
Abstract
Human noroviruses (HuNoVs) are a leading cause of acute viral gastroenteritis worldwide. Infectious outbreaks due to recombinant NoV genotype called GII.P16-GII.2 have been frequently reported since 2016. In this study, we expressed the major capsid protein VP1 from three GII.2 NoV strains using the recombinant baculovirus expression system. The assembly, histo-blood group antigen (HBGA)-binding patterns, and cross-blocking abilities of VP1 proteins were investigated. All the three NoV VP1 proteins successfully assembled into virus-like particles (VLPs). The HBGA-binding assay demonstrated a temporal binding pattern. The latest isolate bound to saliva samples of all blood types. Sequence alignment suggested that the observed gain in HBGA-binding ability was attributed to a limited number of amino acid mutations. Using chimeric VP1 proteins, we demonstrated that synergistic effects resulted in enhanced binding ability. Bile salts increased GII.2 VLP avidity for HBGAs except GII.2-2011/M1. In vitro blockade assay of salivary HBGA-VLP binding demonstrated the presence of cross-blocking effects among different strains. This study provides insight into the evolutionary binding characteristics and cross-blocking effects of GII.2 NoVs to facilitate the development of measures to control this type of viruses.
Similar content being viewed by others
Introduction
Human noroviruses (HuNoVs) have become the leading cause of acute viral gastroenteritis in developed countries. NoVs, which belong to the family Caliciviridae, are a group of non-enveloped, single-stranded, and positive-sense RNA viruses with genome sizes ranging from 7.5 to 7.7 kb in length. The HuNoV genome contains three open reading frames (ORFs). ORF1 encodes a nonstructural polyprotein consisting of at least six mature products: N-terminal protein (p48, NS1/2), NTPase (NS3), 3A-like protein (p22, NS4), viral protein, genome-linked protein (VPg, NS5), protease (Pro, NS6), and RNA-dependent RNA polymerase (RdRp, NS7)1,2,3. ORF2 and ORF3 encode structural proteins, including the major capsid protein VP1 and the minor capsid protein VP2, respectively. In vitro assays of VP1 proteins have been used to assemble virus-like particles (VLPs)4,5. Based on X-ray crystallography and reconstruction of its three-dimensional structure, VP1 has been shown to carry an N-terminal shell (S) and a C-terminal protruding (P) domain. The C-terminal domain comprises the highly conserved P1 and hypervariable P2 subdomains6. The P2 subdomain contains putative neutralization sites or epitopes and mediates interactions with histo-blood group antigens (HBGAs), which serve as the recognition receptors or attachment factors for HuNoVs infection7,8.
Based on the predicted full-length VP1 amino acid sequences, NoVs can be divided into at least 10 genogroups. The viruses in each genogroup can be further classified into polymerase and capsid genotypes. Viruses belonging to the GI, GII, GIV, GVIII, and GIX genogroups infect humans9. GII viruses, particularly GII.4 variants, account for a great majority of outbreaks and sporadic cases10,11,12. In recent years, the number of infections associated with non-GII.4 norovirus has increased significantly. A novel GII.17 variant was prevalent in the population during the winter of 2014–201513,14. During the winter of 2016, the emergence or re-emergence of the GII.2 variant increased the number of acute gastroenteritis outbreaks in China, and these infections rapidly spread to other regions, such as Japan, Italy, and Germany15,16. The earliest GII.2 isolate was traced back to 1971 (GenBank accession number, MF405169), and the Snow Mountain Virus (SMV) strain isolated in 1976 was the most widely studied17,18,19.
The mechanism underlying the sudden increase in GII.2 NoV infection cases have not been confirmed. In this study, in an effort to delineate the possible biological and immunological factors contributing to this phenomenon, we expressed the major capsid protein VP1 of three GII.2 NoV strains using the recombinant baculovirus expression system. The assembly, HBGA-binding patterns, and cross-blocking abilities of the VP1 proteins were investigated. The data provided in this study facilitate our understanding of this group of viruses.
Results
Phylogenetic analysis
Based on the GII.2 NoV VP1 sequences, the phylogenetic tree that constructed using MEGA X revealed that GII.2 NoV strains from different time periods formed distinct clusters, similar to the findings of Li et al.20 (Fig. 1). Namely, GII.2-1978 was Ungrouped, GII.2-2011 and GII.2-2016 were in transmission cluster V and cluster VIII, respectively.

Maximum-likelihood (ML) tree of of NoV GII.2 complete capsid gene sequences (n = 164, 1629 nt, TN93 + G + I). Each strain is labeled based on GenBank accession number, the country and the collection date it was isolated. The strains with VP1 proteins expressed are marked by red circles.
VP1 proteins derived from three representative GII.2 NoV strains assemble into VLPs
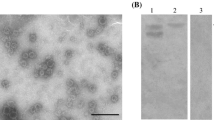
To characterize the biological and immunological relationships among GII.2 strains from different clusters, four GII.2 VP1 proteins (GII.2-1978, GII.2-1989, GII.2-2011, and GII.2-2016) were expressed using the recombinant baculovirus expression system. The expression level of GII.2-1989 VP1 protein was minimal, which led to an inadequate amount of protein for subsequent experiments. The VP1 proteins derived from the other three strains exhibited low-to-high expression levels and were thus purified and analyzed by SDS-PAGE (Supplemental Fig. 1A) and WB (Supplemental Fig. 1B). All three purified GII.2 NoV VP1 proteins were frequently observed as a doublet band. The assembly of the expressed proteins was further verified by TEM. As shown in Supplemental Fig. 2, all three GII.2 VP1 proteins successfully assembled into VLPs.
Salivary HBGA-VLP binding profiles of GII.2 NoV VLPs
To investigate the HBGA-binding profiles of GII.2 NoVs isolated from different time periods and representing different clusters, an in vitro salivary HBGA-VLP binding assay was subsequently performed. Saliva samples collected from healthy individuals with blood types A, B, AB, and O carrying known carbohydrate antigens were used (Supplementary Table 1). As shown in Fig. 2, GII.2 NoV VLPs derived from different clusters and time periods exhibited varied binding abilities against saliva samples. GII.2-1978 NoV VLPs bound strongly to saliva samples from individuals with blood types B and AB, but did not bind to other blood types. GII.2-2011 NoV VLPs did not bind any of the saliva samples tested, whereas GII.2-2016 NoV VLPs bound to all the saliva samples tested.

Salivary HBGA-VLPs binding assays.
Mutant GII.2-2011 VP1 proteins carrying triple mutations bind all saliva samples
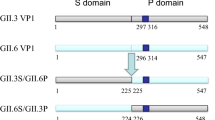
Sequence alignment of GII.2-2011 and GII.2-2016 NoV VP1 proteins revealed a total of eight different residues, with one located in the S domain and seven in the P domain. To determine the residues associated with enhanced binding with the saliva samples, two mutant GII.2-2011 VP1 proteins (GII.2-2011/M1 harboring V335I, T344S, and A354G, and GII.2-2011/M2 carrying D400E, T418I, and T448N) were designed and expressed using the recombinant baculovirus expression system (Fig. 3). The expressed proteins were purified and verified by SDS-PAGE and WB. As shown in Supplemental Fig. 1, both mutant proteins were successfully obtained (EM images in Supplemental Fig. 3). The results of the in vitro salivary HBGA-VLP binding assay indicated that the GII.2-2011/M1 VP1 proteins bound to all saliva samples with similar strengths, while the GII.2-2011/M2 VP1 proteins bound to saliva samples of individuals with blood types B and AB with reduced strength when compared with those of GII.2-2016 NoV VLPs (Fig. 2, Fig. 4). The above results suggest that triple mutations (V335I, T344S, and A354G) in the P2 domain induce binding to all saliva samples.

Schematic representation of the parental and mutant VP1 protein constructs (A) and amino acid mutations harbored by each mutant protein (B). Red and orange represents amino acid mutations harbored by GII.2-2011/M1 and GII.2-2011/M2, respectively.

Salivary HBGA-VLPs binding assays of GII.2 mutants.
Bile salts enhance salivary HBGA-VLP binding except GII.2-2011/M1
According to the study from Mallory et al.21, differences in GII.2 VLP binding to carbohydrates could be mitigated by bile acids. As showed in Fig. 5 in our study, 1% bile salts enhanced the binding of three GII.2 VLP and GII.2 2011/M2 VLP to human type A saliva and enhanced the binding of GII.2-1978 VLP, GII.2-2011 VLP, and GII.2-2011/M2 VLP to B and AB saliva, but decreased the binding of GII.2 2011/M1 VLP to all saliva samples. These data indicate that microvariation in VP1 sequences of GII.2 strains increases or decreases binding to different salivary HBGAs in the presence of bile salts.

Bile salts enhance salivary HBGA-VLP binding except GII.2-2011/M1.
Anti-GII.2-2011 NoV VLP hyperimmune serum exhibits cross-blocking effects
An in vitro salivary HBGA-VLP binding blockade assay was also conducted to determine whether genotype-specific hyperimmune sera exhibit cross-blocking effects. A single saliva sample from a blood type B individual was used because it showed the highest binding signal. The results indicated that the binding of all GII.2 VLPs including GII.2 mutant VLPs against HBGAs were specifically blocked by rabbit anti-GII.2-2011 VLP hyperimmune serum (Fig. 6).

In vitro salivary HBGA-VLP binding blockade assay. VLPs were used at 2 µg/mL. Boiled blood type B saliva sample was coated at 1:500. Rabbit anti-GII.2-2011 was used with a dilution from 1:100 to 1:51,200. OD 450 nm of GII.2-2011 VLP < 0.2.
Discussion
In this study, we expressed the major capsid protein VP1 from three GII.2 NoV strains and their assembly, HBGA-binding patterns, and cross-blocking abilities were investigated. Phylogenetic analysis revealed that GII.2 NoV strains from different time periods formed distinct clusters, consistent with the findings of a previous report20. We then expressed the VP1 protein from the four GII.2 NoV strains representing different clusters to characterize the biological and immunological relationships between these strains. Three GII.2 NoV VP1 proteins were successfully expressed. These proteins exhibited specific cleavage possibly at the N-terminus, as reported previously22. The results of the salivary HBGA-VLP binding assay showed that GII.2-2011 NoV VLPs bound to none of the saliva samples, while GII.2-1978 NoV VLPs bound to blood type B and blood type AB saliva samples. The blood type AB saliva sample has been shown to carry a blood type B carbohydrate antigen22. Surprisingly, GII.2-2016 NoV VLPs exhibited binding ability against all the saliva samples tested. The lack of binding between GII.2-2011 NoV VLPs and salivary HBGAs has yet to be reported, suggesting the possible role of other factors (such as bile salts)21,23. Furthermore, different batches of GII.2-2016 NoV VLPs also showed significantly varied binding activities against salivary HBGAs (data not shown), thus whether the observed lack of binding of GII.2-2011 NoV VLPs against salivary HBGAs is associated with falsely assembled VLPs should be further investigated. Infection of a nonsecretor population by GII.2 NoV has been reported23, further confirming the possible involvement of factors other than HBGAs. In fact, bile acids have been shown to promote the binding of certain types of NoV VLPs to HBGAs, and interactions between NoV VLPs and bacteria have also been described21. Thus, the in vitro HBGA-VLP binding assay might not be fully recapitulate the in vivo binding environment of NoV to HBGAs or other carbohydrate antigens.
A previous study showed that V256 and D382 substitutions might play a role in the expanded HBGA-binding spectrum15. However, no such altered residues were detected in the GII.2-2016 strain. When compared with the GII.2-1976 Snow Mountain Virus (SMV, AY134748) with known crystal structures, these four GII.2 VP1 share ≥ 97.0% similarity in amino acid sequences and a total of 20 amino acid residues located in the VP1 region that differ among them are identified. When analyzed in a chronological order of the HBGA binding interface regions24, residue 354 in site I mutated to A in the GII.2-2011 strain and reversed back to G in the GII.2-2016 strain, which may have led to the regaining of binding ability against HBGAs. Residues in site II and III are conserved among all these four strains, but mutations adjacent to site I, II, and III are observed in both GII.2-2011 and GII.2-2016 strains, which could also have contributed to the changes in HBGA-binding patterns (Supplemental Fig. 4).
In our study, an HBGA non-binder (GII.2-2011) could be converted to an HBGA binder in the presence of bile salts. Furthermore, bile salts enhanced HBGA binding for GII.2-1978. As mentioned by Kilic et al. that bile acid was an important cofactor for GII.1 binding to HBGAs25, bile acid binding to GII.2-2011 might be a critical step that enabled it to bind HBGAs. It is also possible that bile salts, as observed in murine norovirus, cause allosteric conformational changes in the P domain toward a form that binds the receptor or may increase the number of available receptor binding sites26,27.
Sequence alignment demonstrated few amino acid differences between the VP1 proteins of the GII.2-2011 and GII.2-2016 strains. We thus designed two mutant VP1 proteins based on the backbone of GII.2-2011 NoV VP1 to identify the amino acids contributing to the observed HBGA binding. The GII.2-2011/M1 proteins, which harbor the V335I, T344S, and A354G mutations, exhibited binding ability and strength comparable to that of GII.2-2016 VLPs, while GII.2-2011/M2 proteins harboring D400E, T418I, and T448N mutations presented significantly increased binding signals compared with negative controls (P < 0.05) and a binding pattern similar to that of GII.2-1978 VLPs. It should be noted that protein concentrations were obtained using absorbance values and similar amounts of proteins were loaded. However, the GII.2-2011/M2 proteins presented weaker protein band intensities than did the other proteins, and simple comparisons based on protein intensities (SDS-PAGE) revealed that the intensities of the GII.2-1978, GII.2-2011, GII.2-2016, and GII.2-2011/M1 proteins were similar and approximately 0.7-fold greater than those of the GII.2-2011/M2 proteins. This difference might be due to the varied structures formed and the varied absorbance. The results indicate that the limited number of mutations significantly alter the binding profiles against HBGAs; the residues located in different regions might mediate binding against different carbohydrate antigens, and residues in both the N- and C-terminal regions of P2 domains may additively enhance binding to HBGAs.
To further determine whether the phenotype and rapid spread are associated with immune escape, an in vitro salivary HBGA-VLP binding blockade assay was performed. The results indicated that the hyperimmune serum produced against GII.2-2011 VLPs blocked the binding of both GII.2-1978 and GII.2-2016 VLPs, as well as GII.2-2011/M1 and GII.2-2011/M2 proteins to blood type B saliva samples, suggesting the conservation of blocking antigenic epitope(s) among these strains. A one-sided blocking effect was observed for GII.6 hyperimmune serum (data not shown). This phenomenon was reported in our previous study28 and is possibly associated with shared antigenic epitopes that produce steric hindrance when bound by antibodies against the binding of GII.2-2011/M1 proteins to HBGAs. Thus, the outbreaks of GII.2 strains during 2016–2017 was not related to antigenic drift, which was unlike sequential pandemic outbreaks of GII.4 strains21. The factors underlying the sudden appearance and rapid spread of GII.2 to multiple countries remain to be determined, but a multitude of factors might be at play, such as high burden in seafood worldwide29, enhanced stability when compared with other genotypes30, gaining of more replication-efficient RdRp31,32, and expanded binding ability against HBGAs15. It is most likely that the observed spike in GII.2 infection is attributed to a combination of mentioned above factors.
In summary, GII.2 NoVs representing different clusters cocirculate and exhibit different HBGA-binding profiles. While the cross-blocking effects among different GII.2 strains suggest that recent recrudescence might be associated with multiple factors, broad receptor binding might be related to GII.2 sudden outbreaks in this study. The data provided in this study deepen our understanding of the biological and immunological characteristics of GII.2 NoVs and facilitate the development of countermeasures against this type of virus.
Methods
Phylogenetic analysis
The phylogenetic analysis was performed using a total of 164 GII.2 NoV complete VP1 sequences downloaded from the NCBI GenBank database. The VP1 sequence alignment was performed using BioEdit and a maximum-likelihood (ML) tree was constructed by MEGA (version X) using the best-fit model TN93 + G + I (TN93: Tamura-Nei model, G + I: Gamma distributed with Invariant sites). The statistical significance of the constructed phylogenetic tree was estimated by bootstrap analysis with 500 replicates.
Gene synthesis and generation of recombinant baculoviruses
The GII.2 NoV strains under GenBank accession nos. JN699037, X81879, KC464505, and KY457581 were identified, and their corresponding VP1 proteins were designated GII.2-1978 (with an RdRp genotype of P30), GII.2-1989 (with an RdRp genotype of P2), GII.2-2011 (with an RdRp genotype of P16), and GII.2-2016 (RdRp genotype not available), respectively. The full-length and chimeric GII.2 VP1 amino acid sequences were optimized based on the codon usage frequency in Spodoptera frugiperda (Sf9) cells. The sequences were synthesized (Sangon Biotech, Shanghai, China) and ligated into the pFastBac-Dual vector (Invitrogen) under the control of the polyhedrin promoter to generate donor plasmids. Subsequently, they were used to transform competent E. coli DH10B cells to produce bacmids. The bacmids were purified and used to transfect Sf9 cells to generate recombinant baculoviruses as previously described33.
Production of VLPs
VLPs were synthesized and purified as previously described22.The integrity and assembly of the VLPs were analyzed via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transmission electron microscopy (TEM) at 100 kV after negative staining, respectively.
Production of hyperimmune serum
All animal experiments were carried out at Wuhan Institute of Biological Products Co., Ltd., following the guidelines of the Chinese Council on Animal Care and were reported in accordance with ARRIVE guidelines. Purified GII.2-2011 NoV VLPs were used to immunize Japanese big ear rabbits intramuscularly at two-week intervals. A total of four immunizations were conducted at 50 μg/dose mixed 1:1 with Freund’s complete adjuvant (first immunization) or Freund's incomplete adjuvant (subsequent immunizations). One week after the last immunization, rabbits were fasted for at least 4 h and were anesthetized by i.p. injection of 200 mg/kg pentobarbital. Blood was collected from the rabbit cervical artery. Then sera were separated from blood and anti-VLP-specific IgG titers were determined via indirect ELISA using a GII.2 NoV VP1 protein concentration of 0.1 μg/mL.
SDS-PAGE and Western blotting (WB)
For SDS-PAGE analysis, purified VLPs were boiled and then loaded onto a discontinuous 8–16% precast gel. After separation, the proteins were stained with Coomassie blue stain. For WB analysis, the separated proteins were transferred to a nitrocellulose (NC) membrane, which was subsequently incubated with (a) rabbit anti-GII.2-2011 NoV VLP hyperimmune sera diluted at 1:2000, and (b) horseradish peroxide (HRP)-conjugated goat anti-rabbit IgG antibodies. The protein bands were then detected via 3,3′-diaminobenzidine (DAB) staining. The supernatant obtained from wild-type baculovirus-infected Sf9 cells was used as the negative control.
Salivary HBGA-VLP binding assay
Saliva samples were collected from healthy individuals, and HBGAs in saliva samples were determined as described previously using monoclonal antibodies against blood group A antigen, blood group B antigen, Lewis a, Lewis b, Lewis x, Lewis y, blood group H1 (O) antigen, and blood group H2 antigen (Abcam, UK)34. The in vitro VLP-salivary HBGA binding assay was performed as previously described with minor modifications35. Briefly, 96-well microtiter plates were precoated with 100 μL per well of processed blood type A, B, AB, and O saliva samples diluted 1/500 in carbonate-bicarbonate buffer solution (pH 9.6) by incubating at 37 °C overnight and blocked with PBS containing 0.5% (v/v) Tween-20 and 1% (w/v) bovine serum albumin (BSA) by incubating at 37 °C for 1 h before the addition of decreasing two-fold serial dilutions of purified GII.2 NoV VLPs diluted in PBS-T starting at 8 μg/mL. Rabbit anti-GII.2-2011 NoV VLP hyperimmune serum diluted 1:1000 in PBS-T and HRP-conjugated goat anti-rabbit IgG were sequentially added to the above wells and incubated for 1 h and 30 min, respectively. The plate was washed five times with PBS-T after each incubation, and all the incubations were performed at 37 °C. Bound VLPs were detected by the addition of tetramethylbenzidine (TMB) and the peroxide urea. The optical density (OD) at 450 nm was measured on a Multiskan MK3 microplate reader (Thermo Fisher Scientific Inc., Waltham, MA, USA).
Salivary HBGA-VLP binding assay in the presence of bile salts
Bile salts were diluted at 1% in 2 μg/mL VLP solution and then added to blood type B saliva sample coated plates. Bound VLPs were detected with anti-GII.2-2011 rabbit hyperimmune sera, followed by anti-rabbit IgG-HRP, and color developed as above.
Salivary HBGA-VLP binding blockade assay
GII.2 NoV VLPs (2 μg/mL) diluted in PBS-T were preincubated with serially decreasing concentrations of rabbit anti-GII.2-2011 NoV VLP serum34 by incubating at 37 °C for 30 min before they were transferred to blood type B saliva-coated wells. Wells treated with GII.2 NoV VLPs and PBS-T served as positive and negative controls, respectively. The bound VLPs were detected as described above using rabbit anti-GII.2 NoV VLP hyperimmune serum. The blocking index was calculated (as a percentage) using the following formula: (mean OD without sera—mean OD with sera)/mean OD without sera × 100%.
Data availability
All data supporting the findings of this study are available from the corresponding authors on reasonable request and supplementary information files.
References
Zheng, L. et al. Phylogenetic and biological characterizations of a GI.3 norovirus. Infect. Genet. Evol. 85, 104554. https://doi.org/10.1016/j.meegid.2020.104554 (2020).
Karst, S., Wobus, C., Goodfellow, I., Green, K. & Virgin, H. Advances in norovirus biology. Cell Host Microbe 15(6), 668–680. https://doi.org/10.1016/j.chom.2014.05.015 (2014).
Green, K. Fields Virology 582–608 (Lippincott, Williams, and Wilkins, 2013).
Jiang, X., Wang, M., Graham, D. & Estes, M. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J. Virol. 66(11), 6527–6532. https://doi.org/10.1128/jvi.66.11.6527-6532.1992 (1992).
Huo, Y. et al. Expression and purification of norovirus virus like particles in Escherichia coli and their immunogenicity in mice. Mol. Immunol. 93, 278–284. https://doi.org/10.1016/j.molimm.2017.07.014 (2018).
Prasad, B. et al. X-ray crystallographic structure of the Norwalk virus capsid. Science 286(5438), 287–290. https://doi.org/10.1126/science.286.5438.287 (1999).
Tan, M. & Jiang, X. Histo-blood group antigens: A common niche for norovirus and rotavirus. Expert Rev. Mol. Med. 16, e5. https://doi.org/10.1017/erm.2014.2 (2014).
Liu, W. et al. A unique human norovirus lineage with a distinct HBGA binding interface. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1005025 (2015).
Chhabra, P. et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 100(10), 1393–1406. https://doi.org/10.1099/jgv.0.001318 (2019).
Anfruns-Estrada, E. et al. Epidemiological and genetic characterization of norovirus outbreaks that occurred in Catalonia, Spain, 2017–2019. Viruses 14(3), 488. https://doi.org/10.3390/v14030488 (2022).
Yu, F. et al. Norovirus outbreaks in China, 2000–2018: A systematic review. Rev. Med. Virol. 32(6), e2382. https://doi.org/10.1002/rmv.2382 (2022).
Calderwood, L. et al. Norovirus outbreaks in long-term care facilities in the United States, 2009–2018: A decade of surveillance. Clin. Infect. Dis. 74(1), 113–119. https://doi.org/10.1093/cid/ciab808 (2022).
Chan, M. et al. Rapid emergence and predominance of a broadly recognizing and fast-evolving norovirus GII.17 variant in late 2014. Nat. Commun. 6, 10061. https://doi.org/10.1038/ncomms10061 (2015).
Lu, J. et al. Gastroenteritis outbreaks caused by norovirus GII1.7, Guangdong Province, China, 2014–2015. Emerg. Infect. Dis. 21(7), 1240–1242. https://doi.org/10.3201/eid2107.150226 (2015).
Ao, Y. et al. Genetic analysis of reemerging GII.P16-GII.2 noroviruses in 2016–2017 in China. J. Infect. Dis. 218(1), 133–143. https://doi.org/10.1093/infdis/jiy182 (2018).
Wang, J. et al. Norovirus GII.2[P16] strain in Shenzhen, China: A retrospective study. BMC Infect. Dis. 21(1), 1122. https://doi.org/10.1186/s12879-021-06746-9 (2021).
Lindesmith, L. et al. Cellular and humoral immunity following snow mountain virus challenge. J. Virol. 79(5), 2900–2909. https://doi.org/10.1128/jvi.79.5.2900-2909.2005 (2005).
Swanstrom, J., Lindesmith, L. C., Donaldson, E. F., Yount, B. & Baric, R. S. Characterization of blockade antibody responses in GII.2.1976 snow mountain virus-infected subjects. J. Virol. 88(2), 829–837. https://doi.org/10.1128/jvi.02793-13 (2014).
Rouphael, N. et al. Dose-response of a norovirus GII.2 controlled human challenge model inoculum. J. Infect. Dis. 226(10), 1771–1780. https://doi.org/10.1093/infdis/jiac045 (2022).
Li, X., Liu, H., Rife Magalis, B., Kosakovsky Pond, S. L. & Volz, E. M. Molecular evolution of human norovirus GII.2 clusters. Front. Microbiol. 12, 655567. https://doi.org/10.3389/fmicb.2021.655567 (2021).
Mallory, M. L., Lindesmith, L. C., Brewer-Jensen, P. D., Graham, R. L. & Baric, R. S. Bile facilitates human norovirus interactions with diverse histoblood group antigens, compensating for capsid microvariation observed in 2016–2017 GII.2 strains. Viruses 12(9), 989. https://doi.org/10.3390/v12090989 (2020).
Huo, Y., Wan, X., Ling, T. & Shen, S. Biological and immunological characterization of norovirus major capsid proteins from three different genotypes. Microb. Pathog. 90, 78–83. https://doi.org/10.1016/j.micpath.2015.11.022 (2016).
Lindesmith, L. et al. Virus-host interactions between nonsecretors and human norovirus. Cell. Mol. Gastroenterol. Hepatol. 10(2), 245–267. https://doi.org/10.1016/j.jcmgh.2020.03.006 (2020).
Tan, M. & Jiang, X. Norovirus gastroenteritis, carbohydrate receptors, and animal models. PLOS Pathog. 6(8), e1000983. https://doi.org/10.1371/journal.ppat.1000983 (2010).
Kilic, T., Koromyslova, A. & Hansman, G. Structural Basis for human norovirus capsid binding to bile acids. J. Virol. https://doi.org/10.1128/jvi.01581-18 (2019).
Williams, A. et al. A norovirus uses bile salts to escape antibody recognition while enhancing receptor binding. J. Virol. 95(13), e0017621. https://doi.org/10.1128/jvi.00176-21 (2021).
Sherman, M. et al. Bile salts alter the mouse norovirus capsid conformation: Possible implications for cell attachment and immune evasion. J. Virol. https://doi.org/10.1128/jvi.00970-19 (2019).
Huo, Y. et al. Characterization of virus-like particles derived from a GII.3 norovirus strain distantly related with current dominating strains. Virus Genes 52(5), 613–619. https://doi.org/10.1007/s11262-016-1359-1 (2016).
Li, Y. et al. A systematic review and meta-analysis indicates a substantial burden of human noroviruses in shellfish worldwide, with GII.4 and GII.2 being the predominant genotypes. Food Microbiol. 109, 104140. https://doi.org/10.1016/j.fm.2022.104140 (2023).
Tan, M. et al. The globally re-emerging norovirus GII.2 manifests higher heat resistance than norovirus GII.4 and Tulane virus. J. Appl. Microbiol. 132(3), 2441–2449. https://doi.org/10.1111/jam.15379 (2022).
Zheng, G.-l, Zhu, Z.-x, Cui, J.-l & Yu, J.-m. Evolutionary analyses of emerging GII.2[P16] and GII.4 Sydney [P16] noroviruses. Virus Evol. 8(1), veac030. https://doi.org/10.1093/ve/veac030 (2022).
Cheung, S. K. C. et al. Higher viral load of emerging norovirus GII.P16-GII.2 than pandemic GII.4 and epidemic GII.17, Hong Kong, China. Emerg. Infect. Dis. 25(1), 119–122. https://doi.org/10.3201/eid2501.180395 (2018).
Huo, Y., Ma, J. & Liu, J. Identification of a GII.6 norovirus blockade antibody epitope. Virus Res. 334, 199168. https://doi.org/10.1016/j.virusres.2023.199168 (2023).
Huo, Y., Zheng, L., Chen, X., Ge, L. & Wang, Y. Expression and characterization of the major capsid protein derived from a GII.6 norovirus strain isolated in China. Microb. Pathog. 105, 131–137. https://doi.org/10.1016/j.micpath.2017.02.025 (2017).
Huo, Y., Wan, X., Wang, Z., Meng, S. & Shen, S. Production of Norovirus VLPs to size homogeneity. Virus Res. 204, 1–5. https://doi.org/10.1016/j.virusres.2015.04.009 (2015).
Funding
This research was supported by the Scientific and Technological Project of Henan Province (242102310396).
Author information
Authors and Affiliations
Contributions
J.M. and J.L. performed the experiments and participated in data analysis. J.M. wrote the manuscript. Y.H. designed the study and edited the original draft. All authors reviewed the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was approved by the Institutional Ethics Committee of The Sixth People’s Hospital of Zhengzhou, China (IEC-KY-2023-07). All experiments were performed in accordance with relevant guidelines and regulations. Signed informed consent was obtained from each individual before the collection of saliva samples.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, J., Liu, J. & Huo, Y. Biological and immunological characterization of major capsid protein VP1 from distinct GII.2 norovirus clusters. Sci Rep 14, 21035 (2024). https://doi.org/10.1038/s41598-024-72062-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-72062-2
- Springer Nature Limited