
Abstract
Sensory experiences and learning induce long-lasting changes in both excitatory and inhibitory synapses, thereby providing a crucial substrate for memory. However, the co-tuning of excitatory long-term potentiation (eLTP) or depression (eLTD) with the simultaneous changes at inhibitory synapses (iLTP/iLTD) remains unclear. Herein, we investigated the co-expression of NMDA-induced synaptic plasticity at excitatory and inhibitory synapses in hippocampal CA1 pyramidal cells (PCs) using a combination of electrophysiological, optogenetic, and pharmacological approaches. We found that inhibitory inputs from somatostatin (SST) and parvalbumin (PV)-positive interneurons onto CA1 PCs display input-specific long-term plastic changes following transient NMDA receptor activation. Notably, synapses from SST-positive interneurons consistently exhibited iLTP, irrespective of the direction of excitatory plasticity, whereas synapses from PV-positive interneurons predominantly showed iLTP concurrent with eLTP, rather than eLTD. As neuroplasticity is known to depend on the extracellular matrix, we tested the impact of metalloproteinases (MMP) inhibition. MMP3 blockade interfered with GABAergic plasticity for all inhibitory inputs, whereas MMP9 inhibition selectively blocked eLTP and iLTP in SST-CA1PC synapses co-occurring with eLTP but not eLTD. These findings demonstrate the dissociation of excitatory and inhibitory plasticity co-expression. We propose that these mechanisms of plasticity co-expression may be involved in maintaining excitation-inhibition balance and modulating neuronal integration modes.
Similar content being viewed by others


Introduction
Sensory experiences and learning dynamically reshape brain circuits, primarily through modifications of excitatory and inhibitory synapses1,2. Numerous studies have shown that long-lasting synaptic plastic changes in excitatory glutamatergic synapses, specifically long-term potentiation (eLTP) and depression (eLTD), underlie memory formation3. Similarly, many experimental protocols that induce excitatory plasticity often elicit changes in the efficacy of GABAergic synapses, potentially resulting in inhibitory long-term potentiation (iLTP) or depression (iLTD)4,5,6. However, the co-occurrence and putative interdependence of excitatory and GABAergic long-term plasticity remain unclear.
The crucial feature of postsynaptically expressed inhibitory plasticity is its heterosynaptic nature, wherein non-stimulated GABAergic synapses undergo plastic changes in response to stimulation of nearby excitatory synapses7. In brain slices, brief activation of NMDA receptors (20 μM NMDA, 2–3 min) leads to the induction of postsynaptic GABAergic iLTP8,9,10,11. Conversely, extended application of NMDA may result in iLTD induction12,13. This suggests that prolonged or short-term activation of the NMDA receptor may differentially affect the excitatory and inhibitory plasticity that occur simultaneously in postsynaptic neurons.
It is important to note that much of the research on synaptic plasticity has focused on either excitatory or inhibitory mechanisms studied separately. For example, inhibitory synaptic transmission has often been blocked in excitatory plasticity studies14. Similarly, the majority of studies on GABAergic plasticity involves blocking AMPA receptors to facilitate the recording of inhibitory postsynaptic currents. However, a growing body of evidence suggests a complex co-occurrence of plastic changes at GABAergic and glutamatergic synapses, which may stem from the heterosynaptic nature of some forms of inhibitory plasticity15. These processes require thorough examination, as they are expected to shed new light on maintaining an optimal balance of neuronal activity and shaping long-lasting changes in the activity of cell ensembles during learning16.
Recent experimental findings have emphasized the importance of mid-range synaptic plasticity mechanisms that involve the coordination of multiple synapses within a single postsynaptic neuron17. Excitatory synaptic plasticity, for instance, is characterized by cooperativity at the level of postsynaptic calcium elevation or signaling to the nucleus18. Similarly, inhibitory synaptic plasticity is contingent on the local balance between excitatory and inhibitory currents5. Furthermore, inhibitory transmission can significantly impact excitatory plasticity by altering the direction of efficacy changes or by facilitating local disinhibition19,20. Despite these findings, significant gaps exist in our understanding of the molecular mechanisms underlying the co-occurrence and interaction between excitatory and inhibitory plasticity in a single postsynaptic neuron.
The activity of neuronal ensembles depends on how principal neurons integrate excitatory and inhibitory inputs; this process has been studied using various models. In the balanced model, the strength of the inhibitory input closely follows changes in excitatory transmission21, whereas the reciprocal network model suggests that membrane depolarization may locally decrease inhibition22. Empirical evidence indicates that the integration mode may shift during and after learning, with balanced inhibition observed during sharp-wave ripples and reciprocal mode prevalent during theta oscillations and within hippocampal place cell activity23. Heterosynaptic long-term plasticity and the co-occurrence of excitatory and inhibitory plasticity may play key roles in this process. However, the correlation between excitatory LTP/LTD and inhibitory iLTP/iLTD has not been extensively studied, particularly in the context of a diverse array of inhibitory synapses and GABAergic interneurons.
In this study, we used a combination of electrophysiological, optogenetic, and pharmacological approaches to investigate the co-occurrence of NMDA-induced synaptic plasticity at excitatory and inhibitory synapses. We simultaneously recorded excitatory transmission in the hippocampal CA3 → CA1 projection and local inhibitory transmission originating from somatostatin (SST)- or parvalbumin (PV)-positive interneurons. We aimed to address three key questions: (1) does NMDA-dependent GABAergic plasticity in CA1 pyramidal cells depend on the type of presynaptic interneuron; (2) is the sign of inhibitory plasticity the same or different compared to the direction of concurrent excitatory plasticity; and (3) can the co-expression of excitatory and inhibitory plasticity be pharmacologically uncoupled? Our findings provide the first insight into how distinct patterns of inhibitory and excitatory plasticity co-occurrence depend on the timing of NMDAR activity.
Materials and methods
Ethical approval and animals
All procedures involving the use of animals were conducted in accordance with the Act on the Protection of Animals Used for Scientific or Educational Purposes in Poland (Act of January 15, 2015, with subsequent amendments) and EU Directive 2010/63/EU. The Polish Ministry of the Environment granted approval for experiments involving genetically modified organisms (decision numbers 144/2018 and 69/2023). The mice were housed socially under a 12-h alternating light–dark cycle, with food and water provided ad libitum.
The following mouse strains were used in this study: (1) Sst-IRES-Cre (Jackson Labs stock #013044), (2) Pvalb-IRES-Cre (Jackson Labs stock #017320), and (3) Ai32 (Jackson Labs stock #012569). The founding mice for the colony were procured from JAX® and subsequently housed and bred within the Experimental Animal Facility at Wroclaw Medical University. Experiments were conducted using the offspring of crosses between the Cre-driver line (Sst-IRES-Cre or Pvalb-IRES-Cre) and Ai32 reporter mice (floxed-ChR2), resulting in the generation of Sst-Cre::Ai32 and Pvalb-Cre::Ai32 mice expressing channelrhodopsin-2 fused with EYFP (ChR2(H134R)/EYFP) in the targeted population of interneurons. All studies involving animals were conducted in compliance with the ARRIVE guidelines.
Slice preparation
P35-110 mice were deeply anesthetized with isoflurane (Baxter) and subsequently sacrificed by decapitation. The brain was then extracted and sliced into sections in ice-cold cutting solution, which comprised the following components (in mM): 93 N-methyl D-glucamine, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 10 MgSO4, 0.5 CaCl2, with pH adjusted to 7.4 using HCl, and bubbled with carbogen (95% O2/5% CO2). Transverse hippocampal slices (350 µm) were obtained using a stainless-steel blade (Vector) and Leica VT1200 vibrating microtome. After sectioning, the slices were incubated for 15–25 min in cutting solution that had been warmed to 34°C, and the concentration of Na + was gradually increased according to the protocol described by Ting et al.24. Subsequently, the slices were placed in a holding artificial cerebrospinal fluid (aCSF) solution at room temperature for up to 8 h before use in the experiments. The holding aCSF comprised the following components (in mM): 92 NaCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 D-glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 1.3 MgSO4, 2.5 CaCl2, with pH maintained at 7.4 using carbogen and an osmolarity of ~ 295 mOsm.
Electrophysiology
Whole-cell patch-clamp recordings were obtained from CA1 pyramidal cells (PCs) using an Axio Examiner Z1 microscope (Zeiss) equipped with IR-DIC optics. Patch pipettes (borosilicate glass, OD 1.5 mm, ID 0.86 mm, tip resistance 3–5 MΩ) were pulled using a Sutter P-97 micropipette puller. Slices were constantly perfused in a submersion recording chamber with aCSF at a rate of 3–4 mL/min and maintained at 28°C. The aCSF, composed of 119 NaCl, 2.5 KCl, 1.3 NaH2PO4, 26 NaHCO3, 1.3 MgSO4, 2.5 CaCl2, and 11 D-glucose (pH 7.4), was bubbled with carbogen and had an osmolarity of 295 mOsm. To preserve the physiological ionic balance, the internal pipette solution contained physiological concentrations of crucial ions, including chloride (in mM): 135 potassium gluconate, 5 NaCl, 5 Mg-ATP, 0.3 Na-GTP, 10 phosphocreatine, and 10 HEPES, pH 7.3, adjusted with KOH, had an osmolarity of 285 mOsm. The liquid junction potential was not corrected during the recordings. For every cell, the reversal potential of synaptic GABAA receptors was measured by recording the IPSC at different holding potentials.
Voltage clamp experiments were carried out using a Multiclamp 700B amplifier, Digidata 1440 acquisition card and Clampex10.7 software (Molecular Devices). The recorded signals were filtered at 6 kHz and digitalized at 20 kHz. To identify pyramidal neurons, we relied on their large cell bodies and their locations within the pyramidal cell layer. Additionally, the firing patterns of all analyzed neurons were acquired in the current clamp mode. Only neurons with a regularly accommodating train of action potentials and noticeable sag were considered pyramidal cells. If optogenetic stimulation evoked an action potential in patched neurons, the recordings were discarded. The patch solutions were allowed to equilibrate through the cell for more than 5 min before baseline recording was initiated. While this procedure may result in partial washout of essential molecular factors for eLTP and may bias the results towards eLTD, it ensures the recording of stable baseline IPSCs and EPSCs necessary for determining even minor long-term changes in synaptic strength5. Series and input resistances were assessed every 10 s using a 1 s pulse of − 10 mV in the voltage clamp mode. Experiments in which the series resistance was greater than 25 MΩ or was changed by more than 20% were excluded. The series resistance was not compensated.
In order to elicit excitatory synaptic transmission in CA3 → CA1 projection, an extracellular tungsten bipolar stimulating electrode (FHC) was used. The stimuli, consisting of a 300 µs current (50–150 µA) that generated 40% of the maximum EPSP amplitude without causing an action potential, were delivered. EPSCs were recorded every 10 s at − 70 mV holding potential during the hyperpolarizing pulse used to assess the access resistance. To investigate the short-term plasticity of IPSCs, paired stimulation was employed by delivering two stimuli at interstimulus intervals of 50ms, 200ms, 500ms, 2s and 10s. The paired-pulse ratio was calculated as the amplitude of the second response divided by the amplitude of the first response. The burst response was assessed using eight pulses delivered at 200 ms intervals. Burst depression was calculated as the average amplitude from the 5th to the 8th response divided by the amplitude of the first response. To assess the potentiation of synaptic responses, the average postsynaptic current (PSC) amplitude in the first 4 min before NMDA application was compared to the average PSC amplitude 26–30 min after plasticity induction. In a subset of experiments, picrotoxin (1 μM) was administered to validate the purity of optoevoked inhibitory synaptic transmission.
Optogenetic stimulation
To activate the cells and axons expressing channelrhodopsin-2, a fiber-coupled DG-4 high-speed optical switch (Sutter Instruments) with an excitation filter HQ470/40x (Chroma) was used. Blue light was delivered through the back aperture of a microscope objective (40x, 1.0 NA, Zeiss) at a power density of 5–10 mW/mm2. A brief, 2 ms, pulse of light reliably stimulated ChR2-expressing interneurons and elicited IPSCs in CA1 pyramidal neurons. Two different approaches were used to stimulate PV- and SST-positive presynaptic interneurons. To evoke SST → PC synaptic transmission, the microscope objective was centered over the stratum oriens and the aperture of the objective was fully open, resulting in a 250–400 µm diameter disc of light at the focal plane. In contrast, to elicit PV–PC inhibitory transmission, the lens aperture was reduced, limiting the light spot diameter to 20–30 μm over the stratum pyramidale. This approach enabled the precise stimulation of two separate inhibitory inputs to CA1 pyramidal cells with high spatial specificity. The stability of the delivered light power for optogenetic excitation was monitored regularly using a microscope slide power sensor (S170C, Thorlabs).
Pharmacology
In all recordings, the aCSF did not contain any inhibitors of synaptic receptors to preserve excitatory and inhibitory synaptic transmission. In a subset of experiments, specific matrix metalloproteinase (MMP) inhibitors were used, targeting MMP-9 and MMP-3: (1) UK356618, an inhibitor of MMP-3/MMP-13 used at a concentration of 2 μM; (2) SB-3CT, a selective inhibitor of gelatinases MMP-9/MMP-2, applied at a concentration of 10 μM. Both compounds were dissolved in DMSO; thus, all relevant control recordings were conducted in the presence of DMSO (1:1000). All compounds were purchased from Tocris or Merck-Millipore.
Quantification and statistical analysis
To assess all proposed hypotheses, control experiments were systematically interleaved, utilizing slices from the same animal on the same day for comparative analysis. Male and female mice were used in each experiment in similar numbers. Sex differences were tested; however, no differences were found and the data were pooled. Several a priori criteria guided the data analysis: (1) no outliers were excluded, (2) random assignment of slices to treatments was implemented, (3) investigators were not blinded to the treatment during recording/analysis, and (4) the determination of minimal sample sizes was established through power analysis, drawing on insights from prior experiments11 and assuming a minimal power level of 0.80. Electrophysiological recordings were analyzed using Clampfit 10.7. The onset kinetics of the IPSC were determined based on the 10% to 90% rise time, while the decay phase was analyzed by fitting a biexponential function \(y(t)={A}_{1}exp(-t/{\tau }_{fast})+{A}_{2}exp(-t/{\tau }_{slow})\), where τfast and τslow represent the time constants, and A1 and A2 denote the amplitudes of the fast and slow components of the function, respectively. The mean decay time constant (τmean) was calculated using the formula \({\tau }_{mean}={a}_{1}{\uptau }_{fast}+ {a}_{2}{\tau }_{slow}\), where \({a}_{1}={A}_{1}/({A}_{1}+{A}_{2}\)) and \({a}_{2}={A}_{2}/({A}_{1}+{A}_{2}\)).
Normality distribution was assessed using the Shapiro–Wilk test, while equal variance was analyzed using the Brown–Forsythe test. In instances involving multiple groups, one-way analysis of variance (ANOVA) was applied, followed by Tukey’s post hoc test. To evaluate the impact of treatment during recording obtained from the same cell, a two-tailed paired t-test or Wilcoxon test was used, based on the normality distribution. Statistical significance was set at p < 0.05. Data exhibiting a normal distribution are depicted as the mean ± SEM, whereas those demonstrating a non-normal distribution are represented as medians with interquartile ranges. Parameter n denotes the number of cells recorded from separate slices, with an average of 2–3 cells obtained from a single animal. The corresponding statistical analyses are provided in the captions for the data presented in the figures. Statistical details for the other data are specified in the Results section.
Results
Distinct kinetics and short-term plasticity of optogenetically evoked PV → PC and SST → PC synaptic currents in the hippocampal CA1 field
To investigate the activity of specific GABAergic synapses, we employed optogenetic stimulation of parvalbumin (PV) or somatostatin (SST) interneurons expressing channelrhodopsin-2 (ChR2). For this purpose, we crossbred mice expressing Cre recombinase under the PV or SST promoter with mice harboring Cre-dependent ChR2, thereby generating PV-ChR2 and SST-ChR2 mouse lines. Subsequently, we conducted whole-cell patch-clamp recordings from CA1 pyramidal neurons in acute hippocampal slices. During these recordings, we held the postsynaptic pyramidal cells (PC) at a holding potential of -60 mV and elicited inhibitory postsynaptic currents (IPSCs) through light stimulation of presynaptic PV or SST interneurons (Fig. 1A,B). To optimize the stimulation of PV interneurons, we narrowed the objective aperture to 5–10%, confining light stimulation to a 20–30 μm-diameter disk focused on the CA1 stratum pyramidale. In contrast, SST interneurons were stimulated by a fully open objective aperture focused on the CA1 stratum oriens. Under these conditions, we were able to precisely activate two distinct interneuron types that innervate CA1 pyramidal neurons.

Optogenetically evoked inhibitory synaptic transmission and short-term plasticity at CA1 PV → PC and SST → PC synapses. (A) Scheme of recording configuration. IPSCs were recorded from CA1 pyramidal cells (red) using optogenetic stimulation of presynaptic soma targeting PV-INs in the stratum pyramidale (sp, magenta) or dendrite-targeting SST-INs (green) located in the stratum oriens (so) with 2 ms-long pulses of blue light. (B) Exemplary typical IPSC traces recorded from CA1 PCs and evoked by local light stimulation (indicated by the blue mark) of ChR2-expressing presynaptic PV or SST INs. The amplitudes of the IPSCs were normalized for clear visualization of kinetic differences. (C) The amplitudes of recorded IPSCs in PV → PC and SST → PC inhibitory synapses; PV: median 177 pA (interquartile range IRQ 100–241), n = 76; SST: 61.4 pA (IQR 52.2–92.8), n = 64; p < 10−12. (D–E) Summary plot (D) and histogram (E) of mean decay times of recorded PV → PC and SST → PC IPSCs; PV: 23.9 ms (IQR 19.0–31.2), n = 72; SST: 25.4 ms (IQR 22.6–34.3), n = 64; p = 0.008. (F–G) Summary plot of rise times of recorded PV → PC and SST → PC IPSCs (F); PV: 1.84 ms (IQR 1.59–2.45), n = 71; SST: 6.73 ms (IQR 5.68–8.38), n = 64; p < 10−35. The comparison of the rise time histograms (G) indicates that optogenetic stimulation evoked synaptic transmission in two significantly different types of inhibitory synapses. (H–J) Short-term plasticity in response to presynaptic paired-pulse stimulation with interpulse intervals of 50, 200, 500 ms, and 2 s (H). Summary plot (I) of paired-pulse depression in analyzed groups with nearly linear dependence of paired-pulse depression on the logarithm of the stimulation interval in PV → PC synapses. The most pronounced difference in short-term plasticity between the analyzed synapses was evident at the 200ms interval; PV: 0.55 (IQR 0.48–0.61), n = 34; SST: 0.80 (IQR 0.73–0.88), n = 53; p < 10−13. (K–L) Exemplary postsynaptic responses to presynaptic burst stimulation of SST or PV INs at 200ms interval (K). Notably, PV → PC synapses exhibited a significantly deepened burst-induced depression (L); PV: the average depression in response to 6th–8th pulse within a burst − 0.37 (IQR 0.32–0.46), n = 28; SST: 0.51 (IQR 0.45–0.57), n = 53; p < 10−6. Data in (C), (D), (F), and (J) are presented as median ± interquartile range. Data in (I) and (L) are presented as mean ± SEM, Mann–Whitney test, **p < 0.01, ****p < 0.0001.
Considering the specific location of PV synapses at the neuronal soma and proximal dendrites of CA1 PCs, it was anticipated that the currents at these synapses would be weakly affected by electrotonic filtering, showing a faster time course than SST interneurons, which primarily target distal dendrites. The results confirmed these expectations, as we observed not only higher mean amplitudes of light-induced IPSCs (Fig. 1C), but also a faster decay (Fig. 1D–E) and rise times (Fig. 1F–G) in synapses between PV-positive GABAergic interneurons and CA1 PCs compared to those at SST → PC projections. Furthermore, an analysis of the IPSC rise time distributions (Fig. 1G) revealed that the optogenetic stimulation of SST and PV presynaptic interneurons activated two distinct and largely non-overlapping pools of GABAergic synapses formed onto CA1 pyramidal cells25.
Next, we conducted paired-pulse stimulation to evaluate the short-term plasticity of analysed inhibitory synapses (Fig. 1H). Across a broad range of interpulse intervals, including 50 ms, 200 ms, 500 ms, and 2 s, the majority of cells exhibited paired-pulse depression (PPD). Notably, the paired-pulse ratio was smaller in PV → PC synapses, suggesting a more pronounced PPD in this group (Fig. 1I–J). Furthermore, the magnitude of the paired-pulse ratio in PV → PC synapses (but not SST → PC) displayed a nearly linear dependence on the logarithm of the interstimulus intervals (Fig. 1I). Short-term plasticity was not observed at an interstimulus interval of 10 s.
Additionally, we employed optogenetic stimulation at 200 ms intervals over eight repetitions to investigate IPSC amplitudes in response to burst stimulation (Fig. 1K). As shown in Fig. 1L, IPSC amplitude decreased with each successive stimulus. In control conditions for PV → PC synapses, the average IPSC amplitude for the 6th-8th responses in a burst reached 0.38 ± 0.02 (n = 28) of the amplitude observed in the first response. It is noteworthy that burst depression was significantly less pronounced in SST → PC synapses (0.53 ± 0.01, n = 53; p < 10−6, t-test). These findings demonstrate that our methodology, which relies on the local stimulation of PV and SST interneurons in the hippocampal CA1 region using optogenetics and different aperture sizes of the objective, leads to the activation of two distinct types of inhibitory inputs to CA1 PCs, as evidenced by their distinct IPSC kinetics and short-term plasticity.
Unlike the cerebral cortex, in the CA1 region of the hippocampus a relatively small number of somatostatin-expressing interneurons also express parvalbumin26. Our experiments revealed that the rise time of IPSCs is the most reliable parameter for distinguishing between SST and PV inputs to CA1 PCs. We consistently observed that IPSCs in SST → PC synapses had slower rise times, always exceeding 3.2 ms, whereas those in PV → PC synapses displayed a significantly faster onset rate (Fig. 1G). Consequently, we will confidently classify all synaptic recordings in SST-ChR2 slices as originating from dendrite targeting O-LM and bistratified neurons in subsequent studies. In contrast, we will employ a complementary approach in experiments conducted on PV-ChR2 slices, where only IPSCs with rise times shorter than 3 ms will be classified as arising from PV → PC synapses formed by presynaptic axo-axonic cells or PV + basket cells. This approach will allow for the investigation of long-term synaptic plasticity in two fundamentally different and non-overlapping types of inhibitory inputs to hippocampal CA1 PCs.
The activation of NMDA receptors triggers excitatory long-term depression and concurrent input-specific GABAergic plasticity in CA1 PCs
To investigate the co-expression of excitatory and GABAergic plasticity, we used a patch-clamp electrode solution with low physiological chloride concentration, as detailed in the Methodology section. The average reversal potential for the GABAA receptors at PV → CA1 PC synapses was determined as − 70.3 ± 0.62 mV (n = 92). Therefore, by maintaining the CA1 pyramidal cells at a potential of − 60 mV, we were able to record measurable IPSCs, while setting the potential at − 70 mV allowed for the recording of clean EPSCs. Moreover, the amplitude of recorded IPSCs did not correlate with the GABA reversal potential in individual cells (p = 0.76 in SST → PC synapses and p = 0.25 in PV → PC input). In order to simultaneously induce excitatory and inhibitory plasticity, we administered NMDA (20 μM) for 2 min and 30 s (2′30′′) and examined its effects on three inputs to CA1 pyramidal cells, namely, inhibitory SST → PC or PV → PC (stimulated optogenetically) and excitatory CA3 → CA1 (stimulation of Schaffer collaterals with an electrode) (Fig. 2A,E). During and immediately after NMDA infusion, a depression of IPSC (Fig. 2B,F) and EPSC (Fig. 2C,G) amplitudes was observed, likely originating presynaptically27. Additionally, as expected, the activation of NMDARs during NMDA administration transiently decreased the input resistance of the cell and the holding current required to maintain the neuron at − 60mV (Fig. 2D,H).

NMDA application for 2 min 30 s (2′30′′) induces concurrent excitatory LTD in CA3 → CA1 projection and iLTP in SST → PC input. (A–D) Schematic representation of the experimental design (A) and exemplary concurrent recordings of plasticity at inhibitory SST (B) and excitatory CA3 input (C) to CA1 pyramidal cell before and after 20 μM NMDA infusion for 2′30’’. The stability of the recording was assessed by monitoring the series resistance, input resistance, and holding current required to maintain the postsynaptic PC at − 60mV (D) Activation of NMDA receptors during plasticity induction caused transient changes in holding current and input resistance (D). Traces show SST → PC IPSC (B) and CA3 → CA1 EPSC (C) monitored at − 60 mV and − 70 mV, respectively. Scale bars: horizontal − 10 ms, vertical – 20 pA. The bars on the time courses represent the time intervals over which the traces were averaged before (1) and after (2) plasticity induction. (E–H) The experimental design (E) and exemplary recordings of the plasticity at inhibitory PV (F) and excitatory CA3 input (G) to CA1 pyramidal cell before and after 20 μM NMDA infusion for 2′30′′. The stability of the recording was assessed by monitoring the series resistance, input resistance, and holding current required to maintain the postsynaptic PC at − 60mV (H). (I) Summary of plasticity at SST → PC, PV → PC, and CA3 → CA1 synapses after NMDA infusion for 2′30′′. The magnitude of plasticity at 26-30min from induction, SST: 116.3 ± 4.2%, n = 10; PV: 98.9 ± 4.7%, n = 15; CA3 → CA1: 69.0 ± 5.4, n = 25. CA3 → CA1 data were obtained during the measurements in SST → PC and PV → PC groups. Data are presented as the mean ± SEM. (J) Comparison of IPSC and EPSC amplitudes before and 26–30 min after NMDA infusion; SST p = 0.022; PV p = 0.58, CA3 → CA1 p = 0.0007, paired t-test, *p < 0.05, ***p < 0.0001, ns—non-significant.
Interestingly, 30 min after NMDA administration for 2′30′′ excitatory CA3 → CA1 synapses expressed excitatory long-term depression (eLTD) (average EPSC before: 141.6 ± 19.6 pA, after: 109.0 ± 20.8 pA, n = 25, p = 0.002, paired t-test). Conversely, GABAergic SST → PC input developed stable inhibitory long-term potentiation (iLTP) (average IPSC before: 82.5 ± 18.8 pA, after: 92.5 ± 25.0 pA, n = 10, p = 0.022). In contrast, PV → PC synapses failed to exhibit significant long-term plasticity (before: 203.9 ± 28.3 pA, after: 198.7 ± 27.1 pA, n = 15, p = 0.58) (Fig. 2I–J). These findings suggest that the induction of NMDA-dependent eLTD is accompanied by the induction of input-specific GABAergic plasticity.
Concurrent NMDA-dependent excitatory and GABAergic plasticity at different inhibitory inputs to CA1 pyramidal cells
To investigate the relationship between NMDA receptor-mediated excitatory and GABAergic plasticity, we conducted a series of experiments in which we simultaneously assessed both excitatory and inhibitory synaptic transmission. We induced plasticity by varying the duration of NMDA application. Specifically, infusion durations of 1 min and 15 s (1′15′′), 1 min and 45 s (1′45′′), and 2 min and 30 s (2′30′′) were used. NMDA administration offers a notable advantage over electrical stimulation by allowing straightforward adjustment of transient NMDA receptor stimulation across the entire somatodendritic axis of CA1 pyramidal cells. Through varying the duration of NMDA administration, we anticipate the possibility of observing inhibitory plasticity concomitantly induced with eLTP and eLTD. As we have already shown, NMDA infusion for 2′30′′ induces stable excitatory LTD (Fig. 3A,B). Interestingly, we found that shorter NMDA applications for both 1′45′′ and 1′15′′ resulted in a significant potentiation of CA3 → CA1 excitatory synapses (EPSC amplitude, 1′15′′: before: 134.6 ± 16.4 pA, after: 176.7 ± 23.0 pA, n = 21, p = 0.004; 1′45′′: before: 120.0 ± 11.2 pA, after: 147.6 ± 14.2 pA, n = 22, p = 0.00004; paired t-test; Fig. 3A–C).

Co-occurrence of NMDA-dependent excitatory and input-specific inhibitory plasticity in CA1 PCs. Plasticity induced by NMDA infusion for different durations in excitatory CA3 → CA1 (brown, A–C) and inhibitory SST → PC (green, D–F) or PV → PC (violet, G–I) synapses. (A, D, G) Summary of plasticity induced by NMDA administration for 1 min 15 s, 1 min 45 s, and 2 min 30 s. The gray areas represent different durations of NMDA administration. The data corresponding to 2′30'' NMDA treatment were previously depicted in Fig. 2I. (B, E, H) Magnitude of plasticity induced by NMDA infusion for different durations (1 min 15 s, 1 min 45 s, 2 min 30 s) in excitatory (B) and inhibitory (E, H) synapses. Short lines above single bars indicate the significance of plasticity induction assessed by comparing the amplitude of synaptic currents before versus 26–30 min after NMDA infusion (paired t-test). The significant difference between analysed groups is marked with long lines between bars (one-way ANOVA with Tukey post hoc test; (B) 1′15′′: 138.5 ± 15.2%, 1′45′′: 121.2 ± 5.3%, 2′30′′: 69.0 ± 5.4%, F(2,86) = 17.2, p = 0.0009; (E) 1′15′′: 114.8 ± 5.6%, 1′45′′: 112.7 ± 4.7%, 2′30′′: 116.3 ± 4.2%, F(2,42) = 0.13, p = 0.88; (H) 1′15′′: 129.1 ± 9.4%, 1′45′′: 120.2 ± 8.2%, 2′30′′: 98.2 ± 4.3%, F(2,45) = 6.58, p = 0.037). Numbers in the bars denote the number of recordings. The data presented in (H) were acquired during the recordings (B) and (E). (C, F, I) Plasticity at inhibitory SST → PC (C) or PV → PC (F) and excitatory CA3 → CA1 synapses (I) plotted against the duration of NMDA infusion. The black lines indicate the non-linear fit of the data. In contrast to iLTP in SST → PC input, prolonged administration of NMDA for more than 2 min failed to elicit LTP in inhibitory PV and excitatory CA3 inputs to CA1 pyramidal cells. Data are presented as mean ± SEM; *p < 0.05, **p < 0.01, ns—non-significant.
Concurrently, alongside EPSC measurements in the CA3 → CA1 projection, we conducted IPSC recordings by stimulating SST or PV input to the CA1 PC. We found that stable iLTP was consistently induced in SST → PC synapses across all three NMDA administration durations (IPSC amplitude, 1′15′′: before: 69.4 ± 9.3 pA, after: 83.1 ± 16.3 pA, n = 13, p = 0.042; 1′45′′: before: 74.8 ± 7.6 pA, after: 79.3 ± 7.5 pA, n = 22, p = 0.005; 2′30′′: before: 82.5 ± 18.8 pA, after: 92.5 ± 25.0 pA, n = 10, p = 0.022; paired t-test; Fig. 3D–F). We additionally examined potential correlations between the magnitude of SST → PC iLTP and the initial IPSC amplitude, kinetics, short-term plasticity, animal age, and reversal potential for synaptic GABAergic currents (Fig. S1). Our data revealed only one significant positive correlation between the decay time constant and the magnitude of plasticity at SST → PC input (Fig. S1D). Overall, our findings indicate that while NMDA receptor activation in CA1 pyramidal cells leads to the induction of excitatory LTP or LTD, dendritic GABAergic synapses originating from SST interneurons undergo concurrent iLTP.
We further investigated the co-expression of plastic changes at excitatory synapses and inhibitory plasticity in the PV input. Notably, both brief NMDA administration (1′15′′) and medium-duration NMDA administration (1′45′′) resulted in stable potentiation of PV → PC synapses (1′15′′ before: 114.7 ± 20.2 pA, after: 142.1 ± 19.2 pA, n = 7, p = 0.038; 1′45′′ before: 145.5 ± 26.8 pA, after: 167.0 ± 29.9 pA n = 26, p = 0.008; paired t-test; Fig. 3G–H). Statistical comparison of IPSP/EPSC amplitudes before and 24–30 min after plasticity induction shows that excitatory CA3 → CA1 LTP is co-expressed with significant GABAergic iLTP at both PV → PC and SST → PC synapses in response to NMDA infusion for 1′15′′ or 1′45′′ (Supplemental Table 1). Moreover, when prolonged NMDA administration for 2′30′′ was employed to induce excitatory LTD, the strength of PV → PC synapses did not change significantly (before: 203.9 ± 28.3 pA, after: 198.7 ± 27.1 pA n = 15, p = 0.58, paired t-test; Fig. 3G–H). Statistical analysis of IPSC/EPSC amplitude before and after NMDA application for 2′30′′ indicates co-expression of significant CA3 → CA1 eLTD, SST → PC iLTP, and a lack of long-term plasticity in PV → PC synapses (Supplemental Table 1). Thus, while excitatory LTP is associated with GABAergic iLTP, the expression of eLTD does not coincide with plastic changes in PV → PC inhibitory input. Finally, we did not detect any significant correlations between the magnitude of PV → PC iLTP and the initial IPSC amplitude, kinetics, short-term plasticity, animal age, or reversal potential of synaptic GABAergic currents (Fig. S2).
Next, we examined how the duration of NMDA receptor stimulation is related to the direction of plasticity in individual synapses. All data collected during these measurements are presented in Fig. 3C,F, and I. In CA3 → CA1 excitatory transmission, extending the duration of NMDA application to 2 min resulted in a switch from long-term potentiation to long-term depression (Fig. 3C). For SST → PC synapses, successful induction of iLTP was observed with NMDA infusion between 1′ and 2′30′′. Shorter or longer administrations did not result in significant inhibitory long-term plasticity (Fig. 3F). Consequently, the presence of NMDA for 3 min resulted in stable excitatory long-term depression and a lack of plasticity in SST → PC synapses. With regard to PV → PC input, significant iLTP was induced with NMDA infusion for 1′15′′ and 1′45′′. Notably, NMDA administration for 2 min or longer did not result in reproducible inhibitory long-term potentiation or depression in this projection (Fig. 3I).
We directly assessed the co-expression of NMDA-induced excitatory LTP/LTD and GABAergic plasticity by plotting the magnitude of GABAergic plasticity in relation to the concurrent glutamatergic plasticity recorded simultaneously from the same postsynaptic CA1 pyramidal cell, irrespective of the duration of NMDA treatment (Fig. 4A,B). A significant positive correlation was observed between the sign of plasticity in PV → PC and excitatory CA3 → CA1 synapses, suggesting that if NMDA treatment induced excitatory LTP, it was statistically more likely that iLTP in the PV → PC projection would also be established (Fig. 4A). However, no correlation was observed in SST → PC synapses, where GABAergic iLTP was typically induced, regardless of the sign of excitatory plasticity (Fig. 4B). These results suggest that there is a correlation between iLTP at the PV → PC projection and the occurrence of long-term excitatory plasticity.

Correlation between the magnitude of long-term plasticity at excitatory and different GABAergic synapses onto CA1 PCs. (A) The magnitude of inhibitory plasticity at PV → PC synapses in response to NMDA infusion positively correlated with the magnitude of simultaneous excitatory plasticity in CA3 → CA1 pathway (n = 92, p = 0.018). The graph presents data collected from measurements conducted during the induction of plasticity by administering NMDA for durations ranging from 30 s to 3 min. (B) The magnitude of inhibitory plasticity at SST → PC synapses in response to NMDA infusion does not correlate with the magnitude of simultaneous excitatory plasticity in CA3 → CA1 pathway (n = 49, p = 0.67). Plots contain data from simultaneous recordings of CA3 → CA1 EPSC and SST → PC IPSC with NMDA infusion for durations ranging from 30 s to 3 min. Plasticity was assessed 26–30 min after NMDA infusion. Plots contain a linear regression curve (solid line), 95% confidence bands (dotted line), and Pearson correlation coefficient r.*p < 0.05, ns—non-significant. (C–D). The ratio of inhibitory plasticity in PV → PC (C) or SST → PC synapses (D) to the magnitude of simultaneously co-expressed excitatory CA3 → CA1 plasticity in the same postsynaptic CA1 PCs. Plasticity was induced by NMDA application for 1 min 15 s, 1 min 45 s, or 2 min 30 s. Significant differences between the analyzed groups are illustrated by long lines between the bars and were determined using a one-way ANOVA with Tukey post hoc test; (C) 1′15′′: 1.01 ± 0.12, 1′45′′: 0.98 ± 0.091, 2′30′′: 1.69 ± 0.16, F(2,40) = 10.14, p = 0.0003, multiple comparisons: 1′15′′ versus 1′45′′ p = 0.99, 1′15′′ versus 2′30′′ p = 0.011, 1′45′′ versus 2′30′′ p = 0.0003; (D) 1′15′′ 1.21 ± 0.18, 1′45′′: 1.14 ± 0.10, 2′30′′: 1.77 ± 0.21, F(2,36) = 4.55, p = 0.017, multiple comparisons: 1′15′′ versus 1′45′′ p = 0.95, 1′15′′ versus 2′30′′ p = 0.054, 1′45′′ versus 2′30′′ p = 0.018). ***p < 0.00, 1*p < 0.05, ns—non-significant.
To further analyze the relationship between inhibitory and excitatory plasticity co-expressed in individual CA1 PCs, we calculated the ratio of inhibitory to excitatory plasticity when both were measured simultaneously. For shorter NMDA applications (1′15′′ and 1′45′′), resulting in CA3 → CA1 eLTP, this ratio remained close to one for both PV → PC and SST → PC projections (Fig. 4C–D). However, the ratio was significantly higher in groups treated with NMDA for 2′30’’, as this protocol consistently induces eLTD, does not change average PV → PC synapse strength, and leads to iLTP in SST → PC projection. In summary, despite the simultaneous co-expression of excitatory and inhibitory plasticity, the balance between synaptic inhibition and excitation is maintained after brief NMDA infusions (for 1′15′′ or 1′45′′). Conversely, longer NMDA administration shifts the E/I balance in favor of synaptic inhibition.
NMDA-dependent GABAergic plasticity is not accompanied by IPSC short-term plasticity or kinetic changes
Our findings show that the long-term inhibitory plasticity due to NMDA receptor activation shows some differences between SST and PV interneuron synaptic connections onto CA1 pyramidal neurons (Figs. 2, 3). To further explore the features of GABAergic plastic changes, we analyzed short-term plasticity using paired-pulse stimulation before and 30 min after NMDA administration. Although the paired-pulse ratio differed in the SST → PC and PV → PC projections (Fig. 1), the induction of long-term plasticity by NMDA administration (for any duration) did not alter this value in either of these projections (PV → PC iLTP, Fig. S3A; SST → PC, Fig. S3B). Interestingly, for the PV → PC projection, the paired pulse ratio was unaffected after NMDA treatment, irrespective of whether it induced long-term plasticity (1′15′′ and 1′45′′ treatments) or not (2′30′′, Fig. S3C), thus supporting the postsynaptic expression mechanism. We also analyzed the time course of IPSC before and after plasticity induction, and no changes were observed in the decay kinetics (Fig. S3D–F) or rise times (not shown).
Co-occurrence of excitatory and inhibitory plasticity depends on MMPs
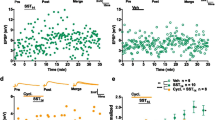
The above data show that the same induction paradigm (transient treatment with NMDA) may give rise to both excitatory and inhibitory plasticity. Thus, it would be interesting to determine whether inhibitory and excitatory plastic changes may share partially common molecular mechanisms. Previous studies from our group and others have demonstrated that both excitatory and inhibitory plasticity may strongly depend on the activity of metalloproteinases, especially MMP-9 and MMP-311,28,29,30,31,32,33. In addition, it would be particularly interesting to determine whether the co-expression of inhibitory and excitatory plasticity can be pharmacologically decoupled. We used SB-3CT (10 μM) to inhibit MMP9, as this protease is known to control the maintenance of excitatory LTP31,32, and its inhibition allowed us to assess the expression of GABAergic plasticity under conditions of eLTP impairment. In another set of experiments, we used UK356618 (2 μM) to inhibit MMP3, a protease that regulates the induction of NMDA-iLTP11,28,34. The use of this inhibitor enabled us to examine plastic changes at excitatory synapses under conditions of impaired GABAergic plasticity. As expected, MMP9 inhibition blocked NMDA-induced eLTP, whereas MMP3 blockade did not affect NMDA-dependent excitatory long-term plasticity (Fig. 5A; Supplemental Table 2). Meanwhile, at PV → PC synapses, the iLTP observed in the control conditions was abolished by inhibition of MMP3 (Fig. 5C), but no statistically significant difference was found in the magnitude of plasticity induced in the control group and the group treated with the MMP9 inhibitor (Fig. 5B). Collectively, these data indicate that the inhibition of MMP9 not only abolished excitatory LTP but also prevented concurrent SST → PC iLTP. Additionally, while MMP3 inhibition did not impact excitatory LTP, it effectively blocked the simultaneous induction of NMDA-iLTP at both SST → PC and PV → PC synapses (Fig. 5F; Supplemental Table 2). Statistical analysis of the IPSP/EPSC amplitudes before and 24–30 min after NMDA administration corroborates these conclusions. The results show that UK3566 prevented the induction of iLTP in both analyzed inhibitory synapses, while SB-3CT impaired SST → PC iLTP and showed strong trend towards normal PV → PC iLTP (p = 0.067; Supplemental Table 2).

Inhibition of extracellular proteolysis decouples the co-expression of excitatory and inhibitory plasticity. (A) MMP2/9 inhibitor (SB3CT 10μM), but not MMP3 inhibitor (UK356618 2μM), impaired CA3 → CA1 PC excitatory LTP induced by NMDA applied for 1 min 45 s. (B) MMP3 inhibitor, but not MMP2/9 inhibitor, impaired PV → PC iLTP induced by NMDA applied for 1 min 45 s. (C) Both, MMP3 inhibitor and MMP2/9 inhibitor impaired SST → PC iLTP induced by NMDA applied for 1 min 45 s. (D) Neither MMP3 inhibitor nor MMP2/9 inhibitor affected CA3 → CA1 excitatory LTD induced by NMDA applied for 2 min 30 s. (E) MMP3 inhibitor, but not MMP2/9 inhibitor, impaired SST → PC iLTP induced by NMDA applied for 2 min 30 s. (F) Summary of inhibitory and excitatory plasticity induced by NMDA infusion for 1 min45 s (one-way ANOVA with Tukey’s post hoc test; (SST → PC) Ctrl: 125.7 ± 8.7%, MMP3 inh: 104.2 ± 2.7%, MMP2/9 inh: 99.2 ± 4.7%, F(2,19) = 5.75, p = 0.011; (PV → PC) Ctrl: 115.4 ± 4.9%, MMP3 inh: 92.9 ± 6.4%, MMP2/9 inh: 110.9 ± 8.2%, F(2,22) = 3.62, p = 0.044; (CA3 → CA1) Ctrl: 123.0 ± 8.2%, MMP3 inh: 116.4 ± 8.0%, MMP2/9 inh: 94.3 ± 8.3%, F(2,32) = 5.14, p = 0.046). (G) Summary of inhibitory and excitatory plasticity induced by NMDA infusion for 2 min 30 s (one-way ANOVA with Tukey post hoc test; (SST → PC) Ctrl: 120.0 ± 5.3%, MMP3 inh: 90.5 ± 2.4%, MMP2/9 inh: 112.6 ± 2.4%, F(2,17) = 5.83, p = 0.012; (CA3 → CA1) Ctrl: 80.1 ± 10.4%, MMP3 inh: 85.1 ± 8.9%, MMP2/9 inh: 70.8 ± 10.3%, F(2,32) = 5.14, p = 0.60). Data are presented as the mean ± SEM; *p < 0.05, **p < 0.01, ns—non-significant.
To more precisely characterize the co-occurrence of plasticity at excitatory and inhibitory synapses, we employed NMDA stimulation for 2′30′′, which induced excitatory LTD and SST → PC iLTP under control conditions. Our results indicate that neither MMP3 nor MMP9 inhibition prevented the induction of excitatory LTD (Fig. 5D; Supplemental Table 2). However, SST → PC iLTP, which was accompanied by excitatory LTD (induced by NMDA stimulation for 2′30''), was abolished by MMP3 inhibition and not by blockade of MMP9 (Fig. 5E,G; Supplemental Table 2). We further compared the ratio of inhibitory to excitatory plasticity in individual experiments, with and without MMP inhibitors (Fig. S4A–C). Although we did not observe significant changes, there was a clear trend towards a reduction in this ratio when plasticity was induced with MMP3 inhibition (Fig. S4B). In conclusion, MMP3 protease participates in the molecular mechanisms of GABAergic plasticity in both analyzed inhibitory synapses. However, MMP9 protease regulates only SST → PC iLTP, interfering at the same time with the induction of excitatory LTP but not eLTD.
Discussion
In the present study, we characterized glutamatergic and GABAergic plasticity induced by varying NMDA administration durations in the hippocampus. We chose classic CA3 → CA1 excitatory and two canonical inhibitory projections, PV → PC and SST → PC, which are known to innervate distinct parts of the principal neuron. The somata of SST interneurons are typically situated in the CA1 stratum oriens and project to the dendrites of principal neurons in both the stratum lacunosum moleculare (O-LM interneurons) and stratum radiatum (bistratified interneurons)35. In contrast, PV-expressing interneurons in the CA1 stratum pyramidale include basket cells that form synapses on the cell bodies of PCs, and axo-axonic interneurons that synapse on the axon initial segment26. Considering these differences, it is not surprising that distinct IPSC properties were observed between PV and SST projections onto excitatory pyramidal neurons in the CA1. The PV → PC input exhibited larger IPSC amplitudes, faster onset and decay kinetics, and more pronounced paired-pulse or burst-induced depression. These differences in the time course of IPSC in PV → PC and SST → PC were expected as a consequence of larger electrotonic filtering in the latter projection25. Moreover, we observed a clear input specificity of GABAergic plasticity in CA1 pyramidal neurons, requiring distinct timing of NMDA administration. NMDA administration for 1′–2′30′′ induced stable iLTP in SST → PC synapses, while in PV → PC input, only a brief NMDA infusion for 1′–1′45′′ resulted in iLTP, whereas longer NMDA applications did not result in reproducible potentiation of IPSCs. Thus, while for considered interneurons there were specific time windows for iLTP induction, beyond which no plasticity was observed, in the excitatory pathway, a reversal of eLTP to eLTD took place for NMDA applications longer than 2′30′′ (Fig. 3C). Moreover, SST → PC synapses can express iLTP concurrently with either depression or potentiation of excitatory transmission, whereas PV → PC synapses exhibit iLTP that co-occurs with eLTP but not with eLTD. The co-occurrence of excitatory and inhibitory plasticities also shows a distinct sensitivity to metalloproteinase blockers. Specifically, MMP3 affected all forms of iLTP, whereas MMP9 influenced eLTP and co-expressed iLTP in SST → PC, but not PV → PC input. These findings suggest the existence of distinct long-term plasticities that co-occur in a single neuron upon NMDAR activation, forming a unique neuronal plasticitome of CA1 PCs36.
GABAergic plasticity in the hippocampus is characterized by input-specificity
Our results align with previous findings of postsynaptically expressed input-specific GABAergic plasticity in various brain regions. For instance, within the medial prefrontal cortex, only synapses targeting layer 2/3 PCs that originate from SST interneurons exhibit NMDA-iLTP in contrast to inputs from PV or VIP interneurons9. In a separate study, repetitive firing from a depolarized membrane potential induced iLTD in PV synapses targeting layer 5 PCs in the rat visual cortex37. Additionally, theta burst stimulation of inhibitory inputs has been shown to trigger T-type calcium channel-dependent homosynaptic iLTD and iLTP in hippocampal CA1 neurons innervated by PV and SST interneurons, respectively25. Moreover, another study focusing on CA1 PCs suggested that only PV and not SST input may express heterosynaptic NMDA-iLTP38. In contrast, our results suggest that NMDA receptor activation alters the strength of both the dendritic (SST) and somatic (PV) CA1 PC synapses. This discrepancy may be attributed to experimental variations, particularly the absence of AMPA receptor blockade in our study. Additionally, the differences among studies suggest that iLTP in CA1 PV → PC synapses can be expressed upon AMPA receptor inhibition, most likely involving dendritic voltage-gated L-type calcium conductance37. By contrast, SST → PC NMDA-iLTP may require AMPAergic activity and neuroligin-dependent transsynaptic adhesion39. Finally, the discrepancies between different studies may be reconciled by assuming that the sign of heterosynaptic GABAergic plasticity depends on the distance from excitatory synapses that undergo eLTP or eLTD7. In summary, the co-expression of glutamatergic and GABAergic plasticity is likely to fine-tune the strength of different inhibitory synapses throughout the entire somatodendritic axis of CA1 PCs, involving distinct molecular mechanisms.
The co-occurrence of excitatory and input specific inhibitory plasticity—implications for network stability and homeostasis
Recent advances in experimental techniques, including multielectrode stimulation5 and optogenetics9,25, have substantially advanced our understanding of GABAergic plasticity. Notably, most forms of postsynaptic GABAergic long-term plasticity exhibit a heterosynaptic nature, with induction occurring beyond inhibitory synapses and often within proximate excitatory synapses7,36. Calcium influx and signaling play pivotal roles in regulating this intricate process, involving CaMKII activation during iLTP40 and calcineurin during iLTD induction12,41. Similar regulatory rules have been reported in excitatory synapses, where CaMKII activation leads to eLTP42, whereas calcineurin drives eLTD43, suggesting shared mechanisms between inhibitory and excitatory plasticity.
The challenges imposed by the heterosynaptic properties of NMDA-dependent inhibitory plasticity significantly complicate the study of this phenomenon. Many forms of inhibitory plasticity have been investigated under conditions that deviate from physiological settings. Common practices include routine blockade of synaptic AMPA receptors or manipulation of chloride ion concentrations, which renders IPSCs excitatory10,25,38. These experimental approaches constrain the concurrent expression of inhibitory and excitatory plasticity, thereby hindering their simultaneous measurement. To address these challenges, we have strategically chosen not to block AMPA receptors and to maintain a physiological chloride ion gradient during whole-cell recordings. Additionally, it is important to note that besides NMDA-dependent plasticity, homosynaptic induction mechanisms of GABAergic plasticity have also been reported, which may require T-type calcium channels25 or GluD1 receptors44.
The intricate relationship between excitatory and inhibitory synapses is crucial for proper functioning of neuronal circuits. The simultaneous induction of excitatory CA3 → CA1 LTP and iLTP in the dendritic and somatic inhibitory synapses (Fig. 3D,G) suggests a mesoscale intersynaptic mechanism that balances excitation and inhibition. As specific excitatory pathways strengthen with learning or experience, the associated inhibitory synapses symmetrically adapt to iLTP to ensure overall network stability5. This adaptability is a key feature of a balanced model of neuronal integration, in which inhibitory synapses continuously adjust to match excitatory input45,46. Our findings also imply that the properties of networks stabilized by inhibition47 are maintained in the presence of excitatory synaptic plasticity through the co-expression of GABAergic plasticity within the postsynaptic principal cell.
In contrast, prolonged NMDA activation weakened excitatory CA3 → CA1 synapses, potentiated dendritic inhibition, and left somatic inhibitory inputs intact (Fig. 2I). This situation resembles the effects observed in the barrel cortex, where whisker deprivation triggered concurrent LTD of excitatory inputs onto layer 2/3 pyramidal cells and LTP of inhibitory drive onto the same neuronal population48. This symmetry-breaking mechanism may redistribute inhibition along the somatodendritic axis in response to NMDAR activation, potentially dampening network activity, and altering information processing. Additionally, such asymmetric co-expression of plasticity likely contributes to pattern separation by strengthening inhibitory control over specific excitatory pathways49,50. Indeed, heterosynaptic plasticity plays a role in refining the tuning of neural circuits by increasing their selectivity in response to specific input patterns16. Overall, our observations underscore the co-expression patterns between excitatory and inhibitory plasticity, suggesting that tightly regulated temporal dynamics between excitatory and inhibitory plasticity are crucial for sustaining network stability and altering dendritic integration.
The molecular coupling between excitatory and inhibitory plasticity
Sophisticated regulatory networks govern homosynaptic and heterosynaptic plastic changes, with extracellular matrix metalloproteinases acting as key modulators33,51. The decision to focus on MMP3 and MMP9 in this study was motivated by their roles in the later stages of plasticity mechanisms52, which presents an opportunity to pharmacologically uncouple the co-expression of excitatory and inhibitory long-term plasticity. Our results (Fig. 5) are consistent with prior research, emphasizing the crucial role of MMP3 in extracellular proteolytic processes related to inhibitory synaptic plasticity11. Notably, the specific inhibition of MMP3, which suppresses iLTP at both SST → PC and PV → PC synapses while leaving eLTP unaffected, is likely to indicate an important experimental avenue for understanding the behavioral implications of inhibitory plasticity in diverse forms of learning in both health and disease22.
Conversely, MMP9 protease was found to be crucial for the induction of NMDA-dependent eLTP but not eLTD. Additionally, while MMP9 inhibition did not impede PV → PC iLTP, it selectively hindered SST → PC iLTP, notably when accompanied by excitatory LTP but not eLTD. This indicates that NMDA-dependent PV → PC iLTP is resilient to pharmacological impairment of co-occurring eLTP. Moreover, our results suggest that, unlike PV → PC synapses, SST → PC input can exhibit a symmetric or asymmetric direction of plasticity in relation to excitatory plasticity, but only symmetric iLTP co-occurring with eLTP requires MMP9 activity. This effect implies the existence of distinct co-expression mechanisms of excitatory and specific inhibitory plasticity along the somatodendritic axis of the CA1 PCs. These putative mechanisms may depend on the synapse-specific functioning of brain integrin-dependent adhesion41,53. Additionally, our findings align with previous studies, which demonstrated that MMP9 inhibition prevents the potentiation of inhibitory events by cholinergic agonists54.
Conclusions
In conclusion, our study emphasizes the detailed balance between excitatory and inhibitory plasticity, revealing a tightly regulated process that co-tunes both types of synaptic transmission at the single-neuron level to maintain network stability. Future research should focus on elucidating the downstream mechanisms governing intermediate-range heterosynaptic signaling17 and exploring the specific subcategories of interneurons involved in these processes55. Additionally, further investigation is required to understand the molecular mechanisms by which postsynaptic NMDA receptor activation drives distinct synapse-specific mechanisms. These findings will be particularly informative in the context of the relationship between heterosynaptic plasticity and local neuronal computations. Moreover, the co-occurrence of excitatory plasticity with diverse forms of GABAergic plasticity in inhibitory synapses formed at other interneurons56 warrants similar investigations. Finally, the potential implications of deregulated inhibitory plasticity in neurological disorders characterized by excitation-inhibition imbalances, such as epilepsy and schizophrenia, should be considered.
Data availability
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
References
Choi, J. H. et al. Interregional synaptic maps among engram cells underlie memory formation. Science (New York, NY) 360(6387), 430–435. https://doi.org/10.1126/science.aas9204 (2018).
Yap, E.-L. et al. Bidirectional perisomatic inhibitory plasticity of a Fos neuronal network. Nature 590(7844), 115–121. https://doi.org/10.1038/s41586-020-3031-0 (2020).
Han, D. H., Park, P., Choi, D. I., Bliss, T. V. P. & Kaang, B. K. The essence of the engram: Cellular or synaptic?. Semin. Cell Dev. Biol 125, 122–135. https://doi.org/10.1016/j.semcdb.2021.05.033 (2022).
D’amour James, A. & Froemke Robert, C. Inhibitory and excitatory spike-timing-dependent plasticity in the auditory cortex. Neuron 86(2), 514–528. https://doi.org/10.1016/j.neuron.2015.03.014 (2015).
Field, R. E. et al. Heterosynaptic plasticity determines the set point for cortical excitatory-inhibitory balance. Neuron 106(5), 842. https://doi.org/10.1016/j.neuron.2020.03.002 (2020).
Ahumada, J., de Sevilla, D. F., Couve, A., Buño, W. & Fuenzalida, M. Long-term depression of inhibitory synaptic transmission induced by spike-timing dependent plasticity requires coactivation of endocannabinoid and muscarinic receptors. Hippocampus 23(12), 1439–1452. https://doi.org/10.1002/hipo.22196 (2013).
Ravasenga, T. et al. Spatial regulation of coordinated excitatory and inhibitory synaptic plasticity at dendritic synapses. Cell Rep. 38(6), 110347. https://doi.org/10.1016/j.celrep.2022.110347 (2022).
Barberis, A. Postsynaptic plasticity of GABAergic synapses. Neuropharmacology 169, 107643. https://doi.org/10.1016/J.Neuropharm.2019.05.020 (2020).
Chiu, C. Q. et al. Input-specific NMDAR-dependent potentiation of dendritic GABAergic inhibition. Neuron 97(2), 368. https://doi.org/10.1016/j.neuron.2017.12.032 (2018).
Marsden, K. C., Beattie, J. B., Friedenthal, J. & Carroll, R. C. NMDA receptor activation potentiates inhibitory transmission through GABA receptor-associated protein-dependent exocytosis of GABAA receptors. J. Neurosci. 27(52), 14326–14337. https://doi.org/10.1523/jneurosci.4433-07.2007 (2007).
Wiera, G. et al. Long-term plasticity of inhibitory synapses in the hippocampus and spatial learning depends on matrix metalloproteinase 3. Cell. Mol. Life Sci. 78(5), 2279–2298. https://doi.org/10.1007/s00018-020-03640-6 (2021).
Muir, J. et al. NMDA receptors regulate GABA A receptor lateral mobility and clustering at inhibitory synapses through serine 327 on the γ2 subunit. Proc. Natl. Acad. Sci. 107(38), 16679–16684. https://doi.org/10.1073/pnas.1000589107 (2010).
Levi, S. et al. Homeostatic regulation of synaptic GlyR numbers driven by lateral diffusion. Neuron 59(2), 261–273. https://doi.org/10.1016/j.neuron.2008.05.030 (2008).
Grover, L. M. & Yan, C. Blockade of GABA receptors facilitates induction of NMDA receptor-independent long-term potentiation. J. Neurophysiol. 81(6), 2814–2822. https://doi.org/10.1152/jn.1999.81.6.2814 (1999).
Chapman, C. A., Nuwer, J. L. & Jacob, T. C. The Yin and Yang of GABAergic and glutamatergic synaptic plasticity: Opposites in balance by crosstalking mechanisms. Front. Synaptic Neurosci. https://doi.org/10.3389/Fnsyn.2022.911020 (2022).
Wu, Y. K., Miehl, C. & Gjorgjieva, J. Regulation of circuit organization and function through inhibitory synaptic plasticity. Trends Neurosci. 45(12), 884–898. https://doi.org/10.1016/j.tins.2022.10.006 (2022).
Chater, T. E. & Goda, Y. My neighbour hetero—deconstructing the mechanisms underlying heterosynaptic plasticity. Curr. Opin. Neurobiol. 67, 106–114. https://doi.org/10.1016/j.conb.2020.10.007 (2021).
Zhai, S., Ark, E. D., Parra-Bueno, P. & Yasuda, R. Long-distance integration of nuclear ERK signaling triggered by activation of a few dendritic spines. Science (New York, NY) 342(6162), 1107–1111. https://doi.org/10.1126/science.1245622 (2013).
Topolnik, L. & Tamboli, S. The role of inhibitory circuits in hippocampal memory processing. Nat. Rev. Neurosci. https://doi.org/10.1038/s41583-022-00599-0 (2022).
Chiu, C. Q. et al. Compartmentalization of GABAergic inhibition by dendritic spines. Science (New York, NY) 340(6133), 759–762. https://doi.org/10.1126/science.1234274 (2013).
Znamenskiy, P. et al. Functional specificity of recurrent inhibition in visual cortex. Neuron https://doi.org/10.1016/j.neuron.2023.12.013 (2024).
Barron, H. C., Vogels, T. P., Behrens, T. E. & Ramaswami, M. Inhibitory engrams in perception and memory. Proc. Natl. Acad. Sci. USA 114(26), 6666–6674. https://doi.org/10.1073/pnas.1701812114 (2017).
Valero, M., Zutshi, I., Yoon, E. & Buzsáki, G. Probing subthreshold dynamics of hippocampal neurons by pulsed optogenetics. Science (New York, NY) 375(6580), 570. https://doi.org/10.1126/science.abm1891 (2022).
Ting, J. T. et al. Preparation of acute brain slices using an optimized N-Methyl-D-glucamine protective recovery method. JOVE 132, 53825. https://doi.org/10.3791/53825 (2018).
Udakis, M., Pedrosa, V., Chamberlain, S. E. L., Clopath, C. & Mellor, J. R. Interneuron-specific plasticity at parvalbumin and somatostatin inhibitory synapses onto CA1 pyramidal neurons shapes hippocampal output. Nat. Commun. 11(1), 4395. https://doi.org/10.1038/s41467-020-18074-8 (2020).
Pelkey, K. A. et al. Hippocampal Gabaergic Inhibitory Interneurons. Physiol. Rev. 97(4), 1619–1747. https://doi.org/10.1152/physrev.00007.2017 (2017).
Banerjee, A., Larsen, R. S., Philpot, B. D. & Paulsen, O. Roles of presynaptic NMDA receptors in neurotransmission and plasticity. Trends Neurosci. 39(1), 26–39. https://doi.org/10.1016/j.tins.2015.11.001 (2016).
Brzdak, P., Wlodarczyk, J., Mozrzymas, J. W. & Wójtowicz, T. Matrix metalloprotease 3 activity supports hippocampal EPSP-to-spike plasticity following patterned neuronal activity via the regulation of NMDAR function and calcium flux. Mol. Neurobiol. 54(1), 804–816. https://doi.org/10.1007/s12035-016-9970-7 (2017).
Stefaniuk, M. et al. Matrix metalloproteinase-9 and synaptic plasticity in the central amygdala in control of alcohol-seeking behavior. Biol. Psychiatr. 81(11), 907–917. https://doi.org/10.1016/j.biopsych.2016.12.026 (2017).
Bijata, M. et al. Synaptic remodeling depends on signaling between serotonin receptors and the extracellular matrix. Cell Rep. 19(9), 1767–1782. https://doi.org/10.1016/j.celrep.2017.05.023 (2017).
Nagy, V. et al. Matrix metalloproteinase-9 is required for hippocampal late-phase long-term potentiation and memory. J. Neurosci.: Off. J. Soc. Neurosci. 26(7), 1923–1934. https://doi.org/10.1523/JNEUROSCI.4359-05.2006 (2006).
Lebida, K. & Mozrzymas, J. W. Spike timing-dependent plasticity in the mouse barrel cortex is strongly modulated by sensory learning and depends on activity of matrix metalloproteinase 9. Mol. Neurobiol. 54(9), 6723–6736. https://doi.org/10.1007/s12035-016-0174-y (2017).
Wiera, G. & Mozrzymas, J. W. Extracellular metalloproteinases in the plasticity of excitatory and inhibitory synapses. Cells 10(8), 2055. https://doi.org/10.3390/Cells10082055 (2021).
Wiera, G. et al. Mechanisms of NMDA receptor- and voltage-gated L-type calcium channel-dependent hippocampal LTP critically rely on proteolysis that is mediated by distinct metalloproteinases. J. Neurosci.: Off. J. Soc. Neurosci. 37(5), 1240–1256. https://doi.org/10.1523/Jneurosci.2170-16.2016 (2017).
Magnin, E. et al. Input-specific synaptic location and function of the α5 GABAA receptor subunit in the mouse CA1 hippocampal neurons. J. Neurosci. 39(5), 788–801. https://doi.org/10.1523/jneurosci.0567-18.2018 (2019).
McFarlan, A. R. et al. The plasticitome of cortical interneurons. Nat. Rev. Neurosci. 24(2), 80–97. https://doi.org/10.1038/s41583-022-00663-9 (2022).
Kurotani, T., Yamada, K., Yoshimura, Y., Crair, M. C. & Komatsu, Y. State-dependent bidirectional modification of somatic inhibition in neocortical pyramidal cells. Neuron 57(6), 905–916. https://doi.org/10.1016/j.neuron.2008.01.030 (2008).
Wu, K., Han, W. & Lu, W. Sleep and wake cycles dynamically modulate hippocampal inhibitory synaptic plasticity. PLoS Biol. 20(11), e3001812. https://doi.org/10.1371/journal.pbio.3001812 (2022).
Horn, M. E. & Nicoll, R. A. Somatostatin and parvalbumin inhibitory synapses onto hippocampal pyramidal neurons are regulated by distinct mechanisms. Proc. Natl. Acad. Sci. USA 115(3), 589–594. https://doi.org/10.1073/pnas.1719523115 (2018).
Petrini, E. M. & Barberis, A. Diffusion dynamics of synaptic molecules during inhibitory postsynaptic plasticity. Front. Cellul. Neurosci. 8, 300. https://doi.org/10.3389/fncel.2014.00300 (2014).
Wiera, G. et al. Integrins bidirectionally regulate the efficacy of inhibitory synaptic transmission and control GABAergic plasticity. J. Neurosci. 42(30), 5830–5842. https://doi.org/10.1523/Jneurosci.1458-21.2022 (2022).
Kennedy, M. B. Synaptic signaling in learning and memory. Cold Spring Harbor Perspect. Biol. 8(2), 1–16 (2014).
Lee, H. K., Kameyama, K., Huganir, R. L. & Bear, M. F. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21(5), 1151–1162. https://doi.org/10.1016/S0896-6273(00)80632-7 (1998).
Piot, L. & Heroven, C. GluD1 binds GABA and controls inhibitory plasticity. Science 382(6677), 1389–1394. https://doi.org/10.1126/science.adf3406 (2023).
Hennequin, G., Agnes, E. J. & Vogels, T. P. Inhibitory plasticity: Balance, control, and codependence. Annu. Rev. Neurosci. 40(40), 557–579. https://doi.org/10.1146/annurev-neuro-072116-031005 (2017).
Bhatia, A., Moza, S. & Bhalla, U. S. Precise excitation-inhibition balance controls gain and timing in the hippocampus. eLife 8, e43415. https://doi.org/10.7554/eLife.43415 (2019).
Sadeh, S. & Clopath, C. Inhibitory stabilization and cortical computation. Nat. Rev. Neurosci. 22(1), 21–37. https://doi.org/10.1038/s41583-020-00390-z (2021).
House, D. R. C., Elstrott, J., Koh, E., Chung, J. & Feldman, D. E. Parallel regulation of feedforward inhibition and excitation during Whisker map plasticity. Neuron 72(5), 819–831. https://doi.org/10.1016/j.neuron.2011.09.008 (2011).
Barron, H. C. Neural inhibition for continual learning and memory. Current opinion in neurobiology 67, 85–94. https://doi.org/10.1016/j.conb.2020.09.007 (2021).
Koolschijn, R. S. et al. The hippocampus and neocortical inhibitory engrams protect against memory interference. Neuron 101(3), 528. https://doi.org/10.1016/j.neuron.2018.11.042 (2019).
Beroun, A. et al. MMPs in learning and memory and neuropsychiatric disorders. Cell. Mol. Life Sci. 76(16), 3207–3228. https://doi.org/10.1007/s00018-019-03180-8 (2019).
Kaczmarek, L. MMP-9 in control of synaptic plasticity: A subjective account. Opera Med. et Physiol. 2(2), 103–111. https://doi.org/10.20388/omp2016.002.0027 (2016).
Favuzzi, E. et al. Distinct molecular programs regulate synapse specificity in cortical inhibitory circuits. Science 363(6425), 413. https://doi.org/10.1126/science.aau8977 (2019).
Salamian, A. et al. Inhibition of matrix metalloproteinase 9 activity promotes synaptogenesis in the hippocampus. Cereb. Cortex 31(8), 3804–3819. https://doi.org/10.1093/cercor/bhab050 (2021).
Müller, C. & Remy, S. Dendritic inhibition mediated by O-LM and bistratified interneurons in the hippocampus. Front. Synaptic Neurosci. 6, 23. https://doi.org/10.3389/fnsyn.2014.00023 (2014).
Brzdąk, P., Lebida, K., Wyroślak, M. & Mozrzymas, J. W. GABAergic synapses onto SST and PV interneurons in the CA1 hippocampal region show cell-specific and integrin-dependent plasticity. Sci. Rep. 13(1), 5079. https://doi.org/10.1038/s41598-023-31882-4 (2023).
Acknowledgements
This work was supported by funding from the National Science Centre (Poland) grant SONATA 2017/26/D/NZ4/00450 (to G.W.); J.W.M. was supported by grant OPUS 2021/43/B/NZ4/01675. We are grateful to Drs. Katarzyna Leida and Patrycja Brzdak for their invaluable contributions in critically reviewing the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization: G.W and J.W.M. Investigation and data analysis: G.W., J.J., A.M.L., and J.W.M. Supervision: G.W. and J.W.M. Funding acquisition: G.W. and J.W.M. Writing—original draft: G.W. and J.W.M. Writing – review and editing: G.W., J.J., A.M.L., and J.W.M.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wiera, G., Jabłońska, J., Lech, A.M. et al. Input specificity of NMDA-dependent GABAergic plasticity in the hippocampus. Sci Rep 14, 20463 (2024). https://doi.org/10.1038/s41598-024-70278-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-70278-w
- Springer Nature Limited