in cadmium-treated NIT-1 cells15. In our present study, CdSO4 treatment elevated oxidative stress indicators, which were reversed by NAC pretreatment. Cadmium-induced apoptosis was also alleviated by NAC, indicating that NAC attenuates cadmium toxicity by reducing oxidative damage. This conclusion is also supported by several studies16,17,18,19. However, the mechanism of this process remains unclear. In recent years, the study of ceRNA has provided a new direction for the study of cadmium toxicity.
The ceRNA interaction pattern, proposed based on miRNAs, can indirectly regulate gene translation by competitively binding microRNAs through response elements20. Previous studies have demonstrated that miR-29a plays a pivotal role in the pathogenesis of diabetes and its associated complications21,22,23,24. Moreover, miR-29a exhibited the greatest variability in the results of the microarray screening (Fig. 1F). Consequently, we selected miR-29a-3p as the foundation for the construction of the ceRNA regulatory axis. Firstly, Col3a1 was identified as the target mRNA. It has been reported that Col3a1 is a key regulator involved in a variety of cellular injuries, and its specific mechanism is closely related to apoptotic induction25,26,27,28. Meanwhile, Col3a1 is frequently regulated by miRNAs, including the miR-29 family, which has an oncogenic role in regulating Col3a1 to control apoptosis27. The maturation of miR-29a-3p inhibits Col3a1 expression and suppresses the progression of gastric cancer by inactivating the Wnt/β-catenin pathway29. Secondly, the focus was on the regulatory mechanism upstream of miR-29a-3p. It has been demonstrated that some circular RNAs or LncRNAs can act as ceRNA to suppress miR-29a-3p levels30,31,32. A study of the DNM3OS/miR-29a/Col3a1 regulatory axis also elucidated the binding sites of each other, thereby demonstrating the feasibility of this model33. In conjunction with database predictions, the 3′ untranslated region of Col3a1 is capable of binding to the sequence of the seed region of miR-29a-3p (Fig. 2E). Similarly, the full-length sequence of NONMMUT036805 is capable of binding to the sequence of the seed region of miR-29a-3p (Fig. 2F). Then, we employed NAC pretreatment to negate the oxidative damage inflicted by CdSO4. The results demonstrated that NONMMUT036805, miR-29a-3p, and Cola31 exhibited disparate trends (Fig. 4). The results of the raw signal analysis lend support to the proposition that NONMMUT036805/miR-29a-3p/Col3a1 regulatory axis has been established.
Nevertheless, there are still some areas that could be enhanced in the construction of the NONMMUT036805/miR-29a-3p/Col3a1 regulatory axis. For instance, luciferase assays, which directly present the binding efficiency between different RNAs, should be conducted in pancreatic β-cells to clarify the binding relationship between NONMMUT036805 and miR-29a-3p or miR-29a-3p and Col3a1. To prove the regulatory relationships between NONMMUT036805 and miR-29a-3p, more specific target interventions should be used. One such intervention would be to directly up-regulate or down-regulate levels of NONMMUT036805 and subsequently observe changes in miR-29a-3p.
Conclusion
In conclusion, a variety of LncRNAs, miRNAs and mRNAs are involved in the oxidative damage induced by cadmium exposure in pancreatic β cells. We established NONMMUT036805/miR-29a-3p/Col3a1 regulatory axis. Although there are some minor flaws, we tentatively suggest that this regulatory axis is involved in the oxidative damage induced by cadmium treatment based on the dose-dependent relationship, the role of NAC pretreatment and bioinformatics analysis. The specific mechanism may be that cadmium reduces the level of NONMMUT036805, which reduces the competitive binding with miR-29a-3p. This may lead to an elevation of miR-29a-3p, which in turn may lead to a reduction of the targeted regulatory expression of Col3a1. Ultimately, this may lead to oxidative damage and apoptosis of pancreatic β cells (Fig. 5).

The mechanisms NONMMUT036805/miR-29a-3p/Col3a1 regulatory axis involved in cadmium-induced oxidative damage. Cadmium reduces the level of NONMMUT036805, which reduces the competitive binding with miR-29a-3p. This may lead to an elevation of miR-29a-3p, which in turn may lead to a reduction of the targeted regulatory expression of Col3a1. Ultimately, this may lead to oxidative damage and apoptosis of pancreatic β cells.
Data availability
All gene expression data have been uploaded to the GEO database with accession number GSE253072. If you would like request the data from this study, you can contact corresponding author or find it in the GEO database. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE253072).
References
Sabir, S. et al. Role of cadmium and arsenic as endocrine disruptors in the metabolism of carbohydrates: Inserting the association into perspectives. Biomed Pharmacother. 114, 108802 (2019).
Li, X. et al. Association of urinary cadmium, circulating fatty acids, and risk of gestational diabetes mellitus: A nested case-control study in China. Environ Int. 137, 105527 (2020).
Shi, P., Yan, H., Fan, X. & Xi, S. A benchmark dose analysis for urinary cadmium and type 2 diabetes mellitus. Environ Pollut. 273, 116519 (2021).
Filippini, T., Wise, L. A. & Vinceti, M. Cadmium exposure and risk of diabetes and prediabetes: A systematic review and dose-response meta-analysis. Environ Int. 158, 106920 (2022).
Camsari, C. et al. Transgenerational Effects of Periconception Heavy Metal Administration on Adipose Weight and Glucose Homeostasis in Mice at Maturity. Toxicol Sci. 168, 610–619 (2019).
Fitzgerald, R. et al. Pancreatic Islets Accumulate Cadmium in a Rodent Model of Cadmium-Induced Hyperglycemia. Int J Mol Sci. 22, 360 (2020).
Hong, H. et al. Cadmium exposure impairs pancreatic β-cell function and exaggerates diabetes by disrupting lipid metabolism. Environ Int. 149, 106406 (2021).
Huang, C.-C. et al. Cadmium exposure induces pancreatic β-cell death via a Ca2+-triggered JNK/CHOP-related apoptotic signaling pathway. Toxicology. 425, 152252 (2019).
Balali-Mood, M., Naseri, K., Tahergorabi, Z., Khazdair, M. R. & Sadeghi, M. Toxic Mechanisms of Five Heavy Metals: Mercury, Lead, Chromium, Cadmium, and Arsenic. Front Pharmacol. 12, 643972 (2021).
Hong, H. et al. Cadmium induces ferroptosis mediated inflammation by activating Gpx4/Ager/p65 axis in pancreatic β-cells. Sci Total Environ. 849, 157819 (2022).
Paithankar, J. G., Saini, S., Dwivedi, S., Sharma, A. & Chowdhuri, D. K. Heavy metal associated health hazards: An interplay of oxidative stress and signal transduction. Chemosphere. 262, 128350 (2021).
Valko, M., Jomova, K., Rhodes, C. J., Kuča, K. & Musílek, K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch Toxicol. 90, 1–37 (2016).
Buha, A. et al. Emerging Links between Cadmium Exposure and Insulin Resistance: Human, Animal, and Cell Study Data. Toxics. 8, 63 (2020).
Stancill, J. S. & Corbett, J. A. The Role of Thioredoxin/Peroxiredoxin in the β-Cell Defense Against Oxidative Damage. Front Endocrinol (Lausanne). 12, 718235 (2021).
Qu, T. et al. Changes and relationship of N6-methyladenosine modification and long non-coding RNAs in oxidative damage induced by cadmium in pancreatic β-cells. Toxicol Lett. 343, 56–66 (2021).
Alizadeh, B., Salehzadeh, A., Ranji, N. & Arasteh, A. Effects of N-Acetyl Cysteine on Genes Expression of c-myc, and Ask-1, Histopathological, Oxidative Stress, Inflammation, and Apoptosis in the Liver of Male Rats Exposed to Cadmium. Biol Trace Elem Res. 200, 661–668 (2022).
Chen, X. et al. Cadmium exposure triggers oxidative stress, necroptosis, Th1/Th2 imbalance and promotes inflammation through the TNF-α/NF-κB pathway in swine small intestine. J Hazard Mater. 421, 126704 (2021).
Wang, Y. et al. Cadmium chloride-induced apoptosis of HK-2 cells via interfering with mitochondrial respiratory chain. Ecotoxicol Environ Saf. 236, 113494 (2022).
Zhaohui, C. et al. ROS-mediated PERK-CHOP pathway plays an important role in cadmium-induced HepG2 cells apoptosis. Environ Toxicol. 38, 2271–2280 (2023).
Li, M. J. et al. Exploring genetic associations with ceRNA regulation in the human genome. Nucleic Acids Res. 45, 5653–5665 (2017).
Dalgaard, L. T., Sørensen, A. E., Hardikar, A. A. & Joglekar, M. V. The microRNA-29 family: role in metabolism and metabolic disease. Am J Physiol Cell Physiol. 323, C367–C377 (2022).
Mohany, K. M., Al Rugaie, O., Al-Wutayd, O. & Al-Nafeesah, A. Investigation of the levels of circulating miR-29a, miR-122, sestrin 2 and inflammatory markers in obese children with/without type 2 diabetes: a case control study. BMC Endocr Disord. 21, 152 (2021).
Sørensen, A. E. et al. The Predictive Value of miR-16, -29a and -134 for Early Identification of Gestational Diabetes: A Nested Analysis of the DALI Cohort. Cells. 10, 170 (2021).
Sun, Y. et al. Expression of miRNA-29 in Pancreatic β Cells Promotes Inflammation and Diabetes via TRAF3. Cell Rep. 34, 108576 (2021).
Liao, Y. et al. Methylmercury cytotoxicity and possible mechanisms in human trophoblastic HTR-8/SVneo cells. Ecotoxicol Environ Saf. 207, 111520 (2021).
Shin, J. et al. Restoration of miR-29b exerts anti-cancer effects on glioblastoma. Cancer Cell Int. 17, 104 (2017).
Xu, W., Li, Z., Zhu, X., Xu, R. & Xu, Y. miR-29 Family Inhibits Resistance to Methotrexate and Promotes Cell Apoptosis by Targeting COL3A1 and MCL1 in Osteosarcoma. Med Sci Monit. 24, 8812–8821 (2018).
Yao, Z. et al. Hydroxycamptothecin Inhibits Peritendinous Adhesion via the Endoplasmic Reticulum Stress-Dependent Apoptosis. Front Pharmacol. 10, 967 (2019).
Han, S., Wang, Z., Liu, J., Wang, H.-M.D. & Yuan, Q. miR-29a-3p-dependent COL3A1 and COL5A1 expression reduction assists sulforaphane to inhibit gastric cancer progression. Biochem Pharmacol. 188, 114539 (2021).
Cui, J., Wang, Y. & Xue, H. Long non-coding RNA GAS5 contributes to the progression of nonalcoholic fatty liver disease by targeting the microRNA-29a-3p/NOTCH2 axis. Bioengineered. 13, 8370–8381 (2022).
Wang, W. et al. Involvement of circHIPK3 in the pathogenesis of diabetic cardiomyopathy in mice. Diabetologia. 64, 681–692 (2021).
Zhang, H. et al. Roles of H19/miR-29a-3p/COL1A1 axis in COE-induced lung cancer. Environ Pollut. 313, 120194 (2022).
Wang, R. et al. Long noncoding RNA DNM3OS promotes prostate stromal cells transformation via the miR-29a/29b/COL3A1 and miR-361/TGFβ1 axes. Aging (Albany NY). 11, 9442–9460 (2019).
Funding
This research was supported by grants from the National Natural Science Foundation of China (NO.82060585) and Yunnan Province Basic Research (NO.202301AT070140).
Author information
Authors and Affiliations
Contributions
Yahao MOU contributed the research idea, completed the experiments and the manuscript. Yifei SUN, Guofen LIU and Nan ZHANG contributed to complete part of the experiments. Zuoshun HE provided advice and edited the manuscript. Shiyan GU conducted the research design and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mou, Y., Sun, Y., Liu, G. et al. Screening of differentially expressed RNAs and identifying a ceRNA axis during cadmium-induced oxidative damage in pancreatic β cells. Sci Rep 14, 18962 (2024). https://doi.org/10.1038/s41598-024-69937-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-69937-9
- Springer Nature Limited