
Abstract
Liver cancer is the fourth leading cause of cancer-related deaths with a steadily increasing rate worldwide, as a well-known hallmark of liver cancer, metabolic alterations are related to liposomal changes, a common characteristic of primary liver cancers based on recent lipidomics studies. Peroxisome proliferator-activated receptor α (PPARα) is a ligand-activated transcription factor with important lipid homeostasis function, therefore we aimed to understand the molecular mechanisms and pathways that activate PPARα after using PPAR-α agonist WY-14643 and identify candidate biomarkers related to PPARα activity and evaluate their effects in liver cancer. The data from differently expressed genes (DEGs) between liver cancer tissue from obese subjects alone and liver tissue after treatment were evaluated by DESeq2 and module genes were analyzed using weighted gene co-expression network analysis (WGCNA). Final candidate genes were identified by intersecting genes among highly ranked DEGs and the brown module, which demonstrated a significant negative correlation with drug treatments. We conducted a protein–protein interaction network, and KEGG enrichment analysis, and core hub genes (CD40, CXCL9, CXCL10, TNFSF14, GBP2, GBP3, APOL3, CLDN1) were identified using the cyto-hubba plugin, among them we focused on GBP2 that plays key roles in oncogenesis and evaluate its expressional with clinical outcomes. In conclusion, the WGCNA-based co-expression network identified GBP2 as one of the hub genes with a negative relation with PPARα agonist treatments. higher expression of GBP2 was closely associated with HCC progression. Therefore, GBP2 might be a potential candidate for the study of PPARα activity in HCC.
Similar content being viewed by others


Background
The incidence of hepatocellular carcinoma (HCC) continues to increase globally, and the lack of early detection and treatment causes a significant burden1,2. It is estimated that, by 2025, more than 1 million people will be affected by primary liver cancer annually3. The common risk factors for liver cancer development are viral infections, alcohol abuse, and metabolic diseases3. Dysregulation of metabolic processes in the liver as the central metabolism organ plays an important role in cancer development4. Lipids are involved in diverse biological processes in the body, including cancer cells, which rely on lipid uptake, intracellular lipid pools, or membrane remodeling5,6. More ever-activated lipid metabolic pathways increase cancer cells’ resistance to immunotherapy agents6,7. People with obesity or liver diseases frequently have chronic inflammatory conditions and lipid alterations such as intrahepatic lipid accumulation have a higher risk of being influenced by the development of liver cancers, so targeting lipid metabolism could also be a promising therapeutic approach for cancer therapy to overcome tumor resistance3,8.
PPAR α is a nuclear receptor that regulates fatty acid oxidation and inflammation through modulation by ligands, PPARα is the most abundant PPAR isotype which contributes to the liver’s remarkable metabolic flexibility9,10. Currently, peroxisome proliferator (PP) agents such as WY-14643 also known as pirinixic acid considered a potent PPARα activator that is widely used in experimental studies affecting lipid and lipoprotein metabolism11. PPARα activators can lower plasma triglyceride levels and increase HDL cholesterol, they can regulate obesity in rodents by increasing hepatic fatty acid oxidation and lowering circulating triglycerides12,13. Mice with PPAR α germline deletion are susceptible to various metabolic defects, including obesity14, hepatic inflammation14, and steatosis15 but not diabetes16. Also, PPARα expression levels were found to be lower in liver tissues of NAFLD/NASH patients as fibrosis progressed17. Recent studies suggest that WY-14643 can inhibit the growth of various cancers, including colon and lung cancers in vivo18,19,20, therefore to better understand the molecular mechanisms of PPARα activation in the liver, particularly in obese individuals, bioinformatics approaches should be combined to identify new genes as clinical biomarkers for PPARα activity. WGCNA (weighted gene co-expression network analysis) is a powerful genetic analysis strategy that relies on a systematic bioinformatics algorithm. This method is used to identify highly and coordinately expressed genes within modules to determine the patterns of genetic association between different samples. In this study, a co-expression network of differentially expressed genes (DEGs) in obese human liver tissue treated with WY-14643 was built using the WGCNA algorithm to identify relevant modules based on their expression profiles for the identification of viable biomarkers in the liver.
Materials and methods
Data source
The data used in this paper was obtained from the GEO database in NCBI (Gene Expression Omnibus http://www.ncbi.nlm.nih.gov/geo), and the experimental data entry number is GSE71731.
Differentially expressed gene identification
First, we used R to read the data from dataset GSE71731. Then we performed DEG analysis screening using the “limma” package. Following the significance analysis of expression levels, the "heatmap" and "ggplot2" R packages were used to create volcano maps and DEG expression heat maps.
Weighted gene co-expression network analysis
The interactive connection among the co-expression modules was studied using the WGCNA algorithm. The WGCNA R software package (http://www.r-project.org/) can be used to determine network construction, the calculation of topological properties, gene selection, module detection, differential network analysis, and network statistics. The soft threshold (β = 20) was used after the standard scale-free network to obtain the weighted adjacency matrix and change it to the topological overlap matrix (TOM) to intensify the connection between genes. We used the average-linkage hierarchical clustering method to cluster genes and identify modules, we set a minimum size of 30 for the gene dendrogram and a cutting line of 0.25 for the modular dendrogram to merge similar modules, Furthermore, a heat map was plotted to exhibit the strength of interaction among the modules.
Screening of candidate hub genes and KEGG analysis
To search for hub genes, we used co-expressed genes between WGCNA-derived brown modules and down-regulated DEGs. We eventually screened 30 overlapping genes as candidate hub genes that may play an important role in the WY-14643 response. Subsequently, by using the “clusterProfiler" R package (version 4.4.4), KEGG analyses (KEGG, https://www.kegg.jp/kegg/kegg1.html) were conducted to assist us in understanding the potential mechanisms and their underlying roles21,22,23,24.
Protein–protein interaction network hub genes
We predicted and visualized protein–protein interaction (PPI) networks using STRING database (https://string-db.org), and Cytoscape software (version 3.9.1). Cytohubba plugin in Cytoscape was used to rank the important genes in PPI networks as our core genes between 30 final hub genes.
Expression analysis of core genes and their prognostic values
The GEPIA2 website, GSCA, and UALCAN database (http://ualcan.path.uab.edu) were used to compare the expression of target genes in tumor tissues and adjacent normal tissues and determine correlation with different tumor stages, age, and immune cell infiltration. The "survival analysis" was used to determine overall survival (OS), and HPA (https://www.proteinatlas.org) was used to obtain immunohistochemistry images of protein expression between liver cancer and normal tissues. TF-gene pairs network was also visualized using the Network Analyst database (https://www.networkanalyst.ca/).
Results
Screening of DEGs
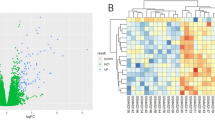
First, we obtained our dataset (GSE71731) from the GEO database to evaluate DEGs, based on the result we identified 220 upregulated and 311 downregulated genes, in the samples treated with WY-14643 compared to non-treated groups. We also draw the heatmap to evaluate the relation between them Fig. 1A–C.

Identification of differentially expressed genes. (A) The volcano plots of DEGs showing the top 20 significant genes with their names, the horizontal axis represents the value of logFC, while the vertical axis represents the mean value of -log 10 (false discovery rate). (B) Heatmap of whole gene expression in all samples related to WY-14643 treatment and age. (C) heatmap of top 20 highly modified genes in the treated group compared to untreated samples.
Co-expression module construction
To identify potential gene clusters involved in human liver tissue response to drugs, we conducted WGCNA with suitable soft-thresholding power to guarantee a scale-free network, Hierarchical cluster analysis indicated samples were well clustered and no outliers or strong clusters were observed. The soft-thresholding powers (soft power = 20) were determined based on a scale-free (R2 = 0.8), The dynamic tree cut method was used for module identification by clustering genes using 1-TOM, with a minimum size cutoff of 30 and a deep split value of 2. A total of 6 different gene modules with different colors were identified based on hierarchical clustering of the calculated dissimilarity. The largest module contains 145 genes, and the smallest module contains 1 gene Fig. 2A–E.

WGCNA was constructed to see gene expression networks of liver samples treated with WY-14643. (A) Sample clustering. (B) Scale independence (left) and mean connectivity (right). (C) A total of 6 modules were obtained. (D) Clustering dendrogram of genes, with dissimilarity based on topological overlap, together with assigned module colors. (E) Expression heatmap of assigned module colors in each sample.
Identification of the modules of interest and enrichment analysis
To obtain specific modules that are related to WY-14643 treatment, and PPAR-α activity in the liver, this study evaluated the modular trait association between the 6 modules and phenotypic traits. Based on the results most modules were negatively associated with WY-14643 treatment, especially two modules the ME brown (r = − 0.94, P = 6e−04), and the ME turquoise (r = − 0.84, P = 0.009). We designed our candidate hub genes between those co-expressed genes from the brown module and those downregulated DEGs with |logFC|> 1, and p < 0.05 and eventually screened 30 overlapping genes as candidate hub genes that may play an important role in the activation of PPAR-α after treatment with WY-14643. The DEGs with logFC < 0 or logFC > 0 were considered downregulated or upregulated genes, respectively (Fig. 3A–C).

- Identification of module trait relationship and KEGG analysis. Module-trait heatmap of the correlation between the clustering gene module and WY-14643 treatment (A). The scatter plot of module brown has the strongest negative correlation with the WY-14643 treatment (B). Venn diagram showing our 30 candidate hub genes from the intersection of genes between the brown module and downregulated DEGs (C). The construction of the PPI network (D). Cytohubba algorithm was used to rank our hub genes, the deeper colors represent the higher-ranking score (E). KEGG pathway analysis of candidate hub genes using the R language cluster Profiler package (F), identifying the TFs related to hub genes expression (G).
We used the STRING online tool to construct a PPI network of overlapping hub genes and then to find our main core genes we visualized highly ranked genes using Cytoscape software, Cytohubba plugins (Fig. 3D,E), (Table1). KEGG enrichment analysis data indicate that our selected hub genes were mostly enriched in viral protein interaction with cytokines, cytokine receptors, toll-like receptor signaling, and NF-κB signaling pathway, and less in primary immunodeficiency and ferroptosis (Fig. 3F), and indicate the transcription factors (TFs) related to each hub genes to gain further insights for understanding gene regulatory networks (Fig. 3G).
Analyzing the correlation of GBP2 with liver cancer clinical outcome
After ranking our 30 hub genes using the cytohubba plugin we especially focused on GBP2 as the first-ranked core genes in our study. GBP2 is a member of Guanylate-Binding Proteins (GBPs) which can be considered as a key indicator of high immunogenicity in most cancer types and predict immunotherapeutic responses25. However, there are few studies on the role of GBP2 in immune properties especially in liver cancer therefore in this study we examined the clinical relevance of GBP2 in HCC and explored its effect on immune properties by evaluating its effect on liver cancer immune cell infiltration. Based on the result GBP2 is highly expressed in liver cancer in both RNA and protein levels and was significantly correlated with tumor stage, and higher age, it is negatively correlated with CPT1A, and its higher expression can increase tumor cell infiltration score, especially by increasing macrophage and downregulating neutrophils and Th17 in tumor tissue microenvironments (Fig. 4A–F).

Higher GBP2 levels are correlated with lower liver cancer clinical outcomes. Higher GBP2 expression is linked with poor patient outcomes, higher stage, grade, and age (A–D) higher GBP2 expression is correlated with higher tumor immune infiltration score via decreasing Th17, neutrophil, monocyte, CD4, CD8 native in HCC (E) immunohistochemistry staining also indicate the higher GBP2 expression in HCC tumors compare to normal tissues (F).
Discussion
Hepatocellular carcinoma (HCC) is considered a highly prevalent cancer globally, necessitating the quick identification of relevant diagnostic biomarkers1,26. Peroxisome proliferator-activated receptor α (PPARα) a nuclear hormone receptor plays pivotal roles in the regulation of fatty acid homeostasis, lipid metabolism as well as inflammation, and immune response in the liver by regulating genes related to fatty acid transport and oxidation, high-density lipoprotein (HDL) metabolism, and ketone synthesis that occur in mitochondria, PPARα preserves lipid metabolism and homeostasis27,28 PPAR-α activation also increases HDL cholesterol and serum triglyceride clearance, along with energy production10,29. Furthermore, there is increasing evidence that PPAR-α can either induce or repress the expression of specific target genes involved in metabolism in different tissues regulates the metabolism of carbohydrates and amino acids, and plays a significant role in the starvation response30, therefore dysregulation of PPAR-dependent pathways is associated with metabolic disorders or cancer13,31,16. At the moment both anti- and pro-tumorigenesis properties have been reported for PPAR-α, but in total, PPARα activities are primarily thought to be anticancer in humans with the ability to suppress oncogenic roles of nuclear factor kappa B (NF-κB) and Akt9,32. Interestingly poor tumor cell adaptation to PPARα-mediated anti-inflammatory response and fatty acid oxidation may lead to reduced proliferation and higher apoptosis and autophagic cell death9,32. Therefore, PPAR agonists and PPAR-α modulators showed great capability to manage the process of cancer33. To gain a comprehensive understanding of the hub genes related to PPARα activity after treatment with PPARα agonist (WY-14643) in the liver tissue, by using WGCNA we created co-expression networks to identify new hub genes that are differentially expressed between treated samples compared to non-treated-groups-and-carry-out-gene-specific-analysis to validate the role of selected core gene in HCC. By using this approach, we discovered 30 intersect genes as the hub genes showing a highly negative correlation with WY-14643 treatment compared to non-treated groups, the KEGG analysis results revealed that key genes were mostly enriched in viral protein interaction with cytokines and cytokine receptors and toll-like receptor and NF-κB signaling pathway. Cytohubba plugin was used to detect highly ranked hub genes as our core genes based on their ranking score Fig. 3E. Finally, we select GBP2 as the member of the GBPs family, a family of interferon-induced GTPases that is highly activated in response to stimulation such as inflammatory cytokines, IFN α/β/γ, tumor necrosis factor–α, and toll-like receptor agonists34, and crucial for autonomous immunity against microbial pathogens, inflammation, and cancer26,35. The molecular mechanism that links GBP2 and cancer is still unknown especially based on its heterogeneous roles in different tissues36. However, elevated GBP2 expression is a marker for esophageal squamous cell carcinoma (ESCC)37,38, and can encourage GBM invasion through the Stat3/FN1 cascade39. Additionally, GBP2 has been demonstrated to be a pancreatic adenocarcinoma prognostic biomarker40. Since very few studies have been done on the role of GBP2in HCC, we selected GBP2 to get a better insight into its role in HCC pathogenesis which indicates higher GBP2 expression in tumors is closely associated with HCC tumor cell proliferation, proving its suggesting role in regulating cancer metastasis25. Therefore, GBP2 is emerges as a novel therapeutic strategy for HCC.
Conclusion
In conclusion, this study through co-expression network analysis, indicates the potential role of GBP2 as the therapeutic target associated with PPARα activity and corelating with lower HCC clinical outcomes. However, more experiments are required to validate its specific relation in the future.
Data availability
The data from the current study can be provided based on the request from the corresponding author. The gene expression data that support our results are available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE71731.
References:
Lee, S., Mardinoglu, A., Zhang, C., Lee, D. & Nielsen, J. Dysregulated signaling hubs of liver lipid metabolism reveal hepatocellular carcinoma pathogenesis. Nucleic Acids Res. 44, 5529 (2016).
AmeliMojarad, M., AmeliMojarad, M. & Cui, X. Prospective role of PD-1/PD-L1 immune checkpoint inhibitors in GI cancer. Pathol. Res. Pract. 244, 154338 (2023).
Paul, B., Lewinska, M. & Andersen, J. B. Lipid alterations in chronic liver disease and liver cancer. JHEP Rep. 4, 100479 (2022).
Calvisi, D. F. et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 140, 1071-1083.e5 (2011).
Peck, B. & Schulze, A. Lipid metabolism at the nexus of diet and tumor microenvironment. Trends Cancer 5, 693–703 (2019).
Olzmann, J. A. & Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 20(3), 137–155 (2018).
Pitso, L., Mofokeng, T. R. P. & Nel, R. Dyslipidaemia pattern and prevalence among type 2 diabetes mellitus patients on lipid-lowering therapy at a tertiary hospital in central South Africa. BMC Endocr. Disord. 21, 1–10 (2021).
Nagarajan, S. R., Butler, L. M. & Hoy, A. J. The diversity and breadth of cancer cell fatty acid metabolism. Cancer Metab. https://doi.org/10.1186/s40170-020-00237-2 (2021).
Cheng, H. S., Yip, Y. S., Lim, E. K. Y., Wahli, W. & Tan, N. S. PPARs and tumor microenvironment: The emerging roles of the metabolic master regulators in tumor stromal-epithelial crosstalk and carcinogenesis. Cancers 13, 2153 (2021).
Auwerx, J., Schoonjans, K., Fruchart, J. C. & Staels, B. Regulation of triglyceride metabolism by PPARs: Fibrates and thiazolidinediones have distinct effects. J. Atheroscler. Thromb. 3, 81–89 (1996).
Wang, R. et al. WY-14643 attenuates lipid deposition via activation of the PPARα/CPT1A axis by targeting Gly335 to inhibit cell proliferation and migration in ccRCC. Lipids Health Dis. 21, 1–18 (2022).
Peters, J. M., Cattley, R. C. & Gonzalez, F. J. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis 18, 2029–2033 (1997).
Evans, R. M., Barish, G. D. & Wang, Y. X. PPARs and the complex journey to obesity. Nat. Med. 10, 355–361 (2004).
Stienstra, R. et al. Peroxisome proliferator-activated receptor alpha protects against obesity-induced hepatic inflammation. Endocrinology 148, 2753–2763 (2007).
Costet, P. et al. Peroxisome proliferator-activated receptor alpha-isoform deficiency leads to progressive dyslipidemia with sexually dimorphic obesity and steatosis. J. Biol. Chem. 273, 29577–29585 (1998).
Guerre-Millo, M. et al. PPAR-alpha-null mice are protected from high-fat diet-induced insulin resistance. Diabetes 50, 2809–2814 (2001).
Francque, S. et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 63, 164–173 (2015).
Pozzi, A. et al. Peroxisomal proliferator-activated receptor-alpha-dependent inhibition of endothelial cell proliferation and tumorigenesis. J. Biol. Chem. 282, 17685–17695 (2007).
Panigrahy, D. et al. PPARα agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc. Natl. Acad. Sci. U. S. A. 105, 985–990 (2008).
Wang, Y., Nakajima, T., Gonzalez, F. J. & Tanaka, N. PPARs as metabolic regulators in the liver: Lessons from liver-specific PPAR-null mice. Int. J. Mol. Sci. 21, 2061 (2020).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Zhang, J. et al. Guanylate-binding protein 2 regulates Drp1-mediated mitochondrial fission to suppress breast cancer cell invasion. Cell Death Dis. 8(10), e3151–e3151 (2017).
AmeliMojarad, M., AmeliMojarad, M., Cui, X. & Shariati, P. Pan-cancer analysis of CTNNB1 with potential as a therapeutic target for human tumorigenesis. Inform. Med. Unlocked 42, 101331 (2023).
Yousefnia, S., Momenzadeh, S., Seyed Forootan, F., Ghaedi, K. & Nasr Esfahani, M. H. The influence of peroxisome proliferator-activated receptor γ (PPARγ) ligands on cancer cell tumorigenicity. Gene 649, 14–22 (2018).
Cheng, H. S. et al. Exploration and development of PPAR modulators in health and disease: An update of clinical evidence. Int. J. Mol. Sci. 20, 5055 (2019).
Pyper, S. R., Viswakarma, N., Yu, S. & Reddy, J. K. PPARalpha: Energy combustion, hypolipidemia, inflammation and cancer. Nucl. Recept. Signal 8, nrs-08002 (2010).
Han, L., Shen, W. J., Bittner, S., Kraemer, F. B. & Azhar, S. PPARs: Regulators of metabolism and as therapeutic targets in cardiovascular disease. Part II: PPAR-β/δ and PPAR-γ. Future Cardiol. 13, 279–296 (2017).
Tan, Y. et al. PPAR-α modulators as current and potential cancer treatments. Front. Oncol. 11, 599995 (2021).
Grabacka, M. & Reiss, K. Anticancer properties of PPARalpha-effects on cellular metabolism and inflammation. PPAR Res. https://doi.org/10.1155/2008/930705 (2008).
Tan, Y. et al. PPAR-α modulators as current and potential cancer treatments. Front. Oncol. https://doi.org/10.3389/fonc.2021.599995 (2021).
Degrandi, D. et al. Murine guanylate binding protein 2 (mGBP2) controls Toxoplasma gondii replication. Proc. Natl. Acad. Sci. U S A 110, 294–299 (2013).
Tretina, K., Park, E. S., Maminska, A. & MacMicking, J. D. Interferon-induced guanylate-binding proteins: Guardians of host defense in health and disease. J. Exp. Med. 216, 482–500 (2019).
Kim, B. H., Shenoy, A. R., Kumar, P., Bradfield, C. J. & MacMicking, J. D. IFN-inducible GTPases in host defense. Cell Host Microbe 12, 432 (2012).
Guimarães, D. P. et al. Interferon-inducible guanylate binding protein (GBP)-2: A novel p53-regulated tumor marker in esophageal squamous cell carcinomas. Int. J. Cancer 124, 272–279 (2009).
Luo, Y., Jin, H., Kim, J. H. & Bae, J. Guanylate-binding proteins induce apoptosis of leukemia cells by regulating MCL-1 and BAK. Oncogenesis 10(7), 1–12 (2021).
Yu, S. et al. GBP2 enhances glioblastoma invasion through Stat3/fibronectin pathway. Oncogene 39, 5042–5055 (2020).
Liu, B. et al. GBP2 as a potential prognostic biomarker in pancreatic adenocarcinoma. PeerJ 9, e11423 (2021).
Acknowledgements
Not applicable
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Ma.A. performed the bioinformatic analysis Ma.A and Me.A contribute equally to drafting, writing, editing, and revising the final submitted version. X.C. supporting the whole process as corresponding author . All the authors read the submitted version and approved it.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
AmeliMojarad, M., AmeliMojarad, M. & Cui, X. Weighted gene co-expression network analysis identified GBP2 connected to PPARα activity and liver cancer. Sci Rep 14, 20745 (2024). https://doi.org/10.1038/s41598-024-70832-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-70832-6
- Springer Nature Limited