
Abstract
Obesity poses a public health threat, reaching epidemic proportions. Our hypothesis suggests that some of this epidemic stems from its transmission across generations via paternal epigenetic mechanisms. To investigate this possibility, we focused on examining the paternal transmission of CpG methylation. First-generation male Wistar rats were fed either a high-fat diet (HF) or chow and were mated with females fed chow. We collected sperm from these males. The resulting offspring were raised on a chow diet until day 35, after which they underwent a dietary challenge. Diet-induced obese (DIO) male rats passed on the obesogenic trait to both male and female offspring. We observed significant hypermethylation of the Pomc promoter in the sperm of HF-treated males and in the hypothalamic arcuate nucleus (Arc) of their offspring at weaning. However, these differences in Arc methylation decreased later in life. This hypermethylation is correlated with increased expression of DNMT3B. Further investigating genes in the Arc that might be involved in obesogenic transgenerational transmission, using reduced representation bisulfite sequencing (RRBS) we identified 77 differentially methylated regions (DMRs), highlighting pathways associated with neuronal development. These findings support paternal CpG methylation as a mechanism for transmitting obesogenic traits across generations.
Similar content being viewed by others


Introduction
Energy intake and balance are controlled on a regular basis by neural circuits in the hypothalamus. Neurons in the hypothalamic arcuate nucleus (Arc) containing the peptides NPY/AgRP signal to the brain to increase feeding, and POMC/CART-containing neurons promote satiety. These neurons monitor signals from the periphery (e.g., leptin, insulin and ghrelin levels), and they are regulated by higher-level neural circuits1,2. Research suggests that dysregulated feeding, resulting in overweight and obesity, is mediated by alterations in these Arc melanocortin neuropeptide circuits3,4. The melanocortin system in the hypothalamus undergoes a prolonged maturation process, during which it is susceptible to external influences5. Furthermore, epigenetic mechanisms have been implicated in dysregulated POMC signaling6,7 and in "programming" susceptibility to obesity in offspring8.
We and others have reported in animal models of obesity (raising rats in small litters or on a high-fat diet (HF)) epigenetic modifications, including DNA hypermethylation of the promotor of the Pomc gene in the Arc9,10,11,12. Furthermore, our studies with Wistar strain rats revealed that maternal, F0, HF-feeding-induced overeating and hypermethylation of the Pomc promoter are inherited by the females of the next generation, F110. Similarly, by extending the findings to a different strain and male offspring, in weaning-age male Sprague–Dawley strain rat offspring of HF-fed mothers, hypermethylation of the Pomc promoter and enhancer areas were found13. From a translational perspective, there is evidence in humans, both children and adults, for a correlation between Pomc methylation in peripheral blood cells and BMI/obesity7. Furthermore, a family cohort study reported that the association between maternal and offspring BMI persisted over 3 generations through the maternal line14, and decreased methylated cytosine levels were found in fetal umbilical cord blood of overweight/obese women compared to normal weight pregnant women15.
Research supports the possibility of environmental epigenetic inheritance through gametes16,17. However, in our above study, the “intergenerational” mode of transfer from mother to offspring18 is not clear, as this could have been either via “direct” epigenetic changes in the dam’s gametes or via the “direct” influence of maternal diet/obesity/behavior on the developing embryo, fetus or suckling pup19,20,21. In contrast, inheritance of environmental experiences from male rodent sires that do not display paternal behavior (and are not present after insemination, i.e., during gestation and lactation) is most likely via epigenetic modifications of the spermatozoa16,22.
Recent research has supported this possibility of paternal epigenetic heredity in obesity23,24. For example, in mice, offspring of HF-induced obese males showed an obese phenotype17, which was suggested to be mediated by sperm microRNA25,26,27. More intricately, maternal HF experience in mice results in a paternally transmitted phenotype (body size and insulin sensitivity) to F3 female offspring, suggesting germline-based transgenerational inheritance28. Even in humans, there are data supporting the relationship between paternal overweight/obesity and (a) sperm DNA methylation percentages29; (b) hypomethylation at differentially methylated regions of imprinted genes in the umbilical cord DNA of newborns30; and (c) increased offspring BMI in childhood31.
Therefore, to increase the likelihood of gametic transfer of diet-induced overeating, in the present study, we examined, in two separate studies, the overeating/obesogenic profile of F1 offspring of F0 HF-induced obese male sires that were mated with chow-fed females. We further examined the hypothesis that the Pomc promoter is hypermethylated in the sperm of F0 males.
Results
Body weight and daily food consumption were affected by HF and transferred to the second generation
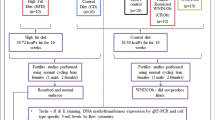
To study paternal heredity, first-generation male rats were raised from the end of the weaning period (PND 21) for 158 days on either a high-fat diet (HF group, 60% fat) or standard rodent chow (CHOW; CH group, 6% fat; Fig. 1a). The body weights (grams) of both diet groups were measured every five days from PND 21–158. Two-way ANOVA with repeated measures (diet group*PND) showed that the HF-fed group weighed significantly more than the chow-fed group did (F(1,19) = 33.93, p < 0.001). Furthermore, there was a significant interaction effect between diet group and PND (F(26,494) = 29.29, p < 0.001), showing that the HF-fed group gained weight at a significantly greater rate over time than did the chow-fed controls (Fig. 1b). Compared with CH-fed rats, HF-fed rats consumed significantly more kcal per day from PND 30 and on (F(1,11) = 129.45, p < 0.001 for the overall difference between the HF- and C-treated groups, see Supplementary Fig. 1a).

Body weight was affected by the HF diet and was transferred by paternal transmission to their offspring. (a) Experimental scheme for the generation of fathers (F0) and offspring (F1). First-generation male Wistar rats were raised from PND 21 until PND 90 and fed HF diet or chow and were subsequently mated with chow-fed females. Significant differences from the chow group are presented with *p < 0.01. (b) F0 body weight was measured every fifth day. (c) Body weights of offspring females from PNDs 1–22. (d) Male offspring body weight from PND 1–22. (e) Body weights of offspring females during food challenge, from PND 35 until PND 70. The offspring received either HF (N = 12 offspring of HF-fed father, N = 14, offspring of chow-fed father) or standard rodent chow (N = 12 offspring of HF-fed father, N = 14, offspring of chow-fed father). Body weight was measured every fifth day. (f) Male offspring body weight during food challenge, from PND 35 until PND 70. The offspring received either HF (N = 12 offspring of HF-fed father, N = 14, offspring of chow-fed father) or standard rodent chow (N = 10 offspring of HF-fed father, N = 14, offspring of chow-fed father). Body weight was measured every fifth day. N = 10–14 for each group. The data are presented as the mean ± SEM. Significant differences in panels e and f are presented with capital letters.
In the first generation, both HF and control male rats were mated between 90 and 100 days with naïve chow-fed females. The mean body weight (grams) of the offspring of both groups was measured at 4 time points during the lactation period, from PND 2 to weaning at PND 21. Two-way ANOVA with LSD repeated measures (paternal diet*PND) showed that paternal HF did not affect newborn offspring body weight for either sex throughout the lactation period (F(1,21) = 0.039, p = 0.845NS for females; F(1,19) = 0.960, p = 0.340NS for males; Fig. 1c, d, respectively).
Since there was no weight effect of the offspring during the lactation period, we predicted that the inheritance effect would become apparent with food challenge. Therefore, the offspring that were born and raised until weaning with their naïve, chow-fed mothers (PND 0–21) were separated, 2 per cage, on the weaning day according to sex and paternal diet (female/male, HF/chow offspring), and they consumed chow diet ad libitum until PND 35. At PND 35, the offspring were split into two different ad libitum diets, HF (60% fat) or chow (6% fat), and were raised until they were sacrificed on day 70.
As a result of the HF challenge, there was a significant increase in female (PND 70, p = 0.05) (Fig. 1e) and male (PND 70, p < 0.05, Fig. 1f) body weight. Under HF challenge (but not chow) conditions, the growth curve was significantly different for the offspring of HF-fed fathers than for those of control fathers (for the interaction (paternal diet group*PND), females, F(7,168) = 3.020, p = 0.005; males, F(10,240) = 2.05, p = 0.03). As expected, the HF groups consumed more calories than the chow fed groups (Supplementary 1 b,c). Two-way ANOVAs with repeated measures (diet group*PND), starting from the HFD challenge (PND35) revealed that under the F1 HF condition, offspring of HF-fed fathers consumed significantly more calories than the paternal-chow-fed group (females, F(1,11) = 6.08, p = 0.031, males, F(1,10) = 5.129, p = 0.041) (Supplementary 1 c). In contrast, under the F1 chow condition, there were no significant differences observed (females F(1,11) = 0.581, p = 0.462NS; males F(1,10) = 0.575, p = 0.466NS).
Thus, the offspring of fathers fed a HF diet gained more weight during the diet challenge period than did the controls.
A high-fat diet is correlated with hypermethylation of the Pomc promoter in the spermatozoa of F0 males, with these epigenetic changes becoming evident by the end of the weaning period
Since we have previously reported that CpG methylation is altered at the Pomc promoter as a result of HF/obesity9,10 and that we observed a transgenerational transfer of the obesogenic trait, we investigated the methylation profile of the Pomc promoter in the spermatozoa of HF- and chow-fed males. We performed a chi-square analysis of 24 CpG sites located between − 62 and − 490 bp from the transcriptional start site and found significantly more methylation in the sperm of HF-fed males than in the sperm of controls at CpG sites 1 (− 490 upstream from the transcriptional start site; p < 0.035), 4 (− 368; p < 0.05), 6 (− 224; p < 0.01), 9 (− 192; p < 0.01), 11 (− 164; p < 0.01), 13 (− 152; p < 0.01), 21 (− 99; p < 0.05) and 24 (− 62; p < 0.05; Fig. 2A). Moreover, over all 24 CpG sites, percent of global methylation was positively correlated with an individual rat's body weight on PND 90 (r(13) = 0.562, p = 0.023) (Fig. 2b).

HF induced hypermethylation at the Pomc promoter in male spermatozoa and their offspring's Arc at PND 21. CpG site-specific methylation of the promoter of the Pomc promoter − 490 bp upstream of the ATG was analyzed using targeted deep sequencing (Chow: n = 7, HF: n = 6; 5–6 clones per animal). The data are presented as the means ± sems percentages of methylation in each group. Differences in methylation patterns were analyzed by chi-square analyses (*p < 0.05). (a) Percentage methylation of the Pomc promoter of spermatozoa. (b) Pearson’s test showed a positive correlation between the percent methylation of 24 CpG sites and body weight at PND 90 (p < 0.05). (c) Percentage methylation of the Pomc promoter of the Arc in the offspring of the treated fathers at PND21. (d) Sum of methylation of the 4 most distal sites − 368 to − 490 bp from the ATG of the Pomc promoter from the Arc in the offspring of the treated fathers at PND21.
Although there were no differences in body weight between offspring of HF- or chow-treated rats fed chow, in the HF-challenged group, a difference in the obesity-prone trait in the offspring was revealed. A possible explanation might involve the methylation pattern at the Pomc promoter in Arc. Therefore, we also analyzed methylation profile of the Arc Pomc promoter at 24 CpG sites located between − 62 and − 490 bp from the transcriptional start site in the offspring on day 21. Similarly, to the methylation pattern in the sperm, we found significantly more methylation in the ARC of offspring of HF-fed fathers than in the offspring of controls at CpG site 3 (− 428; p < 0.05) and site 4 (− 368; p < 0.05; Fig. 2c). In our previous report, the first 4 CpG methylation sites (between − 490 and − 368 bp upstream from the transcriptional start site) were completely methylated in the Arc of both HF and chow fed female adult rats 9,10. Interestingly, in young rats in the current study, these CpG methylation on sites (1–4 between − 490 and − 368 bp upstream from the transcriptional start site) were highly methylated in the offspring of HF fathers (t(166) = 3.972, p < 0.001; Fig. 2d)..
To identify the methylation pathway involved, we examined the mRNA expression of DNMTs. On PND 21, the expression levels of DNMT3b were affected by paternal HF treatment (t(17.56) = 2.4, p < 0.05) (Fig. 3a). However, the expression of DNMT1 (t(15.82) = 1.68, Fig. 3b) and DNMT3a (t(18.17) = 1.068, Fig. 3c) was not significantly affected by HF treatment (p > 0.1).

The male offspring of HF-fed fathers showed increased mRNA expression of DNMT3b but not of DNMT1 or DNMT3a in the Arc. At PND 21, Arc nuclei were dissected and subjected to real-time PCR: (a) DNMT3b (n = 9 for chow and n = 11 for HF animals); (b) DNMT1 (n = 9 for chow and n = 9 for HF animals); (c) DNMT3a (n = 11 for chow and n = 10 for HF animals); The data are presented as the means ± SEMs, * Significantly different from the C group, *p < 0.05.
Obesity-related markers are transgenerationally transmitted
We further examined male offspring at PND 60, which maintained either a steady-state HF or chow feeding, while focusing on the anorexigenic leptin-Pomc pathway. Our analysis revealed notable differences in leptin levels. Specifically, there were significant overall differences between the paternal diet groups (HF vs. CH) (F(1,36) = 8.10, p < 0.01) and between the challenge diet groups (HF vs. CH) (F(1,36) = 28.61, p < 0.001). Moreover, leptin levels were significantly higher in the offspring of the HF group (n = 11) than in those of the other groups: CH-HF (n = 13, p < 0.05), HF-CH (n = 8, p < 0.01), and CH-CH (n = 8, p < 0.001). Significantly higher leptin levels were also found between CH-HF and the CH-CH group (p < 0.001) and a non-significant tendency toward higher leptin levels was evident in the HF-CH group than in the CH-CH group (p = 0.06) (Fig. 4a).

Obesity-related markers are transgenerationally transmitted. At PND 60, rats from all four offspring groups (HF-HF, HF-CH, CH-HF and CH-CH) were sacrificed, their plasma collected and Arc nuclei dissected. Significant differences between groups are indicated with capital letters. (a) Leptin levels were analyzed by ELISA (CH-CH, n = 8; CH-HF, n = 11; HF-CH, n = 8; HF-HF, n = 11). A differs from B (p < 0.05), b (p < 0.01) and C (p < 0.001). B, different from C (p < 0.001). b tendency to differ from C (p = 0.06); (b) Pomc levels analyzed by qPCR (CH-CH n = 8; CH-HF n = 9; HF-CH n = 7; HF-HF n = 9). A Different from B (p < 0.05); (c) DNMT3B levels analyzed by qPCR (CH-CH n = 8, CH-HF n = 8, HF-CH n = 7, HF-HF n = 8), A different from B (p < 0.05).
Moreover, within this pathway, Pomc stands out as a major anorexigenic target expected to respond to elevated leptin levels. As anticipated, Pomc mRNA expression increased in response to HF, significantly differing from the levels observed in the control group (CH-CH). Notably, we observed a significant interaction between the paternal diet group and the challenge diet group (F(1,29) = 4.26, p < 0.05). Furthermore, Pomc mRNA levels were notably lower in the control group (CH-CH, n = 8) than in all other groups: the HF-HF group (n = 9, p < 0.05), the HF-CH group (n = 7, p < 0.05), and the CH-HF group (n = 9, p < 0.05), as shown in Fig. 4b.
Finally, we examined the mRNA levels of DNMT3b due to the notable differences observed in the expression of this enzyme among male offspring at PND 21. Our findings revealed that the significantly elevated DNMT3b mRNA levels observed in the HF-CH group (n = 7) persisted over time, extending into adulthood in F1 offspring of fathers fed a high-fat diet (n = 8, p < 0.05, as depicted in Fig. 4c).
There was a clear reduction in the difference in methylation between male offspring of HF-fed fathers and controls on the Arc Pomc promoter at PND 60
We analyzed the methylation pattern across the Arc Pomc promoter in male offspring during HF challenge at PND 60. Specifically, we analyzed a 540-base pair fragment of the Pomc promoter containing 24 CpGs between − 62 and − 490 upstream from the transcription start site in offspring exposed to a high-fat diet (HF-HF vs. CH-HF). Surprisingly, no differences were detected between the groups in the first four sites (t(166) = 1.095, p > 0.1). However, we did observe significantly higher methylation in the HF-HF group than in the CH-HF group at CpG site 16 (− 128 bp from the transcription start site), with chi-square = 4.79, df = 1, p < 0.05. Conversely, more methylation was found in the CH-HF group than in the HF-HF group at CpG site 7 (− 216 bp from the transcription start site), with chi-square = 4.37, df = 1, p < 0.05, as illustrated in Fig. 5a).

Compared with HF-fed fathers, their offspring exhibit increased methylation at only limited discrete sites on the Pomc promoter after puberty. (a) Comparison between offspring at PND 60 who consumed HF but whose fathers consumed either chow or HF. CpG site-specific methylation of the promoter of the Pomc promoter − 490 bp upstream of the ATG was analyzed using targeted deep sequencing (Chow: n = 7, HF: n = 6; 5–6 clones per animal). The data are presented as the means ± sems percentages of methylation in each group. Differences in methylation patterns were analyzed by chi-square analyses (*P < 0.05). (b) Comparison between offspring at PND60 who consumed chow but whose fathers consumed either chow or HF. CpG site-specific methylation of the promoter of the Pomc promoter − 490 bp upstream of the transcription start site was analyzed using targeted deep sequencing (Chow: n = 7, HF: n = 6; 5–6 clones per animal). The data are presented as the means ± sems percentages of methylation in each group. Differences in methylation patterns were analyzed by chi-square analyses (*p < 0.05).
In the next stage, we examined those differences in the offspring that received chow (no food challenge) (HF-CH vs. CH-CH). We did not find differences between these groups in the first four sites (between -490 and -368 bp upstream from the transcriptional start site) (t(286) = ± 0.957; p > 0.1). We did find significantly more methylation in HF-CH compared to CH-CH group on: CpG (16; − 128 bp from the transcription start site); chi-square = 5.67, df = 1, p < 0.05). In addition, there was more methylation in CH-CH compared to HF-CH group on: CpG (21); chi-square = 3.71, df = 1, p < 0.05, Fig. 5b.
Methylation disparities in females born to fathers exposed to either HF or chow during HF challenge suggest the implication of genes associated with neuronal plasticity
Clearly, the regulation and expression of Pomc alone cannot entirely explain the transgenerational transfer of the obesogenic phenotype. Therefore, to gain a wider perspective on the pathways involved in obesity, we conducted an reduced-representation bisulfite sequencing (RRBS) analysis on the Arc of HF-fed 60-day-old female offspring to fathers consuming either HF or CH. We chose to scrutinize the methylation pattern effect on the paternal transition of the obesogenic trait in the female offspring because the effect on their body weight was greater than that in the male group.
Differentially methylated regions (DMRs) were extracted and analyzed by the software tool metilene32. We found 77 distinct DMRs that were significantly different between the offspring of HF and control fathers at a false discovery rate (FDR) of p < 0.05. Among these DMRs, 67 presented increased methylation in F1 HF offspring compared to F1 CH offspring, designated Hyper-DMRs; 10 DMRs presented decreased methylation in F1 HF offspring compared to F1 CH offspring and were designated Hypo-DMRs (Fig. 6a). Analysis of DMR distribution across different genomic regions revealed that most of them (Hypo and Hyper) reside in promoter regions (34 out of 77), All DMRs in each individual animal with statistical analysis are presented in Table S1). It should be noted that a significant number of the DMRs were in exons (19 out of 77) (It should also be noted that KL568446.1 is a contig with a clear exon that does not annotate to any known gene).

HF induces transgenerational changes in the methylation of neurodevelopmental pathways. The RRBS of DNA from the Arc of HF-challenged offspring of either HF- or chow-fed fathers was analyzed. (a) Genes that were correlated with differentially methylated regions (DMRs) at FDR < 0.05 are shown in a heatmap where the standardized level of methylation for each individual rat is presented. (b) Functional enrichment analysis was performed using Metascape. Bar graph of enriched terms across differentially methylated genes are colored according to p values. (c) Motif enrichment over altered DMRs was determined by STRING (version 12.0). The color code for the interactions: blue – from curated databases; purple – experimentally determined; green—predicted interaction, gene neighborhood; red – gene fusions; dark purple –predicted interaction, gene co-occurrence.
To better understand the functional role of DMRs, we conducted motif enrichment analysis using HOMER tools. Consistent with the observed weight effects (Fig. 1b–e) in the F1 offspring, our data indicated significant enrichment of DMRs around motifs associated with general hypothalamic plasticity parameters, including dendritic development, the telencephalon, and neuron projection development (Fig. 6b). Furthermore, some of the genes involved had previously been correlated with obesity. For instance, neurotrophic receptor tyrosine kinase 2 (NTRK2), known to be associated with severe obesity33, was previously correlated with Pomc34. Additionally, FKBP51, another gene with a hypermethylated DMR in the offspring, was found to confer resistance to diet-induced obesity in null mice35,36.
The gene network analysis, generated from the list of genes differentially methylated by STRING software, and including Pomc, illustrates the pathways involved in the transgenerational transmission of the obesity trait, emphasizing physical connections, coexpression, and genetic interactions (Fig. 6c).
Discussion
Obesity is a rapidly spreading public health crisis that has reached epidemic proportions. We postulated that a significant portion of its rapid spread is attributable to its transmission across generations through epigenetic mechanisms. While demonstrating maternal transmission is challenging due to the maternal environment during pregnancy, we have successfully shown here paternal transmission in rats.
Indeed, previous reports from human research demonstrated both maternal and paternal transmission of the obesogenic trait. Newborns of (preconceptual) parents with obesity were reported to have altered DNA methylation patterns at imprinted genes that are important in normal growth and development37. Associations of maternal obesity with offspring cord blood epigenome-wide DNA methylation were stronger than associations of paternal adiposity38. Accordingly, a study of 912 European American children reported a stronger influence of maternal obesity than paternal obesity on infant and early childhood body mass39. Nevertheless, paternal obesity contributes to the risk for childhood and later obesity in humans30,31 in interaction with the sex of the offspring. For example, a study of predicting adiposity and cardiovascular risk from parental adiposity and smoking reported that regression predictions of BMI from father to son (0.30) and from mother to daughter (0.33) were greater than those from father to daughter (0.23) or from mother to son (0.22)40.
Here, we revealed that male rats with DIO are predisposed toward obesity to both their male and female offspring. This effect is evident when the offspring are exposed to a HF challenge. Surprisingly, in a conceptually similar investigation conducted on Sprague‒Dawley rats, it was reported that F1 and F2 females, whose ancestors had experienced HF-induced obesity, displayed resistance to HF-induced weight gain and exhibited reduced fat mass compared to their respective controls, despite similar energy intake23. The disparity between these findings might allude to a transgenerational modification in energy regulation that can elicit either resilience or susceptibility to obesity, contingent on the intensity of obesity induction in the initial generation or the vigor of the dietary challenge in the subsequent generation. We previously demonstrated this vulnerability/resilience concept through the use of a different environmental concept, heat stress, which is also controlled by the hypothalamus and transmitted to the next generation41. Notably, the results reported by de Castro Barbosa et al.23 were derived from a high-fat diet challenge that was administered to the F1 and F2 offspring as adults for a duration of 12 weeks—a timeframe distinct from our briefer challenge regimen.
Building upon prior research from our laboratory and others, emphasizing the significance of epigenetic malprogramming, particularly regarding the hypothalamic neuropeptide POMC9,12, a key regulator of appetite and energy expenditure within the hunger/satiety center in the Arc. two questions emerged: First, could the second-generation effects we reported be attributed to the maternal environment rather than true heredity? Second, how is the heredity of the obesity trait transmitted from one generation to the next if methylation is erased in the gametes?
To address these questions, in this study we used similar methods to demonstrate paternal heredity (with minimal maternal exposure to the sire) and focused on the methylation in regions of the DNA, both in the sperm and in the Arc nucleus, of the Pomc promoter which previously has been shown to correlate with the obesogenic phenotype. It should be emphasized that since the Arc nucleus is a complex structure with many cell types and diverse physiological roles, the experimental setup, which probes only a limited number of Arc cells, does not allow us to conclude that the changes we are reporting are limited to or specifically involve POMC neurons. On the contrary, since our findings indicate that some of the CpG sites are fully methylated, they probably represent both glial and neuronal cells. Furthermore, even within the neurons, our findings suggest that the methylation differences in the Pomc promoter are a result of changes across many neuronal types probably demonstrating a global shift in the methylation in the ARC nucleus. Moreover, differentially methylated regions in the Arc nucleus as identified by RRBS in the second generation points to genes involved in dendritic and neuronal projection development, indicating that at least part of the changes are occurring in neurons. However, isolation of the Arc cell populations by fluorescence-activated cell sorting (FACS) in future experiments, followed by methylome analysis of POMC neurons as well as other neuronal and/or glial cells. This limitation will be resolved in future experiments by sorting different cell types and comparing the methylation pattern between glial cells and neurons and determining if the methylation in POMC cells is different than in the general neuronal population.
Regarding the question of the mechanism by which epigenetic heredity is transmitted by the sperm, our results revealed distinct hypermethylation of the Pomc promoter in both the sperm of males exposed to a HF and the Arc of their offspring after the weaning period at postnatal day 21 (PND21). This hypermethylation is particularly evident at four CpG sites, spanning from − 398 to − 490 upstream from the transcription start site, showing full methylation in adult rats. The concept of 'epigenetic memory' within sperm has been previously described, especially for traits influenced by environmental factors. A study exploring the correlation between male overweight/obesity and sperm DNA methylation percentages revealed significantly lower methylation levels at 4 of 12 differentially methylated regions in semen from men who were overweight or obese. The authors concluded that male overweight/obesity status leaves a trace in the sperm epigenome42.
In a mouse model of paternal obesity, metabolic disturbances persisted in two generations of offspring. HF-induced DIO in F0 males resulted in a 25% reduction in global DNA methylation and altered microRNA content in the testes and sperm30. As previously mentioned, sperm harbor noncoding RNAs that may influence DNA methylation43,44,45. Studies in overweight male mice revealed altered sperm microRNA and DNA methylation profiles, accompanied by metabolic dysregulation in the offspring, which was partially observable even in F246.
The heightened methylation observed in our study is intricately linked with an increase in DNMT3b expression, as evidenced by a distinct correlation between body weight and DNMT3b mRNA expression. In simpler terms, the data illustrate that greater body weight is associated with elevated expression of DNMT3b (Fig. 3 a). Obesity did not significantly affect DNMT1 or DNMT3a mRNA expression. DNMT3a and DNMT3b function as de novo methyltransferases of cytosine residues, while DNMT1 functions as a maintenance enzyme of methylation. While DNMT3a activates methylation at non-CpG and CpG sites, DNMT3b activates methylation of the CpG dinucleotides in the repeated DNA sequences47. Furthermore, similar to these results, Xia et al.48 reported an increase in DNMT3b and DNMT3a levels in CpG sites on the leptin promoter in mice that received HF for 18 weeks. It should be noted that Lister et al.49 reported very low levels of Dnmt3b expression in the frontal cortex of adolescent mice. The observed differences in expression patterns could be attributed to species-specific variations between rats and mice, or differences between brain regions. Interestingly, species specificity might be involved, as a separate project in our lab involving the chick hypothalamus revealed that stress, specifically heat stress, significantly affected Dnmt3b expression in the hypothalamus during both embryonic development and early post-hatch heat conditioning50,51.
Interestingly, we observed that the differences in methylation along the Pomc promoter between the offspring of HF- and chow-fed fathers diminished as the offspring aged, as evidenced at PND 60. This suggests that other genes may also contribute to the transgenerational transmission of the obesogenic trait. To identify these genes, we conducted RRBS analysis on tissue samples from the Arc at PND 60. This analysis revealed seventy-three differentially methylated regions (DMRs), most of which were hypermethylated in the offspring of HF-fed fathers, primarily affecting gene promoters.
Examining these DMRs revealed notable enrichment around motifs linked to general hypothalamic plasticity, encompassing dendritic development, the telencephalon, and neuron projection development. The observed alterations in methylation within these pathways suggest a potential reorganization of the network governing the set-point that intricately regulates the balance between hunger and satiety.
The key genes identified through methylation comparison in the second generation shed light on significant players in this process. Notably, neurotrophic receptor tyrosine kinase 2 (NTRK2), a gene previously associated with severe obesity33,34 and recognized for its involvement in network reorganization. Additionally, FKBP51, another gene exhibiting hypermethylation in the DMRs of offspring, has been linked to resistance to diet-induced obesity in null mice35,36. These findings underscore the potential influence of epigenetic modifications on critical genes, revealing a molecular framework that may contribute to the intricate regulation of hunger and satiety, with implications for obesity susceptibility.
While this study adhered to many standards of developmental psychobiological research, an important methodological limitation should be noted. Some of our F1 assays (growth curves and ELISA for leptin levels), where all the available data were analyzed, included (equally balanced between groups) pairs of siblings with groups. As Zorrilla52 explained, this has the potential to produce spurious findings and false negatives. Regarding caloric consumption, we averaged the data from the 2 sibling cagemates to one average litter score, as recommended. For the other assays, that used a reduced N per group (methylation analyses and mRNA expression), we used non-sibling offspring representatives from different dams, thus maximizing diverse litter representation. Another limitation to be noted is that we did not control for multiple chi-square comparisons, following an approach supported by several statisticians, in cases such ours in which the comparisons are experimentally planned, complimentary comparisons (No adjustments are needed for multiple comparisons; Multiple Comparison Procedures: The Practical Solution; The Long Way From α-Error Control to Validity Proper: Problems With a Short-Sighted False-Positive Debate53,54,55. This exploratory approach allows for identifying potential promising findings.
In conclusion, this research highlights the possibility of paternal transgenerational transmission of obesogenic traits, emphasizing the role of the regulation of Pomc expression, which is transmitted through the germline. Furthermore, looking at differences in the methylation pattern between offspring of obese and lean chow-fed fathers indicates the possibility of network reorganization at the set-point controlling the balance between hunger and satiety and offers a list of involved genes.
Materials and methods
General subjects
Wistar outbred rats (supplied by Harlan Laboratories Ltd., Jerusalem) were raised in the colony of the Gonda Brain Research Center at Bar-Ilan University, Ramat Gan, Israel. In all of the experiments, polysulfone cages (18.5 cm height, 26.5 cm width, 43 cm length) were used, with stainless steel wire lids and wood shavings as bedding materials. In addition, large plastic tubes were present in all of the cages, which were available for use as a ‘‘house’’ or a hiding place for the rats. Standard chow (2018SCF Teklad Global 18% Protein 6% Fat Rodent Diet HARLAN, Madison, USA) or a high-fat diet (D12492 Research Diets, Inc. Rodent diet with 60% Fat, NJ, USA) and water were freely available. When measuring intake, chow and HF intake in grams were converted to kilocalories (kcal) according to the manufacturer’s data (1 g C diet = 3.1 kcal and 1 g HF diet = 5.24 kcal). The animals were on a 12:12 h light:dark cycle with lights on at 0700 h. Room temperature was maintained at 22 ± 2 °C. On the day of sacrifice, plasma, sperm and hypothalamic regions were collected for hormonal, mRNA and DNA analyses (n per group for each analysis is presented in the results or Figure legends). The research protocols were approved by the Institutional Animal Care and Use Committee, and they adhered to the guidelines of the Society for Neuroscience. All efforts were made to minimize animal suffering and to reduce the number of animals used.
Subjects and study design
Generation F0 Twenty-four male rats were supplied to Bar-Ilan University on PND 21 in two separate cohorts of 12 each. At arrival, each cohort was randomly split into two groups based on their diet—a chow group or a HF group—and housed in pairs. The animals were habituated to the animal house cage for 5 days (acclimation) to minimize the degree of stress after transportation. Next, they were exposed to acclimate to human scents and procedures to minimize the degree of stress. On PND 90–100, each male was mated with one naïve (chow-fed) female (at verified oestrus) in her cage overnight breeding to minimize the degree of stress from another cage mate. The following day, they were returned to their home cages. After verifying the birth of the offspring, the males were sacrificed for sperm and Arc collection at PND 158 (N = 12 chow-fed and 9 HF fed F0 males with complete data).
Generation F1 The matings resulted in 7 litters from chow-fed F0 males and 6 litters from HF-fed F0 males that had a sufficient number of viable pups to be used for the study at weaning. Litter size at birth from the two different experimental cohorts ranged from 6 to 15 (average litter size = 11). Litter size was adjusted randomly (per sex) on PND 2 to a maximum of 8 (aiming for 4 per sex). Pups were weighed on PND 2, 7, 14 and 21. At weaning day (PND 21), one-third of the offspring (males and females) from each experimental paternal group were sacrificed for plasma and brain analyses. Two-thirds of the offspring (males and females) were separated into two siblings of the same sex in a cage fed a chow diet until PND 35. From PND 35, the rats were subjected to diet challenge. Within each sex, half of the paternal chow diet-fed pups and half of the paternal HF diet-fed pups were switched to HF until the end of the experiment (either PND 60 or PND 70; see results and figure legends).
Plasma analysis
Leptin levels were assessed using commercial enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Brain sections
Coronal brain sections of the hypothalamus were cut using a cryostat (− 2.12 to − 4.5 mm from bregma according to Paxinos and Watson, 4th edition), and the Arc was extracted from the brain using a truncated needle. Punches from the left and right hemispheres of each rat were immediately immersed in RNALater (Ambion, Austin, TX, USA) or frozen on dry ice and stored at − 80 °C.
Isolation of cauda epidydimal sperm and density gradient separation of sperm samples
The cauda epididymides of the F0 males were harvested and placed on petri dishes with 5 ml of Ham's F-10 Nutrient Mix. The epididymis was pierced numerous times using a 30-gauge needle, and the sperm were allowed to swim to the media through the holes for 20 min and then transferred to a 15 ml conical tube. Cauda epidydimal semen was purified by centrifugation at 1200 rpm for 10 min at room temperature. After disposal of 4 ml of the supernatant, the semen pellet was resuspended and incubated at 37 °C for 15 min for liquification.
Density gradient separation of sperm samples was performed using 0.5 ml of PureCeption 40% upper phase gradient and 0.5 ml of PureCeption 80% lower phase gradient in a 15 ml conical tube, followed by centrifugation at 1800 rpm for 10 min at room temperature. After centrifugation, there are two layers containing semen: the pellet and the interphase. The interphase semen layer was resuspended in fresh 5 ml of Ham's F-10 Nutrient Mix in a new 15 ml conical tube and washed by centrifugation at 1800 rpm for 10 min at room temperature. The supernatant was discarded, 0.5 ml of the pellet and medium was removed, and the pellet was suspended in the remaining medium for further DNA purification.
RNA isolation and real-time PCR
Total RNA was isolated using Tri-Reagent (Molecular Research Center, Cincinnati, OH, USA) according to the manufacturer’s instructions. Hypothalamic RNA (0.5 µg) was reverse-transcribed to single-stranded cDNA by SuperScript II Reverse Transcriptase and oligo (dT) plus random primers (Invitrogen, Carlsbad, CA, USA). Real-time PCR was performed in duplicate with 10 ng of cDNA on a model 7000 sequence analysis system (Applied Biosystems, Foster City, CA, USA) with Absolute Blue SYBR Green ROX Mix (ABgene, Epsom, UK). Dissociation curves were analyzed following each real-time PCR to confirm the presence of only one product and the absence of primer dimer formation. The threshold cycle (Ct) for each tested gene (X) was used to quantify the relative abundance of that gene using the formula 2—(Ct gene X—Ct standard). It was suggested that hypoxanthine phosphoribosyltransferase-1 (Hprt) is one of the optimal reference genes for reverse-transcription quantitative PCR (RT‒qPCR) in the rat hypothalamus for the study of obesity. This is because it did not change following HF exposure56. Therefore, the mRNA levels of all the genes were normalized to the mRNA expression of Hprt. Primers;
Pomc;F-GCTACGGCGGCTTCATGA, R-CCTCACTGGCCCTTCTTGTG;
DNMT3b; F-GCGGGAGCCCATGTAGCTA, R-CGAGTCCCTCACACCTGGAA.
DNMT3a; F-GGGTAACCTTCCTGGCATGA, R-GTGGTAATGGTCCTCACTTTGCT.
DNMT1: F-GGCTTTTCTACCGCATTTGCT, R-CTGCAAGAACTCGACCACAATC.
Hprt;F- GCGAAAGTGGAAAAGCCAAGT, R- GCCACATCAACAGGACTCTTGTAG.
Sperm and arc-targeted DNA methylation analysis
Total DNA was isolated from the hypothalamic Arc using TriReagent (Molecular Research Center) according to the manufacturer’s instructions. Genomic DNA from the sperm was isolated using the User-Developed Protocol QA03 followed by the QIAamp® DNA Mini Kit (Qiagen, Hilden, Germany). The purified DNA was processed for bisulfite modification using an imprint DNA modification kit (Sigma, St. Louis, MO, USA) according to the manufacturer’s instructions. The quality of the DNA after the bisulfite reaction was evaluated using an agarose gel to verify that the DNA was intact and not nicked. A 540 bp region upstream of the start of transcription from the Pomc promoter region was amplified with specific primers for bisulfite conversion, the forward primer 5′-GTTTTGGGTTGTTATGATTTTTGAT-3′ and the reverse primer 5′-AATCCCTATCACTCTTCT CTCTT-3′. PCR amplification of the fragment was performed using Biotaq DNA Polymerase (Bioline USA Inc., Taunton, MA, USA) under the following conditions: initial incubation at 95 °C for 5 min; 40 cycles of denaturation at 95 °C for 30 s, annealing at 58 °C for 30 s, and elongation at 72 °C for 45 s. The PCR products were separated on agarose gels (1.5%) followed by gel extraction with a QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany) and cloning into PGEMT-easy vectors (Promega, Madison, WI, USA) according to the manufacturer’s instructions. PGEMT-easy vectors were propagated using JM109 competent Escherichia coli cells (Promega) and then purified from the bacteria with the QIAprep Spin Miniprep Kit (Qiagen).
The average %C/T for each CpG per animal was calculated. A total of 24 CpGs sites both in the sperm and in the hypothalamic Arc were analyzed (Chow: n = 6–7, HF: n = 5–6; 5–6 clones per animal).
Reduced-representation bisulfite sequencing (RRBS)
DNA was isolated together with RNA from the same sample by TRI Reagent according to the manufacturer’s instructions (Molecular Research Center). RRBS libraries were prepared using a Zymo-Seq RRBS Library Kit (Zymo) at the Crown Genomics Institute of the Nancy and Stephen Grand Israel National Center for Personalized Medicine, Weizmann Institute of Science. Libraries were prepared from 70 to 140 ng of DNA according to the manufacturer’s protocol. Libraries were quantified by a Qubit (Thermo Fisher Scientific) and Tape Station (Agilent). Sequencing was performed on a NovaSeq 6000 instrument (Illumina) using an SP 100 cycle kit (paired-end sequencing). Trimming of Illumina adapters, as well as quality trimming, was performed with Trim Galore using the following options: –nextseq –non_directional –rrbs. Reads were mapped to the rat genome using Bismark v. 0-22-3, bowtie mode. Next, methylation calls were extracted with Bismark, methylation extractor mode. DMRs were extracted and analyzed by metilene v. 0.2–832. Motif enrichment analysis57 and DMR annotation were performed with HOMER tools58.
Statistical analysis
The body weight of the fathers was analyzed by means of a two-way analysis of variance (ANOVA) mixed design with repeated measures (age) and one between-subjects measurement (group). ANOVA was followed by post hoc Bonferroni correction. In the offspring, group differences in Pomc and DNMT3b mRNA expression levels (RT‒qPCR) and leptin levels (ELISA) were analyzed by Student’s t tests for each sex separately after verifying that the data were normally distributed at PND 21. In addition, we performed Pearson product-moment correlations between DNMT3b expression in each rat and body weight. F1 group differences in body weight were analyzed by means of three-way analysis of variance (ANOVA) with one repeated measure (age) and two between-subjects measures (father and diet in challenge group). Post hoc tests for simple effects with Bonferroni adjustment were used. Differences in methylation patterns were analyzed by chi-square analyses. We did not control for multiple chi-square comparisons, following an approach supported by several statisticians, in cases such ours in which the comparisons are experimentally planned, complimentary comparisons (No adjustments are needed for multiple comparisons; Multiple Comparison Procedures: The Practical Solution; The Long Way From α-Error Control to Validity Proper: Problems With a Short-Sighted False-Positive Debate53,54,55. Significance: *p < 0.05, **p < 0.01, ***p < 0.001, #p < 0.001. Statistical analyses were performed with SPSS 21.0 software (IBM Corp., Armonk, NY, USA).
Data availability
Data is provided within the manuscript or supplementary information files.
References
Tran, L. T. et al. Hypothalamic control of energy expenditure and thermogenesis. Exp. Mol. Med. 54, 358–369 (2022).
Iovino, M. et al. Neuroendocrine modulation of food intake and eating behavior. Endocr. Metab. Immune Disord. Drug Targets. 22, 1252–1262 (2022).
Jais, A. & Brüning, J. C. Arcuate nucleus-dependent regulation of metabolism-pathways to obesity and diabetes mellitus. Endocr. Rev. 43, 314–328 (2022).
Yeo, G. S. H. et al. The melanocortin pathway and energy homeostasis: From discovery to obesity therapy. Mol. Metab. 48, 101206. https://doi.org/10.1016/j.molmet.2021.101206 (2021).
Bouret, S. G. Developmental programming of hypothalamic melanocortin circuits. Exp. Mol. Med. 54, 403–413 (2022).
Benite-Ribeiro, S. A., de Rodrigues, V. A. L. & Machado, M. R. F. Food intake in early life and epigenetic modifications of pro-opiomelanocortin expression in arcuate nucleus. Mol. Biol. Rep. 48, 3773–3784 (2021).
Candler, T., Kühnen, P., Prentice, A. M. & Silver, M. Epigenetic regulation of POMC; implications for nutritional programming, obesity and metabolic disease. Front. Neuroendocrinol. 54, 100773. https://doi.org/10.1016/j.yfrne.2019.100773 (2019).
Zhang, J., Li, S., Luo, X. & Zhang, C. Emerging role of hypothalamus in the metabolic regulation in the offspring of maternal obesity. Front Nutr. 10, 1094616. https://doi.org/10.3389/fnut.2023.1094616 (2023).
Marco, A., Kisliouk, T., Weller, A. & Meiri, N. High fat diet induces hypermethylation of the hypothalamic Pomc promoter and obesity in postweaning rats. Psychoneuroendocrinology. 38, 2844–2853 (2013).
Marco, A., Kisliouk, T., Tabachnik, T., Meiri, N. & Weller, A. Overweight and CpG methylation of the Pomc promoter in offspring of high-fat-diet-fed dams are not “reprogrammed” by regular chow diet in rats. FASEB J. 28, 4148–4157 (2014).
Schumacher, R. et al. Temporary effects of neonatal overfeeding on homeostatic control of food intake involve alterations in POMC promoter methylation in male rats. Mol. Cell Endocrinol. 522, 111123. https://doi.org/10.1016/j.mce.2020.111123 (2021).
Plagemann, A. et al. Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: An epigenetic model of obesity and the metabolic syndrome. J. Physiol. 587, 4963–4976 (2009).
Gali Ramamoorthy, T. et al. Maternal overnutrition programs epigenetic changes in the regulatory regions of hypothalamic Pomc in the offspring of rats. Int. J. Obes. 42, 1431–1444 (2018).
Murrin, C. M., Kelly, G. E., Tremblay, R. E. & Kelleher, C. C. Body mass index and height over three generations: Evidence from the Lifeways cross-generational cohort study. BMC Public Health. 12, 81. https://doi.org/10.1186/1471-2458-12-81 (2012).
Ma, Z. et al. Maternal obesity alters methylation level of cytosine in CpG island for epigenetic inheritance in fetal umbilical cord blood. Hum Genomics. 16, 34. https://doi.org/10.1186/s40246-022-00410-2 (2022).
Wei, Y., Schatten, H. & Sun, Q. Y. Environmental epigenetic inheritance through gametes and implications for human reproduction. Hum. Reprod. Update. 21, 194–208 (2015).
Huypens, P. et al. Epigenetic germline inheritance of diet-induced obesity and insulin resistance. Nat Genet. 48, 497–509 (2016).
Sharp, G. C. & Lawlor, D. A. Paternal impact on the life course development of obesity and type 2 diabetes in the offspring. Diabetologia. 62, 1802–1810 (2019).
Cropley, J. E. et al. Male-lineage transmission of an acquired metabolic phenotype induced by grand-paternal obesity. Mol. Metab. 5, 699–708 (2016).
Breton, C. V. et al. Exploring the evidence for epigenetic regulation of environmental influences on child health across generations. Commun. Biol. 4, 769. https://doi.org/10.1038/s42003-021-02316-6 (2021).
Portha, B., Grandjean, V. & Movassat, J. Mother or father: Who is in the front line? mechanisms underlying the non-genomic transmission of obesity/diabetes via the maternal or the paternal line. Nutrients. 11, 233. https://doi.org/10.3390/nu11020233 (2019).
Heard, E. & Martienssen, R. A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell. 157, 95–109 (2014).
de Castro Barbosa, T. et al. High-fat diet reprograms the epigenome of rat spermatozoa and transgenerationally affects metabolism of the offspring. Mol. Metab. 5, 184–197 (2015).
King, S. E. & Skinner, M. K. Epigenetic transgenerational Inheritance of obesity susceptibility. Trends Endocrinol. Metabol. 31, 478–494 (2020).
Grandjean, V. et al. RNA-mediated paternal heredity of diet-induced obesity and metabolic disorders. Sci. Rep. 5, 18193. https://doi.org/10.1038/srep18193 (2015).
Fullston, T. et al. Paternal obesity initiates metabolic disturbances in two generations of mice with incomplete penetrance to the F2 generation and alters the transcriptional profile of testis and sperm microRNA content. FASEB J. 27, 4226–4243 (2013).
Raad, G. et al. Paternal multigenerational exposure to an obesogenic diet drives epigenetic predisposition to metabolic diseases in mice. Elife. 10, e61736. https://doi.org/10.7554/eLife.61736 (2021).
Dunn, G. A. & Bale, T. L. Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology. 152, 2228–2236 (2011).
Sharp, G. C. et al. Paternal body mass index and offspring DNA methylation: findings from the PACE consortium. Int J Epidemiol. 50, 1297–1315 (2021).
Soubry, A. et al. Obesity-related DNA methylation at imprinted genes in human sperm: Results from the TIEGER study. Clin Epigenetics. 8, 51. https://doi.org/10.1186/s13148-016-0217-2 (2016).
Campbell, J. M. & McPherson, N. O. Influence of increased paternal BMI on pregnancy and child health outcomes independent of maternal effects: A systematic review and meta-analysis. Obes. Res. Clin. Pract. 13, 511–521 (2019).
Jühling, F. et al. Metilene: Fast and sensitive calling of differentially methylated regions from bisulfite sequencing data. Genome Res. 26, 256–262 (2016).
Gray, J. et al. Functional characterization of human NTRK2 mutations identified in patients with severe early-onset obesity. Int. J. Obes. (Lond). 31, 359–364 (2007).
Rask-Andersen, M. et al. Functional coupling analysis suggests link between the obesity gene FTO and the BDNF-NTRK2 signaling pathway. BMC Neurosci. 12, 117. https://doi.org/10.1186/1471-2202-12-117 (2011).
Stechschulte, L. A. et al. FKBP51 null mice are resistant to diet-induced obesity and the PPARγ agonist rosiglitazone. Endocrinology. 157, 3888–3900 (2016).
Häusl, A. S., Balsevich, G., Gassen, N. C. & Schmidt, M. V. Focus on FKBP51: A molecular link between stress and metabolic disorders. Mol Metab. 29, 170–181 (2019).
Soubry, A. et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int J Obes (Lond). 39, 650–657 (2015).
Potabattula, R. et al. Male obesity effects on sperm and next-generation cord blood DNA methylation. PLoS One. 14, e0218615. https://doi.org/10.1371/journal.pone.0218615 (2019).
Linabery, A. M. et al. Stronger influence of maternal than paternal obesity on infant and early childhood body mass index: The Fels Longitudinal Study. Pediatr Obes. 8, 159–169 (2013).
Han, T. S. et al. Contributions of maternal and paternal adiposity and smoking to adult offspring adiposity and cardiovascular risk: The Midspan Family Study. BMJ Open. 5, e007682. https://doi.org/10.1136/bmjopen-2015-007682 (2015).
Cramer, T., Rosenberg, T., Kisliouk, T. & Meiri, N. Early-life epigenetic changes along the corticotropin-releasing hormone (CRH) gene influence resilience or vulnerability to heat stress later in life. Mol Psychiatry. 24, 1013–1026 (2019).
Filkowski, J. N. et al. Hypomethylation and genome instability in the germline of exposed parents and their progeny is associated with altered miRNA expression. Carcinogenesis 31, 1110–1115 (2010).
Sinkkonen, L. et al. MicroRNAs control de novo DNA methylation through regulation of transcriptional repressors in mouse embryonic stem cells. Nat Struct Mol Biol. 15, 259–267 (2008).
Mcpherson, N. O., Owens, J. A., Fullston, T. & Lane, M. Preconception diet or exercise intervention in obese fathers normalizes sperm microRNA profile and metabolic syndrome in female offspring. Am J Physiol Endocrinol Metab. 308, E805-821 (2015).
Wang, Y. et al. Sperm microRNAs confer depression susceptibility to offspring. Sci Adv. 7, eabd7605. https://doi.org/10.1126/sciadv.abd7605 (2021).
Zhang, Y., Shi, J., Rassoulzadegan, M., Tuorto, F. & Chen, Q. Sperm RNA code programmes the metabolic health of offspring. Nat. Rev. Endocrinol. 15, 489–498 (2019).
Chen, Z. & Zhang, Y. Role of mammalian DNA methyltransferases in development. Annu Rev Biochem. 89, 135–158 (2020).
Xia, L. et al. Time-specific changes in DNA methyltransferases associated with the leptin promoter during the development of obesity. Nutr. Hosp. 30, 1248–1255 (2014).
Lister, R. et al. Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905 (2013).
Rosenberg, T. et al. Embryonic heat conditioning induces TET-dependent cross-tolerance to hypothalamic inflammation later in life. Front Genet. 11, 767 (2020).
Cramer, T., Rosenberg, T., Kisliouk, T. & Meiri, N. PARP inhibitor affects long-term heat-stress response via changes in DNA methylation. Neuroscience. 399, 65–76 (2019).
Zorrilla, E. P. Multiparous species present problems (and possibilities) to developmentalists. Dev. Psychobiol. 30, 141–150 (1997).
Fiedler, K., Kutzne, R. F. & Krueger, J. I. The long way from α-error control to validity proper: problems with a short-sighted false-positive debate. Perspect. Psychol. Sci. 7, 661–669 (2012).
Rothman, K. J. No adjustments are needed for multiple comparisons. Epidemiology. 1, 43–46 (1990).
Saville, D. J. Multiple comparison procedures: the practical solution. The Am. Stat. 44, 174–180 (1990).
Li, B. et al. Identification of optimal reference genes for RT-qPCR in the rat hypothalamus and intestine for the study of obesity. Int. J. Obes. (Lond) 38, 192–197 (2014).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 10, 1523. https://doi.org/10.1038/s41467-019-09234-6 (2019).
Benner, C., Heinz, S., & Glass, C. K. HOMER - Software for motif discovery and next generation sequencing analysis. http://homer.ucsd.edu/homer/ (2017).
Acknowledgements
This work was supported by the Israel Science Foundation grant no. 1781/16 to AW, and the Israel Ministry of Science and Technology grant no. 3-15689 which is a collaboration between the labs of AW and NM. Additional thanks to the staff of the Crown Institute for Genomics of the Nancy and Stephen Grand Israel National Center for Personalized Medicine, Weizmann Institute of Science, Israel, and especially to Gilgi Friedlander for her skilled bioinformatics work.
Author information
Authors and Affiliations
Contributions
M.H., T.M., N.C., A.W. and N.M. conceived and designed research; M.H., T.M., N.C., T.K., T.Y., performed research; M.H., T.M., N.C., T.K., A.M. analyzed data; A.W. and N.M. interpreted the data and wrote the paper.All authors reviewed the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Haberman, M., Menashe, T., Cohen, N. et al. Paternal high-fat diet affects weight and DNA methylation of their offspring. Sci Rep 14, 19874 (2024). https://doi.org/10.1038/s41598-024-70438-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-70438-y
- Springer Nature Limited