
Abstract
Liver cancer, classified as a malignant hepatic tumor, can be divided into two categories: primary, originating within the liver, and secondary, resulting from metastasis to the liver from other organs. Hepatocellular carcinoma (HCC) is the main form of primary liver cancer and the third leading cause of cancer-related deaths. The diagnosis and prognosis of HCC using current methods still face numerous challenges. This study aims to develop novel diagnostic and prognostic models while identifying new biomarkers for improved HCC treatment. Diagnostic and prognostic models for HCC were constructed using traditional binary classification methods and machine learning algorithms based on the TCGA database (Downloaded in August 2023). The mechanisms by which APLN (Apelin) affects HCC were investigated using single-cell sequencing data sourced from the GEO database (GSE149614). The diagnostic models yielded by various algorithms could effectively distinguished HCC samples from normal ones. The prognostic model, composed of four genes, was constructed using LASSO and Cox regression algorithms, demonstrating good performance in predicting the three-year survival rate of HCC patients. The HCC biomarker Apelin (APLN) was identified in this study. APLN in liver cancer tissues mainly comes from endothelial cells and is associated with the carcinogenesis of these cells. APLN expression is significantly upregulated in liver cancer tissues, marking it as a viable indicator of endothelial cell malignancy in HCC. Furthermore, APLN expression was determined to be an independent predictor of tumor endothelial cell carcinogenesis, unaffected by its modifications such as single nucleotide variation, copy number variation, and methylation. Additionally, liver cancers characterized by high APLN expression are likely to progress rapidly after T2 stage. Our study presents diagnostic and prognostic models for HCC with appreciably improved accuracy and reliability compared to previous reports. APLN is a reliable HCC biomarker and contributes to the establishment of our models.
Similar content being viewed by others
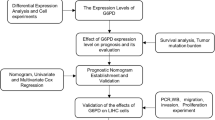


Introduction
Hepatocellular carcinoma (HCC), the major form of primary liver cancer, is the third leading cause of cancer-related mortality, with a 5-year relative survival rate of approximately 18%1. The major risk factors for HCC involve chronic alcohol consumption, hepatitis B or C virus infection, and non-alcoholic fatty liver disease2. Early detection is crucial since it allows for timely intervention and potentially curative treatment options, such as surgical resection, liver transplantation, or local ablation therapies, leading to a high survival rate in HCC patients3. It was revealed that the 5-year survival rate after curative treatment for patients diagnosed in the early stage is more than 50%, whereas the 5-year survival rate for patients diagnosed in the advanced stage remains as low as less than 5%4. However, only 20% of HCC patients are diagnosed in the early stage with the chance to receive curative treatment; leaving the rest diagnosed at advanced stages unsuitable for such interventions5. The poor early-stage detection rate primarily stems from the low sensitivity and specificity of existing diagnostic biomarkers, such as alpha-fetoprotein (AFP)6. Therefore, new biomarkers are in great need to enhance the accuracy of early HCC detection.
The development of a reliable prognostic method that involves the prediction of the progression and survival of patients is significant and important. Such a method can aid in treatment decision-making, provide patients with valuable prognostic information, contribute to personalized medicine approaches, and facilitate both research and clinical trials7. In the case of HCC, up to 70% of patients still experience intrahepatic recurrence within 5 years, even after the curative treatment8. Therefore, there is an urgent need for a reliable prognostic evaluation method to predict the outcomes of HCC patients. Thanks to the remarkable advances in rapid high-throughput sequencing technology and the accumulation of various "omics" data, screening for HCC biomarkers has become feasible and necessary9. More and more gene-based risk score models for prognosis by specific gene screening come to light, further enhancing our understanding of the molecular mechanisms underlying HCC pathogenesis.
The PI3K/Akt pathway plays a significant role in the development and progression of HCC. Activation of this pathway has been observed in HCC and is associated with various cellular processes, including cell growth, survival, migration, and invasion10. It is reported that Apelin (APLN), a gene up-regulated in HCC, promotes HCC progression through the activation of the PI3K/Akt pathway, leading to increased expression of phospho-glycogen synthase kinase 3β (p-GSK3β) and cyclin D1. Inhibition of APLN with a small-molecule inhibitor was found to be effective in inhibiting the APLN-PI3K/Akt cascade and HCC growth11. These findings suggest that APLN could serve as a crucial factor in the diagnosis or prognosis of HCC.
In this study, we conducted an analysis of the data acquired from The Cancer Genome Atlas (TCGA) database to identify potential disease-related genes for the diagnosis and prognosis of HCC. With the help of LASSO Logistic regression, LASSO Cox regression and machine learning algorithms, we have developed several models, many of which exhibited high accuracy and reliability. Furthermore, APLN was identified as a biomarker for HCC in this study. The expression distribution and underlying mechanism were explored in depth through an investigation of the single-cell dataset obtained from the GEO149614 database.
Materials and methods
Data source and availability
All the original data were acquired from public databases such as The Cancer Genome Atlas (TCGA), Genotype Tissue Expression (GTEx), International Cancer Genome Consortium (ICGC) and Gene Expression Omnibus (GEO, RRID:SCR_005012). The TCGA-LIHC dataset consists of 424 samples (374 tumor samples and 50 adjacent normal samples). The GTEx dataset consists of 110 normal samples. The ICGC-LIRI dataset consists of 199 tumor samples. The single-cell dataset, which was obtained from the GEO database (GSE149614), encompasses samples from primary tumors, portal vein tumor thrombus (PVTT), metastatic lymph nodes, and non-tumor liver tissues. The list of 1793 immune-related genes was acquired from the ImmPort database (RRID:SCR_012804, https://www.immport.org/home).
Analysis of differentially expressed genes
The TCGA-LIHC RNA-seq data was filtered to retain only mRNA data and discard genes with zero expression across all samples. Subsequently, the differential expression analysis, using DESeq2 R package (version 1.40.2), was applied to the normalized count data, which was categorized based on normal or tumor status. The criteria for statistical significance was determined as absolute log2 fold change (logFC) greater than 1.05 (or less than − 1.05) combined with a P-adjusted value (Padj) less than 0.05, resulting in the identification of 864 upregulated and 508 downregulated genes. For prognosis-related analysis, a prognostic COX regression analysis was conducted using a variance stabilizing transformation (VST)-normalized expression matrix, which led to the identification of 4272 genes with a significant correlation to prognosis (P < 0.05). Additionally, we incorporated a list of 1793 immune-related genes acquired from the ImmPort database. Genes that were concurrently present in all three categories (differentially expressed, prognosis-related, and immune-related) were classified as key differentially expressed genes, holding potential significance in the context of HCC.
Functional enrichment analysis
GO (gene ontology)/KEGG (Kyoto Encyclopedia of Genes and Genomes)12,13,14 enrichment analysis was conducted using the clusterProfiler (R package; version 4.8.3).
Protein–protein interaction network analysis
Protein–protein interaction (PPI) analysis was carried out using the STRING database (https://string-db.org), which covered almost all functional interactions between the expressed proteins, and interactions with a combined score > 0.4 were considered statistically significant.
Construction of diagnostic models
The TCGA-LIHC dataset and GTEx datasets were divided into a training set and a validation set at a ratio of 7:3 for optimal model training and validation. The identified 36-gene set was used in LASSO (least absolute shrinkage and selection operator) regression analysis to develop LASSO regression models. Additionally, the logistic regression models were developed using the rms R package (version 6.7-1). The LASSO feature screening process is visualized, and the final model is visualized as a nomogram. The calibration curve is used to show the relative relationship between the model risk and the actual risk. Finally, the AUC indicator is used to quantify the training performance or predictive ability of the model in a specific data set. Machine learning models were developed using a spectrum of algorithms, including Lasso, Ridge, Elastic Net (Enet), Stepglm, Support Vector Machine (SVM), glmBoost, Linear Discriminant Analysis (LDA), plsRglm, Random Forest, Gradient Boosting Machine (GBM), Extreme Gradient Boosting (XGBoost), and Naive Bayes. Specifically, 113 combinations were obtained, using one algorithm to select variables and another algorithm to build a classification prediction model under the framework of cross-validation.
Construction of prognostic models
In parallel with our diagnostic models, we employed the same set of key genes for LASSO regression analysis to construct prognostic LASSO regression models. The Cox regression models were developed using the survival R package (version 3.5.5) and rms R package (version 6.7-1). The LASSO feature screening process is visualized, and the final model is visualized as a forest plot and a nomogram. The calibration curve is used to show the relative relationship between the model risk and the actual risk. Finally, the C-index or AUC indicator is used to quantify the training performance or predictive ability of the model in a specific data set. Machine learning models were developed using algorithms such as Lasso, Ridge, Elastic Net (Enet), StepCox, survivalSVM, CoxBoost, SuperPC, plsRcox, Random Survival Forests (RSF), and Gradient Boosting Machine (GBM). Specifically, 101 combinations were obtained, and regression models were constructed after screening for characteristics (some combinations had no feature screening capabilities).
Immune infiltration analysis
CIBERSORT R package (version 0.1.0) was employed to estimate immune cell scores within complex tissue samples. Utilizing the LM22 feature matrix, which comprises 547 genes that distinctly characterize 22 different immune cell phenotypes, we were able to achieve a detailed profiling of the immune landscape. The approach provided insights into the varying levels of immune cell infiltration associated with high-risk and low-risk disease states.
Single-cell RNA-seq analysis
Seurat was used for processing and preparing the data for the following analyses. We filtered out low-quality cells, excluding those expressing less than 500 or more than 8000 total genes, as well as cells with more than 20% mitochondrial RNA contents. The SCTransform, RunPCA, and RunUMAP functions from the Seurat R package (version 4.3.0.1) were used for normalization and dimensionality reduction. The harmony was used to correct batch effects across different arrays. Subsequently, the FindNeighbors and FindClusters functions from Seurat R package (version 4.3.0.1) were used for cell cluster identification and differentiation. To optimize the cluster analysis, we set the dimension parameter to 27 (dims = 27) and the resolution parameter to 0.3. The HGNChelper R package (version 0.8.1) was used to aid in the identification of cell subpopulations. Pseudotime analysis was conducted with the monocle R package (version 2.28.0). We utilized the “DDRTree” method to reduce the dimension and “orderCells” to determine cell differentiation state. CellChat R package (version 1.6.1) was used to process the scRNA-seq data for the inference, visualization, and analysis of cell–cell communication, as well as the depiction of the interactions between ligands, receptors, and their cofactors.
Gene mutation, copy number variation, and methylation analysis
Gene mutation, copy number variation, and methylation analysis were performed using tools such as gene set cancer analysis (GSCA, http://bioinfo.life.hust.edu.cn/GSCA/) and cBioPortal (https://www.cbioportal.org/). These platforms are open resource tools specifically designed for the detailed analysis and visualization of multiple cancer genome datasets.
Statistical analysis
R software (version 4.3.0) along with its associated packages were employed for statistical analysis. The creation of most graphics, including complex visual representations, was achieved using the ggplot2 package, supplemented by the built-in mapping functions of other packages. Unless specified, we defined statistical significance as a p-value less than 0.05. For the TCGA-LIHC RNA-seq data analysis, we utilized the count expression matrix in our differential analysis, as is required by the DESeq2 R package (version 1.40.2). We used the LOG2 (expression + 1) transformation to obtain expression matrices of smaller orders of magnitude.
Results
Establishment and analysis of a geneset for model construction
To construct high-quality diagnostic and prognostic models for HCC, it's imperative to first curate a purpose-specific dataset. To improve the quality of this dataset and limit the number of genes included, we set three criteria for the gene selection: (1) The expressions of the included genes should be significantly different between tumor and normal tissues; (2) The expressions of the included genes should correlate with patient survival in the tumor cohort; and (3) The included genes should be classified as immune-related factors.
Our primary data was sourced from TCGA-LIHC dataset, which comprises 424 samples, including 50 from normal tissues and 374 from tumor tissues. Through Principal Component Analysis (PCA), we observed a distinct separation of samples into two clusters, clearly differentiating normal and tumor tissues, as illustrated in our PCA plot (Fig. 1A). Our differentially expressed genes (DEG) screening identified 1372 genes (coding genes), among which 864 genes were upregulated and 508 genes were downregulated in tumor tissues compared to normal tissues, as depicted in the volcano plot (Fig. 1B). Then, a batch Cox regression analysis was used to explore the relationship between gene expression and patient survival outcome in the tumor samples, leading to the identification of 4527 genes that significantly correlated with patient survival rates. Subsequently, a list of 1793 immune-related genes was obtained from the ImmPort database. Finally, a geneset containing 36 genes was established by the intersection of the above three gene sets, which would be used for the subsequent development of the diagnostic and prognostic models (Fig. 1C).

Establishment of a 36-gene dataset for model construction. (A) Principal component analysis (PCA) on TCGA-LIHC dataset after sample filtering. (B) Volcano plot of DEGs between HCC and normal liver tissues. (C) Venn diagram demonstrating the selection of 36 genes for subsequent model development. (D,E) GO/KEGG enrichment analysis of the identified 36 genes. (F) PPI network analysis of 36 genes, 29 of which were related (combined score > 0.4).
According to the results of GO and KEGG analyses, these 36 genes are involved in biological pathways related to development and growth (such as morphogenesis and axonogenesis regulation), as well as molecular functions such as receptor ligand activity. Most of these genes are implicated in receptor ligand activity, signaling receptor activation, or protein modification processes (Fig. 1D,E). We also conducted a Protein–Protein Interaction (PPI) network analysis and found that 29 of the 36 genes exhibit a moderate to high correlation with a combined score > 0.4, as illustrated in Fig. 1F. Collectively, the enrichment and PPI network analyses not only shed light on the operating mechanisms and intrinsic relationships of these genes but also underscore their potentially critical role in distinguishing between tumor and normal tissues.
Establishment and comparison of the diagnostic models
After obtaining the 36-gene set, we tried to construct an early diagnostic model for HCC. We divided the TCGA-LIHC dataset and GTEx-Liver dataset into training and validation cohorts at a 7:3 ratio, with the former designated for model development and the latter for validation. Our initial analysis focused on the expression correlations of these 36 genes. Based on our experience, genes with low correlations are advantageous for binary classification models (Fig. 2A). Using LASSO regression, we refined the gene set from 36 down to 12 (Fig. 2B,C). We then further narrowed it down to 7 key genes—MAPT, VIPR1, TNFRSF4, CCR1, BIRC5, RFX5, and APLN—through logistic regression analysis (Fig. 2D). The logistic regression model, expressed as Risk Score = (-1.00 × CCR1 expression) + (1.77 × RFX5 expression) + (1.01 × APLN expression) + (0.49 × MAPT expression) + (0.82 × TNFRSF4 expression) + (0.72 × BIRC5 expression) – (0.52 × VIPR1 expression), demonstrated remarkable predictive capacity and accuracy, as evidenced by the decision curve and calibration analysis (Fig. 2E). The model's Area Under Curve (AUC) was 0.996 in both the training and validation cohorts (Fig. 2F), and it maintained robust performance when validated on an external cohort (Fig. 2G).

Evaluation and comparison of the diagnostic models. (A) Correlation of expression levels of 36 genes. (B,C) Gene selection via the Lasso algorithm, focusing on genes selected at the lambda-min for further analysis. (D) Construction of a nomogram model based on seven key genes included in the logistic regression model. (E) Calibration curve for the logistic regression model. (F) Receiver operating characteristic (ROC) curves for the logistic regression model in both training and validation cohorts, with the DeLong test confirming consistency. (G) ROC curves for the logistic regression model across test cohorts (including GSE69715, GSE102079, GSE76427), demonstrating predictive reliability. (H) Top 20 diagnostic models built with machine learning algorithms.
To further refine the diagnostic models for HCC, we embraced the latest popular machine learning algorithms. Experimenting with 113 combinations of 12 different machine learning binary prediction algorithms based on the 36-gene set, we achieved impressive results, as depicted in Fig. 2H and Supplementary Fig. S1. The models ranked first and second, the plsRglm model and Ridge model, involved the full suite of 36 genes. The model ranked third, a combination of Random Forest (RF) and Naive Bayes algorithms, also showed superior predictive performance compared to our logistic regression model. This model utilized a more concise 16-gene set, comprising VIPR1, BIRC5, SEMA3F, GHR, RFX5, APLN, MDK, ESR1, TNFRSF4, MAPT, JUN, CCR1, RFXANK, PIK3R2, ADM, and PSMD4. Impressively, it attained an AUC of 0.993 in the training cohort, 0.991 in the validation cohort, and exceptional performance in three external cohorts with AUCs of 0.999, 0.966, and 0.964 (Fig. 2H).
In conclusion, our research successfully established an early diagnostic model for HCC using a machine learning approach. This model, based on a 16-gene set, is built using the RF + NaiveBayes algorithms. It has demonstrated remarkable performance in both the training and validation cohorts, effectively distinguishing HCC from normal tissue samples with high precision. Utilizing the predict function in the R language environment and our training cohort, this model can be easily applied to clinical diagnostics. In practical terms, when the model predicts a probability greater than 0.5, the sample is classified as tumorous, providing a straightforward and reliable tool for early HCC detection.
Establishment and comparison of the prognostic models
In our research, we extensively evaluated the prognostic significance of genes within our 36-gene set, utilizing the comprehensive clinical data from TCGA. Prior survival correlation analysis underscored the potential of these genes as independent prognostic indicators. However, given patient heterogeneity and varied tumor subtypes, we recognized the need for a multivariate model to enhance prognostic accuracy.
First, we screened out the top 7 survival-correlated genes from the 36-gene set through LASSO regression (Fig. 3A,B), including SPP1, BIRC5, APLN, MAPT, PLXNA1, NDRG1 and CACYBP. Using the stepwise COX regression algorithm, we refined this to a 5-gene set. Yet, as CACYBP didn't meet the proportional-hazards assumption, we established a 4-gene model (Fig. 3C–E), with each gene proving to be an independent prognostic factor. This model, meeting the proportional-hazards assumption (global p = 0.352), is defined as Risk Score = (0.087 × SPP1 expression) + (0.208 × BIRC5 expression) + (0.177 × APLN expression) + (0.276 × PLXNA1 expression). It is shown that this model performed well in the training cohort, with an AUC of 0.748 at 1-year, 0.685 at 2-year, and 0.671 at 3-year (Fig. 3F). It also performed well in the validation cohort and the external test cohort. The validation cohort had an AUC of 0.748 at 1-year, 0.722 at 2-year, and 0.696 at 3-year (Fig. 3G), the external test cohort had an AUC of 0.620 at 1- year, 0.623 at 2-year, and 0.632 at 3-year (Fig. 3H). Overall, the four-factor Cox regression model has practical decision-making value.

Evaluation and comparison of the prognostic models. (A,B) Gene selection via the Lasso algorithm, focusing on genes selected at the lambda-min for further analysis. (C) Assessment of individual factor performance within the multi-Cox model. (D) Calibration curve for the multi-Cox model. (E) Construction of a nomogram model based on four key genes included in the multi-Cox model. (F) ROC curves for the Cox regression model in training cohorts. (G) ROC curves for the Cox regression model in validation cohorts. (H) ROC curves for the Cox regression model in test cohorts ICGC-LIRI. (I) Top 20 prognostic models built with machine learning algorithms. (J) ROC curves for the multi-Cox model across different age groups (youth, middle, and old), with age thresholds set at 45 and 65 years. (K) ROC curves for the multi-Cox model distinguishing between male and female. (L) ROC curves for the multi-Cox model distinguishing between early and advanced HCC stages.
We also tried to explore additional prognostic models with the help of machine learning algorithms, testing 101 combinations of ten machine learning algorithms. While several machine learning-based models outperformed the Cox regression model (Fig. 3I and Supplementary Fig. S2), the top-ranked Ridge model, using numerous genes, demonstrated superior AUC performance across all cohorts. The training cohort had an AUC of 0.775 at 1-year, 0.708 at 2-year, and 0.699 at 3-year, the validation cohort had an AUC of 0.756 at 1-year, 0.771 at 2-year, and 0.716 at 3-year, the external test cohort had an AUC of 0.637 at 1- year, 0.647 at 2-year, and 0.637 at 3-year. Nevertheless, considering the complexity, the streamlined 4-factor Cox model maintains its significance for practical decision-making in prognostics.
Performance evaluation of the prognostic model
We evaluated the performance of the prognostic model by AUC across various conventional clinical subgroups, including T stage, gender, and age. Samples were divided into male or female groups according to gender, and young (below 45 years), middle-aged (45–65 years) or old (above 65 years) groups according to age. Additionally, samples were divided into early-stage (T1 and T2 stages) or advanced-stage (T3 and T4 stages) groups according to T stage. The results show good performance across all these clinical subgroups, suggesting the model's robust discriminatory capability. Notably, we found that the model was particularly effective in young patients, males, and those in advanced stages of HCC (Fig. 3J–L).
APLN is a gene closely associated with HCC
In our DEG analysis and model construction, APLN emerged as a noteworthy gene, frequently featured in various models and integral to our finalized diagnostic and prognostic models, which has received less attention in HCC and plays role in HCC is not enough clear. This led us to a detailed investigation of APLN. Firstly, we reviewed our enrichment analysis findings, focusing on those related to APLN. The results primarily connected APLN to receptor ligand activity, G protein-coupled receptors, and cell proliferation. Considering its linkage with liver cancer, we speculated that APLN-associated receptor ligand activity alterations could lead to endothelial cell proliferation (Fig. 4A). Secondly, we explored APLN's differential expression across a spectrum of cancers. Pan-cancer analysis showed that APLN's expression significantly differed in many cancers relative to normal tissues, predominantly displaying upregulation in tumors. Notable exceptions included kidney renal papillary cell carcinoma (KIRP), lung adenocarcinoma (LUAD), and lung squamous cell carcinoma (LUSC), where APLN was significantly downregulated (Fig. 4B,C), suggesting diverse regulatory mechanisms of APLN across different cancer types. In the context of HCC, we found a significant upregulation of APLN (Fig. 4D), corroborated by our paired sample analysis, indicating APLN upregulation as a potential distinguishing feature of HCC.

Correlation of APLN expression with different factors. (A) APLN-related items in enrichment analysis. (B) Comprehensive analysis of APLN expressions between tumor and normal tissues in a variety of cancers. (C) Specific analysis of APLN expressions between tumor and matched paracancerous tissues in a variety of cancers. (D) Focused analysis of APLN expressions between tumor and matched paracancerous tissues in liver cancer. The correlation analyses of APLN expression with age (E), gender (F), and T stage (G).
We further analyzed APLN expression across various clinical subgroups categorized according to T stage, age, and gender. Samples were divided into three age groups: under 45, between 45 and 65, and above 65 years. Additionally, samples were divided into male and female groups based on gender or four groups representing T1–T4 stages. Our findings revealed stable APLN expression across these subgroups, except for a significant difference within early T stage (T1 and T2 stages). This consistent expression pattern of APLN, irrespective of age, gender, and advanced T stage (T3 and T4), underscores its potential as a robust biomarker for HCC, indicating its stability and reliability independent of conventional clinical factors (Fig. 4E–G).
Investigation of APLN in single-cell sequencing data
To further investigate the potential mechanisms underlying the effects of APLN on HCC and their corresponding cell subsets, we took an in-depth look at single-cell sequencing data. Our analysis is based on the scRNA-seq dataset GSE149614. After data filtration, we analyzed the remaining 62,774 valid cells and 24,879 genes from both tumor and normal tissue samples across multiple cases. Cells were classified into 28 distinct clusters by the FindClusters function and annotated into 9 types by common marker genes (Fig. 5A,B). Notably, we observed a significant reduction of NK cells in tumor tissue compared to normal tissue, while hepatocytes were markedly increased (Fig. 5C). This result is consistent in all seven paired sample analysis, illustrating changes in immune cell content and predominant proliferating cell types in HCC tissue, regardless of potential biases caused by uneven sampling. Next, we examined APLN expression across various cell clusters. In normal tissues, APLN showed minimal expression in endothelial cells. However, in tumor tissues, APLN was highly expressed in endothelial cells and presented weak expression in other cell types. This dramatical difference in APLN expression patterns between tumor and normal tissues, each sample's contribution to assessing APLN expression is depicted in Fig. 5D. Our results indicate that, despite minor variances across samples, the significant upregulation of APLN in tumor endothelial cells was a consistent and reliable observation (Fig. 5E).

Classification of cell types and analysis of APLN expressions across different cell types. (A,B) Identification of nine distinct cell types using common markers. (C) Proportions of different cell types in each sample. (D) Examination of APLN expression across different cell types in the scRNA-seq dataset. (E) Elevated APLN expression in tumor endothelial cells. The left panel is a normal sample, and the right panel is a tumor sample. Visualization of APLN expression in endothelial cells of clusters 5 and 17 from normal (F) and tumor (G) tissues.
During cell clustering, we observed that endothelia cells were divided into two clusters (clusters 5 and 17). Our analysis focused on the expression levels of APLN in these two clusters in both normal and tumor tissues. We found that in normal tissues, APLN expression was low in both clusters. However, in tumor tissues, it was cluster 5 that demonstrated a significantly elevated expression of APLN (Fig. 5F,G). This led us to conduct a DEG analysis of cluster 5 between normal and tumor tissues. Given that the expression level of APLN in cluster 17 showed minimal variation, we concentrated our DEG analysis exclusively on cluster 5. Although APLN wasn't the most significantly altered gene in this cluster, its differential expression was notably distinct and significant, indicating its potential role in the pathogenesis of HCC in specific endothelial cell subsets.
Subsequently, we divided endothelial cells into the APLNPos group (261 cells) and the APLNNeg group (1633 cells), based on the detection of APLN expression (Fig. 6A,B). Pseudotime trajectory analysis showed that APLNNeg cells diverge into two separate evolutionary paths: Branch A, primarily comprised of APLNNeg cells, and Branch B, where APLNPos cells are significantly enriched. Under the hypothesis that the expression level of APLN is related to endothelial cell cancerization, we considered that branch B (APLNPos cells) is a manifestation of endothelial cell carcinogenesis during the occurrence of HCC (Fig. 6C,D). We then examined the differential expression of APLN and established HCC markers from the CellMarker 2.0 database (http://bio-bigdata.hrbmu.edu.cn/CellMarker/index.html), such as VWF and CDH5 (They are known as markers of liver cancer in endothelial cells, as different from our study markers to verify whether APLN can serve as the endothelial cells of liver cancer marker), in all endothelial cells (Fig. 6E). The expression patterns of these genes were similar, suggesting that APLN, akin to VWF and CDH5, could be indicative of the carcinogenic state of endothelial cells. A subsequent DEG analysis of APLNPos and APLNNeg cells, paired with GO enrichment, revealed significant enrichment in biological processes, cellular components, and molecular functions closely linked to immune responses (Fig. 6F). This supports the hypothesis that APLNPos represents a carcinogenic branch of endothelial cells. KEGG pathway analysis underscored that the Apelin pathway, involving APLN, is a principal distinction between APLNPos and APLNNeg groups (Fig. 6G). When cells were grouped based on their origin from normal or tumor tissues and analyzed for differential gene expression with GO and KEGG enrichment, the results, predominantly associated with the extracellular matrix, appeared somewhat disordered (Supplementary Fig. S3A,B). This disarray may hint at the complexities of cell carcinogenesis and could be related to the diverse endothelial cell differentiation pathways observed in our pseudotime analysis.

Detailed analysis of endothelial cell subtypes, differentiation trajectories, gene enrichment, and cell–cell communication. (A) Dimension-reduced visualization of cell types from the scRNA-seq dataset, with endothelial cells categorized into APLNPos and APLNNeg groups. (B) APLN expression across various cell types. (C,D) Differentiation trajectory analysis of endothelial cells based on pseudotime, states, or cell types (APLNPos and APLNNeg). (E) The expression levels of APLN, VWF, and CDH5 in APLNPos and APLNNeg endothelial cells. (F) The biological processes, cellular components, and molecular functions which are enriched in the GO analysis of DEGs identified between APLNPos and APLNNeg endothelial cells. (G) KEGG analysis of DEGs identified between APLNPos and APLNNeg endothelial cells. (H) Cell types involved in Apelin pathway and the extent of their involvement. (I) Expression patterns of APLN and its receptor APLNR across various cell types. (J) Involvement of different cell types in the Apelin signaling pathway.
Finally, we conducted cell–cell communication analysis among endothelial cells (APLNPos and APLNNeg) and other types of cells (Supplementary Fig. S3C–E). After analyzing the Apelin pathway identified in the enrichment analysis, we found that endothelial cells are sole participants in this pathway, with APLNPos cells showing considerably more activity than their APLNNeg counterparts (Fig. 6H). APLN and its receptor APLNR are only highly expressed in APLNPos cells. Meanwhile, a certain amount of APLNR expression is found in APLNNeg cells, which suggests that the evolution of APLNNeg cells may be influenced by APLNPos cells (Fig. 6I,J). In addition, the APLNPos endothelial cells are more capable of communicating with other types of cells than the APLNNeg endothelial cells (Supplementary Fig. S3C).
Investigation into APLN’s role in HCC
We studied the correlation between APLN and liver cancer from multiple aspects. In the immune infiltration assay, we observed a strong correlation between the infiltration levels of specific immune cells and their presence in tumor samples (Supplementary Fig. S4A). We further investigated the relationship between APLN expression and immune infiltration within tumor samples. The result showed no significant correlation between APLN expression and most immune cells, except for a correlation with T cells CD4 memory resting cells and eosinoohils (Supplementary Fig. S4B). In the evaluation of APLN’s impact on the tumor microenvironment (TME), we discovered a mild correlation between APLN expression and tumor purity (Supplementary Fig. S4C,D). The impact of other possible APLN modifications on HCC was also investigated, and the results showed that these modifications (single nucleotide variation, copy number variation, and methylation) were not significantly associated with APLN expression or prognostic outcomes in HCC.
Taken together, our comprehensive analysis led to the conclusion that APLN expression mainly correlates with endothelial cells carcinogenesis. The upregulation of APLN expression during carcinogenesis appears to occur independently of the aforementioned genetic and epigenetic modifications.
Discussion
Hepatocellular carcinoma (HCC) is the most common cancer and the third leading cause of cancer-related mortality worldwide15. Such a high incidence of mortality is a result of the malignant characteristics of HCC, which are manifested in rapid progression, easy metastasis, poor treatment effect, and a high recurrence rate.
Besides the malignant characteristics of HCC, patients' survival is also heavily influenced by late diagnosis. Due to the complicated etiology of HCC and the different genetic backgrounds across individuals, it is difficult to achieve an early diagnosis16. Patients had usually entered the advanced stage of HCC at the time of diagnosis, with an overall 5-year survival rate as low as 10.1%17. Early diagnosis is significant for improving the survival rate of HCC patients. Conventional diagnostic methods encompass clinical symptom assessment, cross-sectional imaging, and the use of molecular biomarkers. With the advent of high-throughput sequencing technology and the accumulation of multiple "omics" data, more and more diagnostic models based on gene expression signatures have come into sight. For instance, Tang et al. developed a diagnostic model with two TMB-related genes (AUCs of 0.928 and 0.950 in training and validation cohorts, respectively)18. Long et al. established a diagnostic model with two DNA methylation-driven genes (AUCs of 0.978 and 0.981 in training and validation cohorts, respectively)19. Cheng et al. designed a diagnostic model based on machine learning algorithms (AUCs of 0.987 and 0.992 in training and validation cohorts, respectively)20. In this study, we have successfully developed a series of diagnostic models using either LASSO logistic regression or machine learning algorithms. After screening, the final model is based on the RF + NaiveBayes algorithm with sixteen genes. To the best of our knowledge, this model could almost perfectly distinguish HCC from normal samples in the training cohort (AUC = 0.993), validation cohort (AUC = 0.991) and test cohort (AUC = 0.999/0.966/0.964), which outperforms all previously reported models. Some machine learning models are better than this model but are less easy to use.
A reliable prognostic model is important for HCC patients by improving clinical outcomes, understanding the molecular mechanisms of the disease, and guiding personalized treatment approaches. In addition to the conventional models based on different clinical staging systems for HCC21, a number of gene-based risk score models have recently emerged. Liu et al. established a prognostic model utilizing six genes to predict the overall survival of HCC patients22. Chen et al. developed a prognostic model with nine immune-related genes that could effectively differentiate HCC from normal samples23. Tang et al. created a prognostic model based on three microvascular invasion-related genes, offering reliable predictions for recurrence-free and overall survival in HCC patients24. In this study, we have built a prognostic model comprising a 4-gene signature, demonstrating strong predictive performance for HCC patient survival over 3 years (in the training cohort, the AUC is 0.748 at 1-year, 0.685 at 2-year and 0.671 at 3-year; in the validation cohort, the AUC is 0.748 at 1-year, 0.722 at 2-year and 0.696 at 3-year). Compared with existing prognostic models, the performance level of our model is in the mid-to-upper range. The model also performed well in the test cohort. Some machine learning models are better than this model but are less easy to use.
We also assessed the association between liver cancer risk and common clinical categories such as age, gender and T stage. The result showed a very low correlation between age, gender and patient survival (p > 0.1 for both age and gender, Supplementary Fig. S5), despite HCC being more common in men than women25. In addition, T stage is significantly associated with cancer risk.
The expression distribution and mechanism of APLN, a gene that simultaneously appears in several diagnostic and prognostic models, were explored in this study. We found that its expression is significantly elevated in a range of tumors, including HCC. Indeed, its elevated expressions have already been reported in many kinds of tumors, such as liver cancer11,26, cholangiocarcinoma27, prostate cancer28, ovarian cancer29, and glioblastoma30. Interestingly, APLN expression is not uniformly upregulated across all tumors. We found that its expression was significantly downregulated in kidney renal papillary cell carcinoma, lung adenocarcinoma, and lung squamous cell carcinoma, indicating diverse roles of APLN in different cancers. Through the analysis of single-cell sequencing data, we further explored APLN's role in HCC. The results showed that APLN was predominantly expressed in endothelia cells of liver tissue, with its upregulation reflecting endothelial cell carcinogenesis in HCC development. Considering Apelin signaling pathway, expression of APLN in endothelial cells and the findings of Chen et al.'s11, we suggest that the enhanced Apelin pathway in APLNPos cells may trigger the downstream PI3K/Akt pathway to promote endothelial carcinogenesis during HCC development. This highlights the important role of APLN in the dynamics of cell communication in the tumor microenvironment. In addition, our results indicate that APLN expression only correlates with endothelial cell carcinogenesis before T2 stage.
Nevertheless, there are some limitations to our study. The potential molecular mechanisms of the genes used in our modeling, especially APLN, require further exploration through functional experiments both in vivo and in vitro to fully understand their roles and impacts in HCC. In addition, sample size or potential biases in data collection, random steps, and bias in data processing may cause potential weak biases.
Conclusions
In this study, we performed an integrated analysis of single-cell and bulk tissue RNA-seq data from HCC patients and established the diagnostic and prognostic models. The diagnostic model demonstrated an almost perfect ability to differentiate HCC from normal samples, while the prognostic model reliably predicted different survival outcomes within a 3-year timeframe with high accuracy. Our findings highlight the critical role of APLN, a diversely sourced and multifunctional secreted protein, within these models. APLN in liver cancer tissues mainly comes from endothelial cells and is associated with the carcinogenesis of these cells. Its upregulated expression in tumor endothelial cells was consistent and reliable and was not affected by self-modifications or conventional clinical factors. Additionally, we observed sustained high APLN expression levels in advanced T stage (T3 and T4 stages), suggesting that liver cancers characterized by high APLN expression are likely to progress rapidly after T2 stage.
When patients undergo transcriptome sequencing as part of their diagnostic process, our model incorporating APLN can facilitate timely diagnosis and assess survival risks. In addition, the consistent upregulation of APLN expression, a characteristic feature of hepatocellular carcinoma (HCC), suggests it as a potential therapeutic target. This opens new avenues for HCC treatment and underscores the critical role of endothelial cell carcinogenesis in liver cancer patients. However, further data and clinical evidence are required to support these implications.
In practice, the application of this model needs requires careful consideration. Firstly, using transcriptome analysis for diagnosis presents challenges in terms of cost and the complexity of data analysis technology. Secondly, models derived from database analyses might confuse batch effects with genuine biological differences when applied clinically. Therefore, these models need to be interpreted with caution and adjusted to reflect real-world conditions accurately, based on concrete data.
Data availability
All the original data were acquired from public databases such as The Cancer Genome Atlas (TCGA), Genotype Tissue Expression (GTEx), International Cancer Genome Consortium (ICGC) and Gene Expression Omnibus (GEO, RRID:SCR_005012). Further information is available from the corresponding author upon request.
Abbreviations
- AFP:
-
Alpha-fetoprotein
- APLN:
-
Apelin
- DEG:
-
Differentially expressed genes
- GEO:
-
Gene expression omnibus
- GO:
-
Gene ontology
- GSCA:
-
Gene set cancer analysis
- GTEx:
-
Genotype tissue expression
- HCC:
-
Hepatocellular carcinoma
- ICGC:
-
International cancer genome consortium
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- KIRP:
-
Kidney renal papillary cell carcinoma
- LASSO:
-
Least absolute shrinkage and selection operator
- LUAD:
-
Lung adenocarcinoma
- LUSC:
-
Lung squamous cell carcinoma
- p-GSK3β:
-
Phospho-glycogen synthase kinase 3β
- PCA:
-
Principal component analysis
- PPI:
-
Protein–protein interaction
- PVTT:
-
Portal vein tumor thrombus
- TCGA:
-
The cancer genome atlas
References
Vogel, A., Meyer, T., Sapisochin, G., Salem, R. & Saborowski, A. Hepatocellular carcinoma. The Lancet. 400, 1345–1362 (2022).
Yang, J. D. et al. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 16, 589–604 (2019).
Lurje, I. et al. Treatment strategies for hepatocellular carcinoma—a multidisciplinary approach. IJMS. 20, 1465 (2019).
Zhu, W.-W. et al. Evaluation of midkine as a diagnostic serum biomarker in hepatocellular carcinoma. Clin. Cancer Res. 19, 3944–3954 (2013).
Gu, Y. et al. CCL14 is a prognostic biomarker and correlates with immune infiltrates in hepatocellular carcinoma. Aging. 12, 784–807 (2020).
Nakagawa, T. et al. Glycomic analysis of alpha-fetoprotein L3 in hepatoma cell lines and hepatocellular carcinoma patients. J. Proteome Res. 7, 2222–2233 (2008).
Yang, C. et al. Prognosis and personalized treatment prediction in TP53-mutant hepatocellular carcinoma: An in silico strategy towards precision oncology. Brief. Bioinform. 22, 164 (2021).
Wang, L., Liu, B.-X. & Long, H.-Y. Ablative strategies for recurrent hepatocellular carcinoma. World J. Hepatol. 15, 515–524 (2023).
Liu, X.-N. et al. Multiple, “Omics” data-based biomarker screening for hepatocellular carcinoma diagnosis. WJG. 25, 4199–4212 (2019).
Saxena, N. K. et al. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 67, 2497–2507 (2007).
Chen, H. et al. APLN promotes hepatocellular carcinoma through activating PI3K/Akt pathway and is a druggable target. Theranostics. 9, 5246–5260 (2019).
Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 71, 209–249 (2021).
Fu, J. & Wang, H. Precision diagnosis and treatment of liver cancer in China. Cancer Lett. 412, 283–288 (2018).
Zeng, H. et al. Cancer survival in China, 2003–2005: A population-based study. Int. J. Cancer. 136, 1921–1930 (2015).
Tang, B. et al. Diagnosis and prognosis models for hepatocellular carcinoma patient’s management based on tumor mutation burden. J. Adv. Res. 33, 153–165 (2021).
Long, J. et al. DNA methylation-driven genes for constructing diagnostic, prognostic, and recurrence models for hepatocellular carcinoma. Theranostics. 9, 7251–7267 (2019).
Cheng, B., Zhou, P. & Chen, Y. Machine-learning algorithms based on personalized pathways for a novel predictive model for the diagnosis of hepatocellular carcinoma. BMC Bioinformatics. 23, 248 (2022).
Tellapuri, S., Sutphin, P. D., Beg, M. S., Singal, A. G. & Kalva, S. P. Staging systems of hepatocellular carcinoma: A review. Indian J. Gastroenterol. 37, 481–491 (2018).
Liu, G.-M., Zeng, H.-D., Zhang, C.-Y. & Xu, J.-W. Identification of a six-gene signature predicting overall survival for hepatocellular carcinoma. Cancer Cell Int. 19, 138 (2019).
Chen, W., Ou, M., Tang, D., Dai, Y. & Du, W. Identification and validation of immune-related gene prognostic signature for hepatocellular carcinoma. J. Immunol. Res. 2020, 1–14 (2020).
Tang, Y. et al. Identification and validation of a prognostic model based on three MVI-related genes in hepatocellular carcinoma. Int. J. Biol. Sci. 18, 261–275 (2022).
Villanueva, A. Hepatocellular carcinoma. N. Engl. J. Med. 380, 1450–1462 (2019).
Muto, J. et al. The apelin-APJ system induces tumor arteriogenesis in hepatocellular carcinoma. Anticancer Res. 34, 5313–5320 (2014).
Hall, C. et al. Inhibition of the apelin/apelin receptor axis decreases cholangiocarcinoma growth. Cancer Lett. 386, 179–188 (2017).
Wan, Y. et al. Dysregulated microRNA-224/apelin axis associated with aggressive progression and poor prognosis in patients with prostate cancer. Hum. Pathol. 46, 295–303 (2015).
Hoffmann, M., Fiedor, E. & Ptak, A. Bisphenol A and its derivatives tetrabromobisphenol A and tetrachlorobisphenol A induce apelin expression and secretion in ovarian cancer cells through a peroxisome proliferator-activated receptor gamma-dependent mechanism. Toxicol. Lett. 269, 15–22 (2017).
Harford-Wright, E. et al. Pharmacological targeting of apelin impairs glioblastoma growth. Brain. 140, 2939–2954 (2017).
Funding
This work was supported by grants from National Key Research and Development Program of China (2018YFE0204503).
Author information
Authors and Affiliations
Contributions
Conceptualization, Y.L. and X.J.; data curation, X.M. and X.Z.; validation, T.P., Z.L., and Y.Z.; investigation, X.M., X.Z., and P.S.; writing—original draft preparation, X.M. and X.Z.; writing—review and editing, Y.L. and Q.Z. All authors have read and agreed to the published version of the manuscript. The work reported in the paper has been performed by the authors, unless clearly specified in the text.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mao, X., Zhu, X., Pan, T. et al. Apelin (APLN) is a biomarker contributing to the diagnosis and prognosis of hepatocellular carcinoma. Sci Rep 14, 20441 (2024). https://doi.org/10.1038/s41598-024-71495-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-71495-z
- Springer Nature Limited