
Abstract
The natural populations of the marine mussel Mytilus chilensis and the associated aquaculture industry forms a sensitive social-ecological system that relies on the released propagules for cultivation in the highly heterogeneous environment (temperature, productivity, and salinity) of northern Patagonia (42–44 °S). We assessed spatial genetic structure, signals of local adaptation, and population assignment of M. chilensis analyzing 5963 SNPs from 125 individuals across six natural populations sampled over two consecutive years along the southeast Pacific coast (39° 25′ to 43° 07′ S, ~ 430 km). Neutral and putatively adaptive loci revealed high genetic diversity and low genetic differentiation among populations. Of the whole dataset, less than 1% (50) of loci were identified as putatively adaptive through multiple approaches, with only 0.1% detected in by all of them, and only two loci of them were correlated with environmental variables. No evidence of Isolation by Environment (IBE) was found, albeit a slight differentiation in the southern sampling location (Yaldad). These results suggest that the genetic structure observed is primarily shaped by neutral processes with weak signals of local adaptation. Gene-flow appears to be the main evolutionary force influencing the species’ population genetic structure. Because of the importance for the industry, the probability of correct assignment of individuals to their population of origin using allelic frequencies was evaluated. Analyses exhibited relatively low probabilities (< 50% for four out of six sites) of accurately assigning individuals to their geographic origin, with a limited success of SNP markers the for such purposes. Likely, species' high dispersal capacity, seed translocation, and the spill-over effect of mussel aquaculture prevents population genetic differentiation through high effective gene flow, hindering local genetic adaptation.
Similar content being viewed by others

Introduction
The Chilean blue mussel Mytilus chilensis Hupé 1854 is an endemic marine species that inhabits highly heterogeneous environments in estuaries and protected areas1,2,3 and at depths between 10 and 25 m4 in the southeast Pacific. It is distributed from the Tirúa River (38 °S) to Punta Arenas (53 °S)5,6 where it forms dense benthic wild populations or natural beds of sessile adults. The species is an economically important mollusk in Chile with a production of ~ 1300 annual tons in the last six years (SERNAPESCA, 2018–2023). Most of the production in Chile is provided by the mussel aquaculture industry, representing ~ 2.1% of the worldwide production of Mytilus spp., being the second largest producer after China (FAO 2020). Mytiliculture depends on spat that is obtained from wild populations and placed in suspended cultures in coastal waters, which is known as «capture-based aquaculture»7. The mussel aquaculture is a fundamental social-ecological system located mainly in the Chilean northern Patagonian area (42–44 °S). This area harbors over 65% of the total aquaculture of M. chilensis and the production of ~ 99% of both seed production and mussel biomass (SERNAPESCA, 2021–2023). The temporally stable and persistent wild populations of M. chilensis are in the proximity of mussel aquaculture centers around the Chiloé archipelago (42–43 °S), the inner sea of Chiloé, and in the protected continental Chilean coast to the east of Chiloé.
Geographic population assignment of individuals and authentication is a main goal for marine resource species in high demand for human consumption such as marine bivalves8. The availability of tools for the population assignment of individuals emerges as a critical necessity for the mussel aquaculture industry in northern Patagonia, as they could enable the authentication of the geographical origin of both the seeds and the marketed individuals, ensuring traceability and enhancing consumer confidence9,10,11. However, implementing population assignment using genetic markers may be complex due to the biological characteristics of the species. Mytilus chilensis, like many mussels, has a free-living planktonic larval stage lasting 30–45 days12,13, which confers a high dispersal potential14,15. This high dispersal potential contributes to low-to-moderate spatial and temporal genetic differentiation among wild populations in the southeast Pacific coast including northern Patagonia, as reported by several studies5,16,17,18,19. However, even in the presence of high levels of gene flow, genetic structure can persist, especially in species that exhibit responsiveness to environmental heterogeneity at a genetic level20,21,22. This persistence is a consequence of robust selective processes that occur after dispersal, recruitment, settlement, and post-settlement stages, resulting in elevated mortality rates before reproduction and subsequently reducing effective functional connectivity20,21,22,23. In M chilensis, local adaptation in response to seascape has been suggested17 but correlation between genetic structure and environmental variables has not been explicitly evaluated. Assessing these correlations is crucial to understand the population genetic diversity and differentiation of wild mussel populations, and the association between genotypes and environmental variables, can provide insights into how populations locally adapt to different environmental conditions. By identifying putatively adaptive genetic variations, it may be also possible to develop strategies that enhance the resilience and sustainability of aquaculture operations, and eventually identify highly differentiated markers, which could provide a potential tool for DNA population assignment in M. chilensis. Therefore, detailed knowledge of the neutral and adaptive genetic diversity is essential for management of wild populations and the advancement of aquaculture practices.
The north Patagonia area is considered an estuary zone with freshwater input from rivers and ice melting that interacts with oceanic masses24, leading to drastic changes in environmental features, mainly in salinity, chlorophyll, temperature, pH, and Oxygen25,26. Consistent with the heterogenous seascape inhabited by M. chilensis, experimental studies have revealed that populations have a broad plastic response to environmental stressors such as salinity27, temperature28, ocean pH -acidification29,30,31,32, and to the interaction of different climatic variables33,34,35. In M. chilensis, changes in salinity, pH, temperature, and ocean acidification36,37,38,39affects respiration, growth, calcification, and reproduction rates. Additionally, differences on gene expression in response to environments’ variability have been also reported35,40,41,42,43,44. Therefore, populations of the species may respond to changes through a combination of phenotypic plasticity and local genetic adaptation45,46,47. If natural populations are locally adapted to different environments, forming distinct adaptive units, significant changes in environmental conditions could jeopardize the stability of the system, potentially compromising its ability to support the aquaculture of M. chilensis. Additionally, mixing of individuals should be avoided when there are locally adapted genotypes, as the introduction of foreign genotypes, even if maladapted, could lead to short-term disruptions in local populations48,49. Although maladapted genotypes may eventually be removed from the population, the presence of recessive deleterious alleles could persist through introgression.
Because M. chilensis is a species with a high social-ecological relevance that sustains a relevant economic activity and given that natural populations are the genetic reservoir and source of spat for the industry, it is relevant to further understand the population and seascape genomics of the wild populations of the species. The goal of this study was to contribute with information for the sustainability of M. chilensis through a population genetic structure assessment and a seascape genomics approach. This approach includes the integration of environmental variables to detect genetic markers putatively linked to local adaptation, inferring the population genetic structure of the species in the area, and determining the feasibility of population assignment of the geographic origin of individuals using allele frequencies of thousands of SNPs scattered throughout the genome.
Results
Quality filtering and demultiplexing
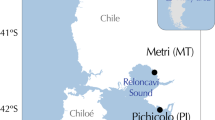
In this study, 125 individuals from 6 natural populations of Mytilus chilensis from the Southeast Pacific including the inner sea of Chiloé in northern Patagonia (Fig. 1) were used to assess genetic structure using Genotyping-by-Sequencing. A total of 263,487,800 reads were generated by GBS with a mean quality score of 34.34. After bioinformatic filtering (missing data, MAF, HWE) a total of 5963 SNPs were obtained. Sampling was conducted during two consecutive years; only juvenile individuals (< 1 year old) were sampled to avoid overlapping cohorts. Differentiation between years was evaluated because there is evidence for the species in the study area of inter-annual changes in genetic diversity, likely due to sweepstake reproductive success. There was a high genetic homogeneity between years, with no significant differences observed at the same site among years. FST values were non-significant with the only exception of Mehuín (ME) (p = 0.001) (Table S1). Despite this difference, the value was low (0.001) and confirmed by a clustering analysis that showed data of both years was overlapping (data now shown). Therefore, the sites were grouped across years for subsequent analyses.

Sampling map of wild populations of Mytilus chilensis in the southeast Pacific coast. In each sampling site, the acronym of each natural bed and the final number of sequenced individuals is indicated.
Population structure and seascape genomics
In all wild populations the effective number of alleles (corrected by sampling size) were similar and ranged from 1.396 to 1.416, being Pichicolo (PI) the site with the lower value and Los Molinos (LM) the one with higher values (Table 1). Observed and expected heterozygosities showed similar values, with the lowest values in the PI, and the highest in LM. In general, observed heterozygosities were lower than expected with positive and significant values of FIS indicating an overall heterozygote deficiency (Table 1).
Several approaches were used to detect putatively adaptive loci. BAYESCAN identified 11 outliers (Table S2) and PCADAPT 46, with seven loci detected in common and 50 outliers detected in total (Fig. 2). On the other hand, Genotype environment association to detect putatively adaptive loci in response to environmental variables (e.g., Table S2). were performed using LFMM and BayEnv. Each of the environmental association approaches individually detected from 0 to 69 putatively adaptive loci depending on the environmental variable (Fig. 2). Only two of them were detected in common by both analyses, one associated to the maximum sea surface temperature (SST max) and the other to the minimum sea surface temperature (SST min). Extreme values (max and min) of SST and chlorophyll-a were the variables associated with the largest number of outlier loci (Fig. 2).

Venn diagrams of outlier detection of loci putatively under local adaptation for Mytilus chilensis using Bayescan and PCAdapt, and environmental association analyses (LFMM and BayEnv) correlated with four environmental variables: Chlorophyll-a (Chl-a max, Chl-a min, Chl-a mean), Sea Surface Temperature (SST max, SST min, STT mean), pH and Salinity.
The patterns of spatial genetic structure were assessed with the 50 outlier loci detected through Population Differentiation Analysis (PDA). Low levels of genetic differentiation were found among populations of M. chilensis in the study area, with significant FST values that ranged from 0.004 to 0.015 for neutral loci and from 0.032 to 0.209 for outlier loci (Table 2). Clustering analysis using STRUCTURE detected two and three genetic groups for neutral and outlier loci, respectively (Fig. 3). For neutral loci, the two main clusters were homogeneously distributed along the study area except for Mehuín (the northernmost site) that was slightly differentiated from the rest (Fig. 3a). The predominant cluster at the northernmost locality, ME, appears throughout the study area with high levels of admixture, and is most abundant particularly in LM and Yaldad (YA). The second cluster, also widely distributed in the study area, predominates in the natural inner sea of Chiloé populations of MT, PI and PU (Fig. 3a). With outlier loci, the clusters detected by STRUCTURE were overall consistent with neutral data with a third differentiated cluster in the southernmost locality of the study area, YA. In ME, there was a predominant cluster (Cluster 1) with ~ 60% admixture, while the other cluster (Cluster 2) accounted for about 40%, according to Mapmixture (Fig. 3b) and a widely distributed cluster that predominates in LM, and in most of the localities of the interior sea of Chiloé (MT, PI, PU). DAPC was consistent with STRUCTURE detecting two and three groups for neutral and outlier loci, respectively (Fig. 3c,d). For neutral data, the horizontal axe showed two groups with ME and YA in the same group but slightly differentiated from each other, and a second a cluster was formed with MT, PU, and LM. PI was somewhat separated from this cluster but with several overlapping individuals (Fig. 3c). For outlier loci, ME and YA were somewhat differentiated from the rest of the sites and there was a highly overlapped group formed by LM, MT, PI, PU (Fig. 3d).

Genetic clustering of SNPs obtained from six sites on the southeast Pacific of Mytilus chilensis. Probabilistic assignment of individuals to each genetic group detected with, (a) Bayesian clustering implemented in STRUCTURE; (b) average admixture per natural bed estimated with construct, and (c) clustering using the information of the geographic origin of the individuals using Discriminant Analysis of Principal Components (DAPC).
Redundancy analysis to evaluate the relative contribution of spatial and environmental variation was performed using the first two principal components (PC) of a PCA of the environmental variables (Table S2). Due to the relatively simple spatial structure of the sampling area, only two spatial vectors (dbMEMs) were sufficient to estimate the spatial variation using the coordinates of each sampling site. The full RDA model (including environmental and spatial variables) was highly significant (F = 2.1, p < 0.001), and the first two axes of the full model explained 77% of the total variance (Fig. 4a). In this model, both environmental PCs were significant (PC1 F = 2.72, P < 0.001; PC2 F = 2.093, p < 0.001), as well as the dbMEM2 vector (F = 2.5, p < 0.001) (Table 3). PC1 corresponded mainly to differences in extreme values of temperature, chlorophyll, and primary productivity, and PC2 was positively correlated with salinity and negatively correlated with temperature and maximum chlorophyll (Figure S1). In the case of spatial vectors, on the one hand, dbMEM2 vector values were mapped to each sampling site, revealing that sites located at the northern and southern edges of the sampling range shared similar values. This indicates that dbMEM2 captured specific spatial variation (i.e. coastline-distance) at the geographic extremes of the sampling range. This was also suggested by both, an analysis of variance evaluating dbMEM2 values between sites located in the Chiloé Inner Sea with respect to the sampling edges (Anova F = 58.6, p < 0.001) (i.e. Mehuín, Yaldad) (Anova F = 58.6, p < 0.001) and in the RDA plot in which MT, PI, PU, and some ML individuals cluster in the same RDA space as dbMEM2, while the edge locations ME and YA appear in a different space, indicating distinct spatial influences (Fig. 4a). On the other hand, dbMEM1 vector, was strongly negatively associated with the latitude (R = − 0.98) and was not significant (F = 1.08, p = 0.32), which may indicate that latitude does not significantly influence genetic structure.

Effect of environment on the spatial genetic structure of loci putatively subject to selection (outlier loci) in Mytilus chilensis from six sites on the southeast Pacific. (a) Plot of the Full the RDA model considering the environment through the first two components of the PCA (Figure S1) and spatial vectors, and (b) Mantel test of the joint effect of environmental distance and geographic distance (coastline distance) on genetic distance using multiple matrix regression with randomization analysis (MMRR). Results of partial Mantel estimation of isolation by distance and isolation by environment also showed (See methods).
Partial models, corresponding to environmental distance controlled by coastline distance and vice-versa, showed the same pattern as the full model (Table 3). Despite these patterns, the adjusted R2 value in the full model was low (0.03) and no significant spatial clustering was observed, which is consistent with the low genetic differentiation detected with the rest of the analyses (Fig. 4b). When exploring isolation by distance (IBD) and isolation by environment (IBE), no significant patterns were detected (Fig. 4b), although the best model was the Partial Mantel using environmental distance controlled by coastline distance, and the Mantel test using the joint effect of environmental distance and coastline distance (Fig. 4b) (Mantel R: 0.49, p = 0.1).
Population assignment
Maximum Likelihood based assignment probability for population assignment showed that individuals sampled in ME and YA, the two sites that showed a slight differentiation, were in general the most likely to be assigned to their geographic origin, with both neutral and outlier loci. For ME, 75.8% of individuals could be correctly assigned with neutral loci and 58.6% with putatively adaptive loci, with 41.4% of individuals misassigned. In the case of YA, both markers were able to correctly assign the sampled individuals to their population of origin (With neutral markers, 84% of individuals were correctly assigned and 96% with outliers) (Fig. 5). Assignment was inconsistent between markers for other sites. For example, LM had a relatively high percentage of assignment with neutral loci (62.5%) but a low percentage with putatively adaptive loci (37.5%), and ME had a relatively high percentage of assignment using putatively adaptive loci (63.2%) and less than a half of the individuals correctly assigned with neutral loci (47.4%). The two populations from the Inner Sea of Chiloé had the lowest assignment success. PI had a 0% and 20% success rate with neutral and putatively adaptive loci, respectively, while PU had a 7.6% and 30.6% of individuals correctly assigned to their population of origin with neutral and putatively adaptive loci, respectively.

Assignment of individuals of Mytilus chilensis to their site of origin using maximum likelihood approach. For each site, the percentage of correct assignation with neutral and outlier loci to the geographic origin of individuals is shown.
Discussion
Our data from 125 individuals of six wild populations of Mytilus chilensis using multiple approaches showed high genetic diversity, low genetic differentiation and weak influence of the environment and geographic distance on genetic structure patterns. These results suggest overall population genetic homogeneity in the study area, albeit slight detected differentiation. Fifty loci putatively subject to selection (0.83%) were detected in total. Seven of them (0.11%) detected by the different approaches used for outlier loci identification, and only two were consistently correlated to relevant environmental variables for the species. The low level of genetic differentiation in neutral loci, in addition with the low number of outlier loci, suggests that local adaptation may not be the primary driver of this species’ genetic structure, at least at the scale and geographic area studied as observed in other marine species50,51,52,53. For example, Saenz-Agudelo et al.52 studied two closely related limpet species of the genus Scurria on the Southeastern Pacific coast. Based on the number of outliers, they suggest that the genetic structure in both species is shaped by distinct and contrasting evolutionary mechanisms, indicating that the genetic structure of the species with the least amount of putatively adaptive loci (Scurria viridula) is mainly modeled by neutral processes. A similar pattern has been observed in the blackfin tuna, Thunnus atlanticus, in the western Atlantic Ocean, where low genetic structure, high gene flow, and a small number of outlier loci suggest a limited impact of diversifying selection and local adaptation53. In contrast, studies conducted in the same biogeographic region as our study, using SNPs, have shown strong local adaptation, with high spatial genetic structure and the consistent identification of 3–5% outlier loci across different approaches, even with smaller datasets22. This could mean that effective connectivity appears to outweigh the effects of natural selection in wild populations. Likely, the high genetic diversity of M. chilensis19 underlies the observed variation in phenotypic responses to the environment25,28,30,35,40,43,44. Additionally, the large effective population sizes may also play a significant role in how this species adjusts to and copes with environmental changes, alongside genetic differentiation.
The analyses of spatial genetic structure unveiled two and three genetic clusters for neutral and putatively adaptive loci, respectively. The two northernmost sites, that are outside of the Chiloé area to the north, showed higher contribution of one of the clusters with both datasets, compared to the rest of the sites located in the Inner Sea of Chiloé area. A low number of loci were detected in common by all different approaches used, indicating an overall weak signature of local adaptation. Despite this, spatial analysis of putatively adaptive loci detected identified across multiple approaches reveals subtle differentiation of some sampling locations. For example, the redundancy analyses showed a slight separation between three sites of the inner Sea of Chiloé (Metri, Pichicolo, Putemún) and some individuals of Los Molinos with the rest of the populations. This pattern was associated with temperature, chlorophyll-a, and primary productivity. These were also relevant oceanographic features in genotype-environment association approaches. The analyses also showed a significant influence of one of the spatial vectors (MEM2), likely indicating that the weak genetic variation is partly explained by either unmeasured environmental factors or by the spatial separation of the most distant locations, as evidenced by the significantly higher MEM2 values in the Inner Sea of Chiloé sites compared to the North and South edges of the sampling area (Mehuín and Yaldad). However, we did not find any significant patterns of Isolation by Distance (IBD) or Isolation by Environment (IBE).
With neutral loci, all the sampling sites of the Chiloé area (Metri, Pichicolo, Putemún and Yaldad), displayed a higher prevalence of the most widely distributed genetic cluster. Since adults of M. chilensis are sessile, the primary intrinsic dispersal phase is the long-lived planktonic larvae. Larvae, mediated by their behavior and ocean currents can favor gene flow along broad geographic areas15,54,55,56. High genetic connectivity also coincides with the lack of significance of the Mantel analyses, albeit a slight positive but non-significant relationship of environment-and-distance with genetic differentiation. The slight spatial structure with both markers (neutral, outliers) found between the inner Sea of Chiloé (Metri, Pichicolo, Putemún and Yaldad) and the exposed coast of the South Pacific (Mehuín, Los Molinos) could be explained by the different hydrodynamics patterns in both regions. Differences among Pacific Ocean and Inner Sea of Chiloé could provoke potential dispersal barriers for connectivity. For example, a study that analyzed the abundance of floating algae in the area showed a tendency to decrease towards the interior channels (i.e. inner sea of Chiloé), possibly due to the influence of currents and a greater supply of freshwater that would limit the availability of algal beds attached to the substrate57. In addition, the northern exposed coast has, comparatively, higher temperatures and more stable salinity58, which could favor faster development for larval and juvenile stages. Larval transport processes, and consequently the effective dispersion of individuals in southern Chile are likely modulated by intense ocean currents influenced by both the resonance of the tidal wave and the direction and intensity of the wind and seasonal an interannual variability of ocean currents26,59 Ocean circulation around the bifurcation of the Humboldt Current and the Cape Horn Current (38°–46 °S), is seasonally dynamic. During winter, northerly winds could increase in intensity and change the direction of currents, influencing ocean circulation in the region59,60. This means a greater influence of the Humboldt Current in winter (south to north) and a greater influence of the Antarctic Circumpolar Current in summer (north to south)58. On the other hand, tidal currents in the Inner Sea of Chiloé are intense and could reach speeds of up to 1 knot58 which could favor gene-flow, however, resonance of tidal waves which produce large variations in the tidal range, especially in the Reloncaví Sound (i.e. Metri), where the amplitude of the sea level reaches up to about 8 m58, potentially being a disadvantage for connectivity. Highly temporal and spatial dynamic oceanographic processes of the study area, could differentially influence the intrinsic dispersal processes of the species, explaining the slight patterns of differentiation found particularly between the sites of the southwest Pacific Ocean and the inner sea of Chiloé.
Positive values of inbreeding coefficients in all the analyzed populations with both data sets indicate a heterozygote deficiency. This could be explained by natural variation in reproductive success among sites and years, provided by the dispersal of planktonic larval stages, and by anthropogenic transportation. Translocation of seeds for mytiliculture occurs from localities with higher larval production, which may vary over time but are usually somewhere in the inner sea of Chiloé or the Reloncaví Sound, to the mytiliculture centers. This human-mediated spat movement, enhances genetic connectivity within the study area promoting genetic homogenization, as has been previously suggested19, and, together with the natural temporal variation on reproductive success19, could lead to a Wahlund effect, i.e., the sampling of two or more populations as if they were one. In our study, this could be further exacerbated by seeds for aquaculture deriving from different sources, followed by external fertilization process after widespread spawning events. The role of aquaculture facilities in enhancing population connectivity has been studied in other mussel species, such as M. edulis, M. galloprovincialis, and Perna canalicus, in which cultivated populations contribute to the restoration of wild populations through larval spill-over61,62,63. Seed collection has been prevalent in the Reloncaví Sound and Fjord, and the seed collection centers located in these areas supply seeds to all the aquaculture centers in northern Patagonia64. Both larval transport and anthropogenic active seed translocation serve as vectors for gene flow of M. chilensis in the study area, likely preventing genetic diversity loss and genetic differentiation in the study area.
Using putatively adaptive loci, the southernmost sampling site Yaldad, located in the south of Chiloé island, had a predominant third genetic cluster, suggesting mild signals of local adaptation of that sampling location and weak signatures of selection the overall sampling area. Even though the genetic structure detected with putatively adaptive loci was low, it is consistent with the environmental and oceanographic scenario. The sites in the Chiloé area are in protected coasts that have strong influence of freshwater interacting with oceanic waters24. The geomorphological configuration of the coast around Yaldad, the tidal currents and advection processes26,55,56, and the interactions between salinity, carbonate system, temperature26 and the geographic separation, could favor larval retention, reduce gene flow and contribute to the observed pattern of slight local adaptive genetic differentiation of Yaldad.
A desired application of genetic markers for the mytiliculture industry and social-ecological system, is the possibility of performing population assignment of individuals. Because of the low temporal and spatial genetic differentiation among most wild populations, and the low density of adaptive loci (potentially highly differentiated among sampling sites), population assignment using genetic markers has a low probability of being accurate in M. chilensis. Evaluation of neutral and putatively adaptive markers showed different results when assigning individuals successfully to their site of origin. A likely assignment of geographic origin was possible only for the Mehuín and Yaldad, with 75.8% and 84% of accuracy with neutral markers and 58.6% and 96% with putatively adaptive markers, respectively. Some of the probabilities of assignment are, however, higher than reported by other population assignment studies using DNA in M. chilensis, with less than 50% of correct assignment with microsatellites65. Population assignment has been successfully achieved in the genus for monitoring mislabeling of European food trade66,67,68,69 but at large geographic scales, including different oceans66, in comparison to the search for markers between populations within the northern Patagonia area. The relatively small study area in northern Patagonia (5° of latitude) and the high interpopulation connectivity of M. chilensis challenges the possible development of reliable population assignment tools to identify the geographic origin based on genetic markers. The genetic structure found could also indicate that the population of origin may not be play a significant role for the mytiliculture industry, given the high gene flow both larval and anthropogenic. The large effective population size could be beneficial for the maintenance of long-term population stocks in the species.
Conclusions
As expected for species with high larval and anthropogenic dispersal potential, the genetic structure of Mytilus chilensis of northern Patagonia based on genome-wide SNP markers revealed low levels of genetic differentiation among wild populations. This suggests that gene flow, high genetic diversity and high effective population size are underlying factors that could favor the phenotypic plasticity previously reported for M. chilensis in the study area as a main evolutionary driver allowing populations to cope with environmental heterogeneity. Accordantly, there is little influence on spatial genetic structure by local adaptation at the genotype level. These findings contribute to our understanding of the drivers of diversity that underlie the success of M. chilensis in a variable environment and have important implications for the conservation and management of this resource species and the associated social-ecological system.
Methods
Sample collection and DNA extraction
Samples of Mytilus chilensis were extracted from natural beds located in management areas (TURFs) or concessions for aquaculture in the Southeast pacific coast between 39 and 43 °S from two consecutive years (2014 and 2015). Individuals were purchased directly from fishers who have extraction rights of marine resources. The procedures and protocols performed were evaluated by a specialized ethics committee of the Universidad Católica del Norte (Chile) (CEC UCN RES N°5) and were in accordance with ethical conflicts of both national and international legislation. Six natural beds were considered; two of the sites were along the more exposed pacific coast while the other 4 were in protected areas in the Inner Sea of Chiloé (Fig. 1). Individuals were placed in seawater with oxygen provision until processing to maintain DNA integrity. For each sampled individual, adductor muscle tissue was preserved in absolute ethanol at − 20 °C. Ten mg of adductor muscle tissue were used for DNA extraction with the QIAGEN DNAeasy tissue and blood kit (QIAGEN, USA) following the manufacturer's instructions. The quality/purity of DNA was measured in a Nanodrop ® Spectrophotomer (Thermo Fisher Scientific) and a QUBIT 4 (Thermo Fisher Scientific).
Genotyping by sequencing and SNP calling
SNPs were obtained through reduced-representation sequencing, using paired-end Genotyping-by-Sequencing70. Samples were sequenced in the Biotechnology Center of the University of Wisconsin, USA, in a Ilumina NovaSeq6000 platform. Genomic libraries were constructed, after optimization, using ApeKI restriction enzyme with a unique barcode adapter of 4–8 bp of length for each individual and quality check was evaluated using FASTQC71. Raw dataset was demultiplexed and filtered in the pipeline Universal Network-Enabled Analysis Kit (UNEAK) from suite of the TASSEL software version 5.072. An error tolerance rate (ETR) of 0.03 was used, potentially reducing the number of sequencing errors and the number of retained SNPs. A standard minor allele frequency (MAF) of 5% and a 20% missing data was allowed in the first sets of filters. After this, we calculated Hardy–Weinberg Equilibrium (HWE) in each population and loci separately, and those loci that consistently deviated from HWE in at least 60% of the populations (4 of 6) were excluded from further analyses73. Given the evidence of inter-annual dynamics in genetic diversity in the species19, prior to all the rest of the analyses, we first evaluated the possibility of grouping the different years of sampling. For this, interannual differences between sampling years were evaluated by calculating the pairwise FST between the same site among years in ARLEQUIN v.3.574 with 1000 permutations. The significance levels were corrected with an FDR of 5% to avoid false positives due to multiple comparisons.
Outlier detection and genotype-environment association analysis
Determination of putative signals of local adaptation were based on population differentiation and genotype-environment association/correlation, both using the significant differences in allele frequencies, and departures from what is expected under neutrality. The identification of putatively adaptive and neutral SNPs from the whole dataset was performed using two different approaches. First, we used population differentiation analyses to detect loci significantly differentiated based on the expected degree of differentiation under a neutral distribution. For this, two methods were used. First, a Bayesian approach implemented in BAYESCAN 2.175 that is based on logistic regressions that identify SNPs putatively affected by diversifying selection from population-specific demographic processes (null model). We used as a priori model (pr_odds) a value of 1000 based on number of SNPs75 in a total of ten separate runs that were performed from 50,000 to 500,000 iterations with a 10% burn-in period to assure the convergence of the Markov chain Monte Carlo (MCMC)75. In parallel, we used PCAdapt76, in R (R Core Team, 2022) to detect SNPs loci putatively under selection based on a principal component analysis (PCA). This approach estimated an optimal number of genetic clusters (k) based on the percentage of variance explained by each component. Once the optimal k value was determined, using a Mahalanobis distance test, outlier loci were determined based on each loading value, assuming that those loci that were the most correlated with the optimal k value and, in consequence, explained the most of genetic differentiation, were the most likely under diversifying selection. In both, BAYESCAN and PCAdapt, outlier loci were identified after a False Discovery Rate (FDR) of 5%.
Sea surface temperature (SST), chlorophyll-a/primary productivity, pH, and salinity and their variations were used as environmental variables for genotype-environment associations. These variables were chosen according to bibliographical revision (Table S2) and were obtained from Bio-Oracle 2.0 (Terra-MODIS and SeaWifs)77. The database used had a resolution of 4 km2 for all variables, except for salinity and pH, which had a resolution of 9.2 km2. Data was monthly and over a period of 12–18 years, depending on the variable. Extreme values were calculated within the time frame available for each variable (Table S2). To estimate loci under putatively diversifying selection in response to environmental variables two different approaches were used. First, the Latent Factor Mixed Model (LFMM)78, algorithm implemented in the LEA package in R. An environmental file with standardized environmental variables was used to correlate with individual genotypes. This approach uses neutral genetic structure as a latent or unobserved factor. This latent factor was based on the number of estimated genetic clusters determined by STRUCTURE and DAPC results (these methods are detailed in section ahead). Five repetitions were conducted with 200,000 iterations per latent factor (k) and a burn-in period of 100,000. Adjusted P-values were obtained using the genomic inflation factor (λ) and a False Discovery Rate (FDR) of 5%. A second approach involved using BAYENV279 to correlate individual genotypes to environmental variables. This approach tests if the correlation between individual SNPs and environmental variables that best fit a neutral/null model. For this, BAYENV2 uses a Bayesian method that estimates a pairwise covariance matrix of neutral allele frequencies between populations. This covariance matrix was used as a neutral/null model to perform a parametric test of the effect of each environmental variable on the frequency of an allele at a given SNP79. The determination of SNP loci candidates for local adaptation was performed by calculating their Bayes Factors, filtering those with values greater than 10, or with strong evidence of being correlated, according to the Jeffery’s criteria, with their corresponding environmental variable80.
To assess the influence of the environment on the putatively adaptive genetic structure, a PCA on the environmental variables was performed to reduce the dimensionality of the dataset. Components that explained more than 90% of the variance were retained for further analyses. A redundancy analysis (RDA) was performed using vegan in R (4.2.0) to determine the relative contribution of the geographic position and each environmental variable to the genetic structure of putatively adaptive loci. Spatial genetic structure was estimated using the geographic coordinates of the sampling sites based on distance-based Moran's eigenvector maps, dbMEMs81 using the adespatial package in R. To evaluate the spatial structure estimated by the dbMEM vectors, the resulting values were mapped to each sampling site to correlate them with latitude and longitude. Additionally, using a one-way ANOVA, the relationship between vector values with the most distant locations in the sampling area (i.e. Mehuín, Yaldad) respect to the other sampling sites was evaluated.
Prior to the analysis, genotype data were standardized by removing the broadscale trend using the decostand function with the Hellinger's method in vegan. Three RDA models were tested. One full model, using spatial vectors and environmental variables (PC), and two partial models. A partial environmental model, using environmental variables as fixed factors and spatial vectors as conditional variables, and a partial spatial analysis, with spatial vectors as fixed factors and environmental variables as conditional variables. Finally, marginal ANOVA by term were conducted for each model with 10,000 permutations to evaluate the significance of each variable.
Additionally, to assess possible patterns of isolation by distance (IBD) or isolation by environment (IBE), geographic distance matrices were first estimated using the coordinates of the sampling sites. To estimate the coastal distance between sampling sites, we used GSHHG shoreline data v. 2.3.782. The coastline was smoothed using spline interpolation in Python with the geopy library, and the nearest points on the smoothed coastline were identified. We calculated the subline between these points using the shapely library. For distances between the sampling sites in mainland and sites in the Chiloé Island, depending on the pairwise comparison, we used linear geodesic distance (Metri-Chiloé, Pichicolo-Chiloé) or added the distance of the Chacao Channel or Reloncaví Fjord (Mehuín-Chiloé, Los Molinos-Chiloé). The environmental distance matrix was calculated using the values contained from the main principal components of the PCA analysis, and then transformed to inter-site distance using the dist function in Vegan. Using the genetic differentiation matrix (FST) of putatively adaptive loci, two partial Mantel analyses were also run. One to assess IBD controlling for environmental distance, and another to assess IBE controlling for coastline distance. Finally, to evaluate the joint influence of environmental and geographic distances, a Multiple matrix regression with randomization analysis (MMRR)83 was performed using random permutations (10,000) to assess the statistical significance of the regression coefficients obtained. This allowed assessing the relationship between the weighted influence of environmental and geographic distance matrices with the genetic dissimilarity matrix and determining the significance and magnitude of environmental and geographical effects on genetic dissimilarity. Considering the specific influence of each matrix, a joint effect-difference matrix was calculated and correlated with the FST using a Mantel analysis with 10,000 permutations in vegan.
Genetic diversity and genetic structure analysis
Analyses were performed with data from both sampled years and by dataset, of neutral and putatively adaptive (outlier) loci. Number of polymorphic sites, allelic richness, expected heterozygosity and observed heterozygosity of each population was estimated using GenoDive 3.084. Fixation index FST was calculated in ARLEQUIN v.3.574 with 1000 permutations. The significance levels obtained in FST were corrected with an FDR of 5% to avoid false positives due to multiple comparisons.
Genetic clusters were estimated using three approximations that used different methods to obtain an optimal k-groups value. First, a Bayesian analysis in STRUCTURE v.2.3.485, in the parallelized version STRAUTO86 was used to evaluate the probability of assignment of a given individual to a genetic cluster. For this, ten independent runs for each cluster (K) (1 to 6) were performed in parallel with 500,000 MCMC and a burn-in of 10%. The optimal number of K was determined independently for each data set (neutral and outliers) using Evanno et al.86,87 proposed delta K method implemented in STRUCTURE HARVESTER88. By evaluating the rate of change in the log likelihood of the data between different theoretical values of K, the value that maximizes the delta K in each data set was identified. Only the optimal K data determined by Evanno's was presented. As a second approach, a Discriminant Analysis Principal Component (DAPC) using the adegenet package89, were conducted in R with 100,000 iterations. This method uses principal component analysis and the information of the population of origin of individuals to discriminate among estimated genetic clusters. The optimal number of clusters was determined using the find.clusters function with a Bayesian Information Criterion (BIC)89 with 100,000 iterations, 20 principal components and 3 discriminant functions. Finally, pie charts of relative frequencies of admixture per sampling site in the study area were projected using the package Mapmixture90 in R.
Population assignment of individuals of M chilensis
A maximum likelihood analysis-based assignment probability, implemented in GENODIVE84 was used to estimate the feasibility of performing population assignment of individuals to their population of origin using multi-locus allele frequency information. The purpose of this analysis was to assign individuals to population of origin by calculating the likelihood of an individual genotype being present in a wild population, based on the average allele frequencies of that local population91.
Data availability
All the data used in this study is openly available. VCF files, environmental files, and metadata used in this study has been uploaded to DataDryad (https://datadryad.org/stash/share/RpN5LodBrvN4eQc2EJzh669bQorVvAO0WAGFSZsk5ME).
References
Barria, A., Gebauer, P. & Molinet, C. Variabilidad espacial y temporal del suministro larval de mitílidos en el Seno de Reloncaví, sur de Chile. Rev. Biol. Mar. Oceanogr. 47, 461–473 (2012).
Molinet, C. A. et al. Spatial distribution pattern of Mytilus chilensis beds in the Reloncaví fjord: Hypothesis on associated processes. Rev. Chil. Hist. Nat. 88, 1–12 (2015).
Campos, B. & Landaeta, M. F. Moluscos planctónicos entre el fiordo Reloncaví y el golfo Corcovado, sur de Chile: Ocurrencia, distribución y abundancia en invierno. Rev. Biol. Mar. Oceanogr. 51, 527–539 (2016).
Krapivka, S. et al. Shell-shape variation along the latitudinal range of the Chilean blue mussel Mytilus chilensis (Hupe 1854). Aquacult. Res. 38, 1770–1777 (2007).
Astorga, M. P. Complex spatial genetic connectivity of mussels Mytilus chilensis along the southeastern Pacific coast and its importance for resource management. J. Shellfish Res. 39, 77–86 (2020).
Gardner, J. P., Oyarzún, P. A., Toro, J. E., Wenne, R. & Zbawicka, M. Phylogeography of Southern Hemisphere blue mussels of the genus Mytilus: Evolution, biosecurity, aquaculture and food labelling. Oceanogr. Mar. Biol. 59, 139–228 (2021).
Lovatelli, A., & Holthus, P. F. (Eds.). Capture-based aquaculture: global overview (p. 298). Rome: Food and Agriculture Organization of the United Nations (2008).
Santos, A., Ricardo, F., Domingues, M. R. M., Patinha, C. & Calado, R. Current trends in the traceability of geographic origin and detection of species-mislabeling in marine bivalves. Food Control 152, 109840 (2023).
Villacorta-Rath, C. Outlier SNPs enable food traceability of the southern rock lobster. Jasus Edwardsii. Mar. Biol. 163, 1–11 (2016).
Davis, R. P., Boyd, C. E. & Davis, D. A. The utility of discriminant analysis to determine the geographic origin of commercially important seafood and aquaculture species: A meta-analysis. Rev. Fish. Sci. Aquacult. 29, 791–799 (2021).
Mamede, R. et al. Assessing the use of surrogate species for a more cost-effective traceability of geographic origin using elemental fingerprints of bivalve shells. Ecol. Indic. 130, 108065 (2021).
Hilbish, T., Carson, E., Plante, J., Weaver, L. & Gilg, M. Distribution of Mytilus edulis, Mytilus galloprovincialis, and their hybrids in open-coast populations of mussels in southwestern England. Mar. Biol. 140, 137–142 (2002).
Toro, J., Innes, D. J. & Thompson, R. J. Genetic variation among life-history stages of mussels in a Mytilus edulis- Mytilus trossulus hybrid zone. Mar. Biol. 145, 713–725 (2004).
Weersing, K. & Toonen, R. J. Population genetics, larval dispersal, and connectivity in marine systems. Mar. Ecol. Prog. Ser. 393, 1–12 (2009).
Haye, P. A. Phylogeographic structure in benthic marine invertebrates of the southeast Pacific coast of Chile with differing dispersal potential. PLoS One 9, e88613 (2014).
Larraín, M. A. et al. Heterologous microsatellite-based genetic diversity in blue mussel (Mytilus chilensis) and differentiation among localities in southern Chile. Lat. Am. J. Aquat. Res. 43, 998–1010 (2015).
Araneda, C., Larraín, M. A., Hecht, B. & Narum, S. Adaptive genetic variation distinguishes Chilean blue mussels (Mytilus chilensis) from different marine environments. Ecol. Evol. 6, 3632–3644 (2016).
Astorga, M. P., Vargas, J., Valenzuela, A., Molinet, C. & Marín, S. L. Population genetic structure and differential selection in mussel Mytilus chilensis. Aquacult. Res. 49, 919–927 (2018).
Haye, P. A. & Segovia, N. I. Shedding light on variation in reproductive success through studies of population genetic structure in a Southeast Pacific Coast mussel. Heredity 130(6), 1–12 (2023).
Benestan, L. Seascape genomics provides evidence for thermal adaptation and current-mediated population structure in American lobster (Homarus americanus). Mol. Ecol. 25(20), 5073–5092 (2016).
Carreras, C. East is East and West is West: Population genomics and hierarchical analyses reveal genetic structure and adaptation footprints in the keystone species Paracentrotus lividus (Echinoidea). Divers. Distrib. 26(3), 382–398 (2020).
Segovia, N. I., González-Wevar, C. A. & Haye, P. A. Signatures of local adaptation in the spatial genetic structure of the ascidian Pyura chilensis along the southeast Pacific coast. Sci. Rep. 10(1), 1–14 (2020).
Marshall, D. J., Monro, K., Bode, M., Keough, M. J. & Swearer, S. Phenotype–environment mismatches reduce connectivity in the sea. Ecol. Lett. 13(1), 128–140 (2010).
Sievers, H. A., & Silva, N. Masas de agua y circulación en los canales y fiordos australes. Avances en el Conocimiento Oceanográfico de las Aguas Interiores Chilenas, Puerto Montt a Cabo de Hornos, eds N. Silva & S. Palma (Valparaíso: Comité Oceanográfico Nacional—Pontificia Universidad Católica de Valparaíso), 53–58 (2006).
González, H. E. Seasonal plankton variability in Chilean Patagonia fjords: Carbon flow through the pelagic food web of Aysen Fjord and plankton dynamics in the Moraleda Channel basin. Cont. Shelf Res. 31(3–4), 225–243 (2011).
Narváez, D. A. et al. Dominant scales of subtidal variability in coastal hydrography of the Northern Chilean Patagonia. J. Mar. Syst. 193, 59–73 (2019).
Jahnsen-Guzmán, N. Environmental refuges increase performance of juvenile mussels Mytilus chilensis: Implications for mussel seedling and farming strategies. Sci. Total Environ. 751, 141723 (2021).
Lagos, L., Uriarte, I. & Yany, G. Assessment of the reproductive potential of the mussel (Mytilus chilensis) from two natural populations subjected to different conditioning temperatures. Lat. Am. J. Aquat. Res. 40(2), 389–397 (2012).
Navarro, J. M. Impact of medium-term exposure to elevated pCO2 levels on the physiological energetics of the mussel Mytilus chilensis. Chemosphere 90(3), 1242–1248 (2013).
Duarte, C. Intraspecific variability in the response of the edible mussel Mytilus chilensis (Hupe) to ocean acidification. Estuaries and Coasts 38, 590–598 (2015).
Diaz, R., Lardies, M. A., Tapia, F. J., Tarifeño, E. & Vargas, C. A. Transgenerational effects of pCO2-driven ocean acidification on adult mussels Mytilus chilensis modulate physiological response to multiple stressors in larvae. Front. Physiol. 9, 1349 (2018).
Mellado, C. Ocean acidification exacerbates the effects of paralytic shellfish toxins on the fitness of the edible mussel Mytilus chilensis. Sci. Total Environ. 653, 455–464 (2019).
Duarte, C. Combined effects of temperature and ocean acidification on the juvenile individuals of the mussel Mytilus chilensis. J. Sea Res. 85, 308–314 (2014).
Navarro, J. M. Ocean warming and elevated carbon dioxide: Multiple stressor impacts on juvenile mussels from southern Chile. ICES J. Mar. Sci. 73(3), 764–771 (2016).
Osores, S. J. Plasticity and inter-population variability in physiological and life-history traits of the mussel Mytilus chilensis: A reciprocal transplant experiment. J. Exp. Mar. Biol. Ecol. 490, 1–12 (2017).
Gazeau, F. et al. Impact of elevated CO2 on shellfish calcification. Geophys. Res. Lett. 34, L07603 (2007).
Duarte, C. The energetic physiology of juvenile mussels, Mytilus chilensis (Hupe): The prevalent role of salinity under current and predicted pCO2 scenarios. Environ. Pollut. 242(part A), 156–163 (2018).
Ruiz, M., Tarifeño, E., Llanos-Rivera, A., Padget, C. & Campos, B. Efecto de la temperatura en el desarrollo embrionario y larval del mejillón, Mytilus galloprovincialis (Lamarck, 1819). Rev. Biol. Mar. Oceanogr. 43(1), 51–61 (2008).
Vargas, C. A. Species-specific responses to ocean acidification should account for local adaptation and adaptive plasticity. Nat. Ecol. Evol. 1(4), 0084 (2017).
Núñez-Acuña, G., Tapia, F. J., Haye, P. A. & Gallardo-Escárate, C. Gene expression analysis in Mytilus chilensis populations reveals local patterns associated with ocean environmental conditions. J. Exp. Mar. Biol. Ecol. 420–421(582), 56–64 (2012).
Jarque, S. et al. Seasonal variations of gene expression biomarkers in Mytilus galloprovincialis cultured populations: Temperature, oxidative stress and reproductive cycle as major modulators. Sci. Total Environ. 499, 363–372 (2014).
Molinet, C. A. Spatial distribution pattern of Mytilus chilensis beds in the Reloncaví fjord: hypothesis on associated processes. Rev. Chil. Hist. Nat. 88(1), 1–12 (2015).
Kapsenberg, L. Molecular basis of ocean acidification sensitivity and adaptation in Mytilus galloprovincialis. iScience 25(8), 104677 (2022).
Belivermiş, M. Physiological and gene expression responses of the mussel Mytilus galloprovincialis to low pH and low dissolved oxygen. Mar. Pollut. Bull. 187(4), 114602 (2023).
Kawecki, T. J. & Ebert, D. Conceptual issues in local adaptation. Ecol. Lett. 7(12), 1225–1241 (2004).
Sanford, E. & Kelly, M. W. Local adaptation in marine invertebrates. Annu. Rev. Mar. Sci. 3, 509–535 (2011).
Wadgymar, S. M., DeMarche, M. L., Josephs, E. B., Sheth, S. N. & Anderson, J. T. Local adaptation: Causal agents of selection and adaptive trait divergence. Annu. Rev. Ecol. Evol. Syst. 53(1), 87–111 (2022).
Shi, Y. Gene flow influences the genomic architecture of local adaptation in six riverine fish species. Mol. Ecol. 32(7), 1549–1566 (2023).
Grummer, J. A. et al. The immediate costs and long-term benefits of assisted gene flow in large populations. Conserv. Biol. 36(4), e13911 (2022).
Uthicke, S. et al. Little evidence of adaptation potential to ocean acidification in sea urchins living in “Future Ocean” conditions at a CO2 vent. Ecol. Evol. 9(17), 10004–10016 (2019).
Villacorta-Rath, C. et al. Disentangling genetic from environmental effects on phenotypic variability of southern rock lobster (Jasus edwardsii) postlarvae. Ecol. Evol. 12(11), e9519 (2022).
Saenz-Agudelo, P. et al. Population genomic analyses reveal hybridization and marked differences in genetic structure of Scurria limpet sister species with parapatric distributions across the Southeastern Pacific. Ecol. Evol. 12(5), e8888 (2022).
Dimens, P. et al. Weak range-wide population structure in the blackfin tuna (Thunnus atlanticus) revealed by analysis of genome-wide SNPs. ICES J. Mar. Sci. 80(4), 941–952 (2023).
Lara, C., Saldías, G. S., Tapia, F. J., Iriarte, J. L. & Broitman, B. R. Interannual variability in temporal patterns of Chlorophyll-a and their potential influence on the supply of mussel larvae to inner waters in northern Patagonia (41–44 °S). J. Mar. Syst. 155, 11–18 (2016).
Pérez-Santos, I. et al. Synoptic-scale variability of surface winds and ocean response to atmospheric forcing in the eastern austral Pacific Ocean. Ocean Sci. 15(5), 1247–1266 (2019).
Linford, P. et al. Recent deoxygenation of patagonian fjord subsurface waters connected to the peru-chile undercurrent and equatorial subsurface water variability. Global Biogeochem. Cycles 37(6), e2022GB007688 (2023).
Hinojosa, I., Gonzalez, E., Macaya, E. & Thiel, M. Macroalgas flotantes en el mar interior de Chiloé, Chile y su fauna asociada con énfasis en peracarida y estados. Ciencia y Tecnología del Mar 33(2), 71–86 (2010).
Strub, P. T., James, C., Montecino, V., Rutllant, J. A. & Blanco, J. L. Ocean circulation along the southern Chile transition region (38–46 °S): Mean, seasonal and interannual variability, with a focus on 2014–2016. Prog. Oceanogr. 172, 159–198 (2019).
Valle-Levinson, A., Jara, F., Molinet, C. & Soto, D. Observations of intratidal variability of flows over a sill/contraction combination in a Chilean fjord. J. Geophys. Res. Oceans 106(C4), 7051–7064 (2001).
Cáceres, M., Valle-Levinson, A., Molinet, C., Castillo, M. & Bello, M. Lateral variability of flow over a sill in a channel of southern Chile. Ocean Dyn. 56, 352–359 (2006).
Coolen, J. W. Marine stepping-stones: Connectivity of Mytilus edulis populations between offshore energy installations. Mol. Ecol. 29(4), 686–703 (2020).
Norrie, C., Dunphy, B., Roughan, M., Weppe, S. & Lundquist, C. Spill-over from aquaculture may provide a larval subsidy for the restoration of mussel reefs. Aquacult. Environ. Interact. 12, 231–249 (2020).
Simon, A. et al. Replicated anthropogenic hybridisations reveal parallel patterns of admixture in marine mussels. Evol. Appl. 13(3), 575–599 (2020).
Larraín, M. A., Díaz, N. F., Lamas, C., Vargas, C. & Araneda, C. Genetic composition of Mytilus species in mussel populations from southern Chile. Lat. Am. J. Aquat. Res. 40(4), 1077–1084 (2012).
Larraín, M. A., Díaz, N. F., Lamas, C., Uribe, C. & Araneda, C. Traceability of mussel (Mytilus chilensis) in southern Chile using microsatellite molecular markers and assignment algorithms. Exploratory survey. Food Res. Int. 62, 104–110 (2014).
del Rio-Lavín, A. et al. Population structure and geographic origin assignment of Mytilus galloprovincialis mussels using SNPs. Aquaculture 550, 919–930 (2022).
Wenne, R. et al. Provenance of Mytilus food products in Europe using SNP genetic markers. Aquaculture 554, 738135 (2022).
Giusti, A. et al. Mussels (Mytilus spp.) products authentication: A case study on the Italian market confirms issues in species identification and arises concern on commercial names attribution. Food Control 118, 107379 (2020).
Giusti, A. et al. Mislabeling assessment and species identification by PCR-RFLP of mussel-based products (Mytilus spp.) sold on the Italian market. Food Control 134, 108692 (2022).
De Donato, M., Peters, S. O., Mitchell, S. E., Hussain, T. & Imumorin, I. G. Genotyping-by-sequencing (GBS): A novel, efficient and cost-effective genotyping method for cattle using next-generation sequencing. PloS One 8(5), e62137 (2013).
Andrews, S. FastQC: A quality control tool for high throughput sequence data. Available online. Retrieved May, 17, (2023).
Bradburd, G. S., Ralph, P. L. & Coop, G. M. A spatial framework for understanding population structure and admixture. PLoS Genet. 12(1), e1005703 (2016).
Pearman, W. S., Urban, L. & Alexander, A. Commonly used Hardy-Weinberg equilibrium filtering schemes impact population structure inferences using RADseq data. Mol. Ecol. Resour. 22(7), 2599–2613 (2022).
Excoffier, L. & Lischer, H. E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10(3), 564–567 (2010).
Foll, M. & Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180(2), 977–993 (2008).
Luu, K., Bazin, E. & Blum, M. G. pcadapt: an R package to perform genome scans for selection based on principal component analysis. Mol. Ecol. Resour. 17(1), 67–77 (2017).
Assis, J. et al. Bio-ORACLE v2.0: Extending marine data layers for bioclimatic modelling. Global Ecol. Biogeogr. 27(3), 277–284 (2017).
Frichot, E. & François, O. LEA: An R package for landscape and ecological association studies. Methods Ecol. Evol. 6(8), 925–929 (2015).
Günther, T. & Coop, G. Robust identification of local adaptation from allele frequencies. Genetics 195(1), 205–220 (2013).
Lee, M. D. & Wagenmakers, E. J. Bayesian cognitive modeling: A practical course (Cambridge University Press, 2014).
Legendre, P. & Legendre, L. Numerical Ecology (Elsevier, 2012).
Wessel, P. & Smith, W. H. F. A global, self-consistent, hierarchical, high-resolution shoreline database. J. Geophys. Res. Solid Earth 101(B4), 8741–8743 (1996).
Wang, I. J., Glor, R. E. & Losos, J. B. Quantifying the roles of ecology and geography in spatial genetic divergence. Ecol. Lett. 16(2), 175–182 (2013).
Meirmans, P. G. Genodive version 3.0: Easy-to-use software for the analysis of genetic data of diploids and polyploids. Mol. Ecol. Resour. 20(4), 1126–1131 (2020).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155(2), 945–959 (2000).
Chhatre, V. E. & Emerson, K. J. StrAuto: Automation and parallelization of STRUCTURE analysis. BMC Bioinf. 18, 192 (2017).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 14(8), 2611–2620 (2005).
Earl, D. & VonHoldt, B. Structure harvester: A website and a program for visualizing structure output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Jombart, T., & Collins, C. A tutorial for discriminant analysis of principal components (DAPC) using adegenet 2.0. 0. London: Imperial College London, MRC Centre for Outbreak Analysis and Modelling (2015).
Jenkins, T. L. MAPMIXTURE: An R package and web app for spatial visualisation of admixture and population structure. Mol. Ecol. Resour. 24(4), e13943 (2024).
Paetkau, D., Slade, R., Burden, M. & Estoup, A. Genetic assignment methods for the direct, real-time estimation of migration rate: A simulation-based exploration of accuracy and power. Mol. Ecol. 13(1), 55–65 (2004).
Acknowledgements
Authors acknowledge Natalia Muñoz, Francisca Gálvez, Daniel Aliste, Paulina Gyorgy and particularly Raúl Vera for their valuable contributions in the field, sample processing and DNA extractions and other laboratory procedures. We also thank Dr. Bernardo Broitman for his valuable comments. We would like to acknowledge the local fishers in the study area for help and assistance with sample collection. This research was funded supported by the Chilean National Fund for Scientific and Technological Development (ANID) through Grants FONDECYT N°1140862, FONDECYT N° 11220913, FONDECYT N°1231579, and the Millennium Science Initiative Program (Code ICN2019_015), Chile.
Author information
Authors and Affiliations
Contributions
P.A.H. conceived the study, P.A.H. and N.I.S. wrote the manuscript. N.I.S. and D.C.S. performed biostatistical and statistical analysis and D.C.S. performed laboratory work. All authors provide feedback on the manuscript and have approved the final version prior to submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Segovia, N.I., Coral-Santacruz, D. & Haye, P.A. Genetic homogeneity and weak signatures of local adaptation in the marine mussel Mytilus chilensis. Sci Rep 14, 21081 (2024). https://doi.org/10.1038/s41598-024-71944-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-71944-9
- Springer Nature Limited