
Abstract
Following an injury, the liver embarks on a process that drives the accumulation and reformation of the extracellular matrix, leading to hepatic fibrosis. Type I interferons (IFNs), including IFN-α and IFN-β, play a crucial role in averting chronic liver injury through the activation of IFN-stimulated genes (ISGs), which are instrumental in sculpting adaptive immunity. The role of 2ʹ–5ʹ-oligoadenylate synthase-like protein 1 (OASL1), an antiviral ISG, in the context of liver fibrosis remains to be elucidated. To elicit liver fibrosis, a diet containing 0.1% diethoxycarbonyl-1,4-dihydrocollidine (DDC) and carbon tetrachloride (CCl4) were employed to induce cholestatic- and hepatotoxin-mediated liver fibrosis, respectively. Histological analyses of both models revealed that OASL1−/− mice exhibited reduced liver damage and, consequently, expressed lower levels of fibrotic mediators, notably α-smooth muscle actin. OASL1−/− mice demonstrated significantly elevated IFN-α and IFN-β mRNA levels, regulated by the IFN regulatory factor 7 (IRF7). Additionally, OASL1−/− ameliorated chronic liver fibrosis through the modulation of nuclear factor-κB (NF-κB) signaling. The effect of OASL1 on type I IFN production in acute liver damage was further explored and OASL1−/− mice consistently showed lower alanine transaminase levels and pro-inflammatory cytokines, but IFN-α and IFN-β mRNA levels were upregulated, leading to amelioration of acute liver injury. Additionally, the study discovered that F4/80-positive cells were observed more frequently in OASL1−/− CCl4 acutely treated mice. This implies that there is a significant synergy in the function of macrophages and OASL1 deficiency. These results demonstrate that in instances of liver injury, OASL1 inhibits the production of type I IFN by modulating the NF-κB signaling pathway, thereby worsening disease.
Similar content being viewed by others


Introduction
Chronic liver diseases, affecting approximately 850 million individuals globally, pose a significant health challenge1. Liver fibrosis, a manifestation of chronic and progressive liver ailments, arises from various liver insults that prompt the activation of hepatic stellate cells (HSCs). This process results in a disruption of extracellular matrix production, marking a pivotal stage in the development of liver fibrosis and potentially cirrhosis2. Escalation of this condition can lead to life-threatening complications such as portal hypertension, liver failure, or hepatocellular carcinoma3. Without intervention, liver fibrosis and its consequential disorders can culminate in death due to compromised liver functionality, underscoring the critical demand for antifibrotic treatments4. Thus, a profound comprehension of the molecular mechanisms driving liver fibrosis is imperative for devising novel therapeutic strategies.
The 2′–5′-oligoadenylate synthetase (OAS) family, activated by type I interferons (IFN)5, includes OAS1, OAS2, OAS3, and the OAS-like protein (OASL) of IFN-stimulated genes (ISGs)6. Despite being part of the OAS family, OASL diverges by lacking the conventional OAS activity and instead showcases two ubiquitin-like (UBL) domains7. Humans possess two OASL isoforms: OASLa (p59) and OASLb (p30), in contrast to mice that harbor two OASL1 genes situated on chromosome 5, OASL1 and OASL28,9. Specifically, OASL1 is modulated by type I IFN and is implicated in bolstering antiviral defenses while simultaneously attenuating robust type I IFN synthesis through the inhibition of IFN translation via IFN regulatory factor 7 (IRF7)10. Additional research has illuminated OASL1’s capacity to diminish the type I IFN response by modulating deoxyribonucleic acid (DNA) virus infections through its interaction with the cyclic guanosine monophosphate–adenosine monophosphate synthase and stimulator of IFN genes pathway11. In models of hepatic ischemia/reperfusion injury, the absence of OASL1 or nuclear factor erythroid 2-related factor 2 intensified the inflammatory response, mediated by Ras GTPase-activating protein-binding protein 1 and TANK-binding kinase 1, alongside elevating caspase-3-initiated apoptosis and receptor-interacting serine/threonine-protein kinase 3 mediated necroptosis12. Conversely, nullifying OASL1 precipitated antitumorigenic immune responses through the activation of type I IFN13. Furthermore, investigations into multiple sclerosis, an autoimmune demyelinating condition, disclosed that OASL1 deficiency mitigated the emergence of autoimmunity14. It has been demonstrated that inhibiting OASL1 augments antiviral defense against genital herpes simplex virus type 2 infection by amplifying type I IFN production15. Accordingly, OASL1’s function significantly oscillates based on the disease environment.
Type I IFNs, comprising the broadly expressed IFN-α and IFN-β, exhibit anti-inflammatory properties as constituents of innate immunity16. Similarly, activating dendritic cells via Toll-like receptor 7 signaling engenders the production of type I IFN, which subsequently triggers antifibrogenic interleukin (IL)-1 receptor antagonist synthesis in kupffer cells within fibrotic livers3. Hence, type I IFNs might mitigate chronic liver injury through anti-inflammatory actions. Nevertheless, despite OASL1’s established role in modulating type I IFN, its involvement in liver fibrosis remains to be thoroughly explored. This study posits that inhibition of OASL1 activity ameliorates liver injury via enhancement of type I IFN production.
OASL1 enhances the severity of biliary fibrosis in mice
After a 4-week regimen of 0.1% diethoxycarbonyl-1,4-dihydrocollidine (DDC) feeding (Fig. 1A), mice subjected to the DDC diet demonstrated notable loss in body weight (BW). Figure 1B illustrates that there were no significant disparities in BW between wild-type (WT) and OASL1 knockout mice (abbreviated as OASL1−/−) on the DDC diet. Intriguingly, a period of 4 weeks on the DDC diet led to a marked increase in OASL1 mRNA expression in the livers of WT mice (Fig. 1C). Concurrently, OASL1−/− mice on the DDC diet exhibited significantly lower serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) compared with WT mice subjected to the same diet (Fig. 1D). Considering that cholangiopathies induced by DDC and related ductular proliferation responses are intimately linked with inflammation in the progression of biliary fibrosis17, we further explored the role of OASL1 in hepatic inflammation. As depicted in Fig. 1E, OASL1−/− mice fed with DDC displayed significantly diminished tumor necrosis factor-alpha (TNF-α) levels in comparison to WT mice on the DDC diet. Subsequently, a double immunofluorescence staining of alpha smooth muscle actin (αSMA) and glial fibrillary acid protein (GFAP) in tissue sections was performed to analyze HSC immunoreactivity18. Staining revealed significantly lower αSMA (red fluorescence) and GFAP (green fluorescence) immunoreactivity mainly in portal tracts of OASL1−/− mice on the DDC diet compared to DDC-fed WT mice (Fig. 1F). GFAP was colocalized with αSMA but additionally showed immunoreactivity in perisinusoidal stellate cells. Next, mRNA expression levels of αSMA and collagen type I alpha 1 (Col 1α1) were assessed to gauge fibrogenic activities. OASL1−/− mice on the DDC diet showed reduced αSMA expression levels and significantly lower Col 1α1 expression relative to their WT counterparts on the DDC diet (Fig. 1G). Consistent with these findings, OASL1−/− mice on the DDC diet manifested significantly diminished αSMA protein levels in their livers (Fig. 1H and Supplementary Fig. S2).

OASL1 increases the effects of DDC on biliary fibrosis severity in mice. (A) Schematic diagram of the experimental design used to determine effects of OASL1 on DDC feeding. (B) Changes in BW of WT and OASL1−/− mice, measured in grams (g), following 4 weeks of DDC feeding compared to the control chow diet (NC), with each group consisting of 7 mice. (C) Quantification of OASL1 mRNA expression patterns via real-time polymerase chain reaction (RT‒qPCR). (D) Plasma biochemistry analyses reveal the extent of cholestasis (AST in IU/L) and parenchymal liver damage (ALT in IU/L), with groups of 6 mice each, assayed in duplicate. (E) mRNA levels of the TNFα proinflammatory cytokine. (F) Immunofluorescence visualization of αSMA (red-white arrows) and GFAP (green-arrow heads) within liver sections of WT and OASL1−/− mice fed with DDC. (G) αSMA, and Col 1α1 fibrogenic markers, showing relative mRNA expression. (H) Cropped western blots for liver tissues from WT and OASL1−/− mice, depicting αSMA protein levels relative to β-actin (n = 3/group). Original blots are presented in Supplementary Fig. S2. Each data point represents the mean ± standard deviation (SD). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
OASL1−/− affects DDC diet-induced biliary fibrosis phenotypes
Histopathological analysis assessing biliary fibrosis revealed that OASL1−/− mice on the DDC diet had significantly less bile duct proliferation and collagen accumulation in liver tissues than WT mice on the DDC diet (Fig. 2A,B). Additionally, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining for apoptosis demonstrated a marked reduction in the TUNEL-positive area in the livers of OASL1−/− mice fed the DDC diet compared to WT mice (Fig. 2C). During cholangitis aberrant expression of epithelial cell marker cytokeratin 19 (CK19) is induced by DDC-feeding19. Thus, CK19-positive cells were significantly less pronounced in OASL1−/− mice fed with DDC than WT mice on the DDC diet (Fig. 2D). Moreover, elevated serum bilirubin levels, commonly associated with obstructive jaundice and cholestasis20, were significantly lower in DDC-fed OASL1−/− mice than in WT mice. We also measured the amount of hydroxyproline in liver lysates, and we found that OASL1−/− mice fed with DDC diet had significantly lower levels of hydroxyproline in their livers (Fig. 2E). Subsequent analysis of the F4/80 mRNA expression levels revealed a slightly rising trend in OASL1−/− mice fed the DDC diet, but no statistically significant difference was noted (Fig. 2E). Utilizing a mouse IFN-α enzyme-linked immunosorbent assay (ELISA) kit, it was quantitatively determined that OASL1−/− mice fed with DDC diet had considerably higher levels of IFN-α expression (Fig. 2F). However, IFN-α and IFN-β mRNA levels in OASL1-deficient mice showed no statistical significance (Fig. 2G). Collectively, these results underscore a pivotal role for OASL1 in advancing biliary fibrosis.

OASL1−/− reduces DDC diet-induced biliary fibrosis phenotypes. (A) Histological analysis of liver sections from diet-fed WT and OASL1 −/− mice, visualized through haematoxylin and eosin (H&E) staining. (B) The presence of Sirius red-positive collagen fibers was quantified as a percentage of the staining area. (C) Enumeration of TUNEL-positive cells represented by yellow arrows and black arrows representing bile pigments in the zoomed-in image. Total magnification utilized: 200×. Scale bar measures 100 µm. (D) Representative images of CK19 positive cells (black arrows) at total magnification: 400×. Scale bar measures 50 µm. (E) Measurement of serum bilirubin levels using a bilirubin ELISA kit, quantification of hydroxyproline content in μg/mg, and F4/80 mRNA levels. (F, G) Analysis of IFN-α via ELISA (ng/mg) and relative mRNA expression for IFN-β and IFN-α. The number of samples is n = 6–7/group. Data is presented as the mean ± SD. Significance indicated by *p < 0.05, **p < 0.01; ***p < 0.001.
OASL1 enhances the severity of carbon tetrachloride (CCl4)-induced liver fibrosis in mice
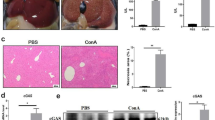
To further investigate the role of OASL1 in liver fibrosis, we conducted additional experiments using CCl4 (Fig. 3A). The chronic liver injury model induced by CCl4 is known to provoke type I IFN stimulation21. Interestingly, OASL1 expression was elevated in CCl4-treated WT mice, as illustrated in Fig. 3B. In contrast, the BW of the OASL1−/− mice treated with CCl4 remained stable over time, whereas the weights of the CCl4-challenged WT mice started to decline markedly from the first to the second week (Fig. 3C). The serum ALT levels in CCl4-treated OASL1−/− mice were significantly lower than those in WT mice exposed to CCl4 (Fig. 3D). Further histopathological analyses using H&E and Sirius red staining revealed that CCl4 -treated OASL1−/− mice exhibited significantly reduced collagen deposition in their livers compared to WT mice subjected to CCl4 (Fig. 3E,F). Additionally, immunohistochemistry (IHC) staining of αSMA-positive cells in Fig. 3G further demonstrated significantly lower expression levels in CCl4-treated OASL1−/− mice than in WT mice exposed to CCl4, corroborating the observations in Fig. 3F. TUNEL staining to detect apoptosis under CCl4 treatment showed that the proportion of apoptotic cells was markedly lower in OASL1−/− mice than in WT mice treated with CCl4 (Fig. 3H). Collectively, these results suggest that the absence of OASL1 mitigates CCl4-induced chronic liver fibrosis.

OASL1−/− reduces CCl4-induced fibrosis phenotypes. (A) Schematic diagram of the experimental design used to determine effects of OASL1 on CCl4-induced fibrosis. (B) A bar graph depicts the relative mRNA expression levels of OASL1. (C) Changes in BW, presented as a percentage (%). (D) Levels of Serum ALT (IU/L). (E) H&E staining of liver sections embedded in paraffin is illustrated through photomicrographs. (F) Quantification of the Sirius red staining area, shown as a percentage. (G) α-SMA immunoreactive cells. Total magnification utilized: 100×. Scale bar measures 200 µm. (H) TUNEL positive cells (black arrows showing apoptotic cells). The total magnification is 200×. Scale bar = 100 µm (n = 6–7/group). Data are presented as the mean ± SD. *p < 0.05, **p < 0.01.
OASL1−/− decreases liver damage by promoting type I IFN production upon CCl4-induced fibrosis in mice
CCl4 administration, inducing liver fibrosis and subsequent inflammation22, led us to analyze the protein levels of αSMA and IFN-β via western blot. Results showed no significant differences in αSMA protein levels in CCl4-treated OASL1−/− mice compared to WT livers injured by CCl4 (Fig. 4A). Remarkably, IFN-β protein expression was substantially higher in CCl4-treated OASL1−/− livers at 4 weeks relative to WT mice (Fig. 4A). The level of liver total collagen hydroxyproline was significantly low in CCl4-treated OASL1−/− mice compared to WT CCl4-treated livers (Fig. 4B). Transforming growth factor beta 1 (TGF-β1), crucial in hepatic fibrosis development, prompts extracellular matrix overexpression and upregulates fibrogenes such as tissue inhibitor of metalloproteinases-1 (TIMP-1), primarily in HSCs23,24. In the CCl4 model, OASL1−/− mice demonstrated markedly lower TGF-β and TIMP-1 mRNA levels than WT mice (Fig. 4B). IL-1β mRNA levels were reduced in OASL1−/− livers treated with CCl4 (Fig. 4C). Serum IL-1β, serum IL-6 and tissue lysate TNF-α levels, assessed via ELISA, were significantly lower in CCl4-treated OASL1−/− mice than in WT mice (Fig. 4D,E). As IRF7 governs type I IFN production and can be inhibited by OASL125, a notable increase in IFN-α and IFN-β levels were observed in OASL1−/− mice, correlating with slight increase in IRF7 mRNA levels (Fig. 4E,F). Interestingly, western blot analysis revealed significantly high levels of IRF7 protein levels in CCl4-treated OASL1−/− mice compared to WT livers injured by CCl4 (Fig. 4F and Supplementary Fig. S3). All together, these findings point to OASL1's role as an IRF7 translation inhibitor that negatively regulates type I IFN.

OASL1−/− decreases liver damage by promoting type I IFN production in response to CCl4-induced fibrosis in mice. (A) Displays cropped western blot images, illustrating the relative αSMA and IL-1β protein levels normalized against β-actin levels (n = 3/group). Original western blots are presented in Supplementary Fig. S3. (B) Hydroxyproline content in liver lysates measured in μg/mg. RT‒qPCR measurements of mRNA expression levels of TGF-β, TIMP-1, and (C) IL-1β. (D, E) IL-1β levels, IL-6 serum levels, and TNF-α (in liver lysates) of proinflammatory cytokines were quantified using sandwich ELISA. Panel (E) also presents RT‒qPCR measurements of mRNA expression levels of IFN-α, and IFN-β. (F) IRF7 mRNA expression and cropped western blot analysis of IRF-7 protein levels. Original western blots are presented in Supplementary Fig. S3. Data are represented as mean ± SD. * signifies p < 0.05, **p < 0.01, while *** denotes p < 0.001.
OASL1 modulates liver fibrosis via nuclear factor-κB (NF-κB) activation
The activation of NF-κB is recognized as a sufficient condition for the induction of liver fibrosis under various stressors26. Consequently, we investigated OASL1’s role in modulating extracellular signal-regulated kinases (ERK), mitogen-activated protein kinase p38 (p38), and NF-κB activation in CCl4-induced liver fibrosis. Western blot analysis of protein expression revealed a marked reduction in phosphorylated NF-κB protein levels in CCl4-treated OASL1−/− mice. Conversely, mice lacking OASL1−/− treated with CCl4 exhibited decreased levels of phosphorylated P38 and ERK (Fig. 5A). Moreover, NF-κB activation in liver fibrosis has been implicated in the increased release of TGF-β27. Hence, we isolated primary HSCs and employed siOASL1 to diminish OASL1 expression. Remarkably, TGF-β levels were significantly lowered in HSCs with reduced OASL1 expression, without substantially altering fibrotic marker levels one-day post-transfection (Fig. 5B). Therefore, the deficiency of OASL1 attenuates NF-κB activation and its associated inflammatory response, thereby mitigating CCl4-induced liver fibrosis.

OASL1 influences liver fibrosis through the activation of NF-κB. (A) Cropped western blot images and analysis illustrate the impact of OASL1 deletion on p38, ERK1/2, and NF-κB signaling pathways. β-Actin served as the loading control (n = 3/group). Original western blot gels are presented in Supplementary Fig. S4. (B) HSCs were extracted using a combination of collagenase and pronase digestion, purified with Opti -prep densities of 60% and 11.5%, and subsequently cultivated following a final rinse in Dulbeccos Modified Eagle Medium (DMEM). Each value is presented as the mean ± SD. Statistical significance is denoted by **p < 0.01 and *p < 0. 05.
OASL1−/− mice resist CCl4-induced acute liver injury (ALI)
Type I IFN acutely facilitates ALI resolution, usually by priming macrophage polarization28. Also, type I IFNs are renowned for their dual effects: they can either enhance inflammation or inhibit pro-inflammatory processes29. Thus, to further add credence to the potential of OASL1 as a therapeutic target and explore the role of the macrophage in the development of liver injury, we then challenged mice with CCl4 for 24 h (Fig. 6A). Evidence from Fig. 6B shows that serum ALT levels were similar to the chronic models Figs. 1D and 3D. In addition, the TUNEL-positive cells were remarkably lower in CCl4-challenged OASL1−/− than WT CCl4 livers (Fig. 6C). The levels of pro-inflammatory cytokines TNF-α, IL-1β and mRNA expression of TGF-β in liver lysates were significantly lower in mice lacking OASL1−/− treated with CCl4 acutely than WT CCl4 challenged mice (Fig. 6D,E). Interestingly, OASL1−/− mice challenged with CCl4 maintained significantly high mRNA expression levels of IFN-α, IFN-β, and IRF7 (Fig. 6E,F). However, there were no significant changes to the mRNA expression levels of IRF3 (Fig. 6F).

OASL1−/− mice resist acute CCl4-induced liver injury. (A) Schematic illustration of the experimental setup that was utilized to ascertain the acute effects of OASL1 on CCl4 induction. (B) Analysis of liver damage ALT in IU/L using plasma biochemistry (C) Histopathological images of H&E staining and TUNEL staining of apoptotic cells quantified in (%). (D) A sandwich ELISA assay was used to determine TNF-α and IL-1β in ng/mg. (E, F) TGF-β, IFN-α, IFN-β, IRF7 and IRF3 gene expression levels were determined by RT-qPCR analysis. Each value signifies the mean ± SD. * signifies p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Abundant F4/80 positive macrophages in the CCl4-challenged OASL1−/− mice
Macrophages are crucial regulators of homeostasis and host defense immune response, primarily engulfing apoptotic cells and foreign products without causing severe inflammation30,31. We then further examined the role of OASL1 in the hepatic cell population of F4/80+ macrophages. Interestingly, via immunofluorescence staining of F4/80+ cells (red), macrophages were significantly abundant upon 24 h CCl4-induction in OASL1−/− liver sections compared to CCl4-treated WT mice and this data was supported by F4/80 mRNA expression data (Fig. 7A). Although it was also noted via immunofluorescence that the F4/80+ cells in untreated OASL1−/− livers were slightly more abundant, the difference was not significant.

F4/80 positive macrophage population was elevated in CCl4-induced OASL1−/− mice. (A) White arrowheads show F4/80 positive macrophage assessed via immunofluorescence (red). (B) Phase contrast of seeded untreated cells and CCl4 treated RAW macrophages. RT‒qPCR analysis revealed increased OASL1 mRNA expression levels upon 12 h treatment with CCl4. (C) IFN-α and IFN-β expression levels in control or siOASL1 transfected on RAW macrophages treated with CCl4. (D) IFN-α obtained via ELISA assay (pg/mL). (E) IRF7 mRNA expression levels. (F) TNF-α obtained via ELISA assay (pg/mL). Each value signifies the mean ± SD. **Indicates p < 0.01 and * signifies p < 0.05.
We then sought to investigate the effects of direct OASL1 knockdown on macrophages. Firstly, RAW 264.7 macrophages were treated with 0.2% concentration of CCl4 for 12 h and an increase of OASL1 mRNA expression was observed (Fig. 7B). RAW cells were transfected with siOASL1 or a control silencer and treated with CCl4. CCl4 treatment of siOASL1 cells resulted in significantly higher levels of IFN-α, IFN-β mRNA expression, and IFN-α assessed via ELISA (Fig. 7C,D). IRF7 levels only showed an increasing trend (Fig. 7E). Pro-inflammatory cytokine TNF-α was lower in siOASL1 (Fig. 7F). Collectively, these observations suggest that OASL1 deficiency sustains type I IFN in the liver via macrophages.
Discussion
Excessive extracellular fibrotic tissue accumulates due to repeated damage, leading to liver fibrosis as the body’s repair mechanisms become counterproductive32. Such protective responses to liver insults may induce adverse outcomes, like tissue dysfunction due to scar formation and cirrhosis33. In our study, we employed OASL1−/− mice to assess OASL1’s role in biliary and hepatic fibrosis caused by DDC feeding and CCl4 treatment, respectively. Our results demonstrate that in WT mice, the hepatic OASL1 gene is upregulated in both models of chronic liver disease, contributing to biliary injury, liver inflammation, and the expression of fibrogenic genes. Furthermore, OASL1 deletion reduced levels of the proinflammatory cytokines IL-1β and IL-6 during CCl4 induction, thereby decreasing fibrosis and modulating downstream NF-κB signaling. Given that OASL1 functions as an ISG, its activation leads to a significant influx of various cytokines, ultimately causing liver inflammation and fibrosis. While certain studies have linked ISG expression with therapeutic outcomes in chronic hepatitis C patients34, our observations suggest liver injury exacerbation due to an excessive immune response, promoting liver fibrosis. Mouse OASL1 disrupts type I IFN regulation by interacting with IRF7 mRNA expression35. Consistent with prior research10,36,37, our study finds that OASL1 ablation enhances IRF7 activity, markedly maintaining IFN-α and IFN-β levels, crucial in mitigating chronic fibrotic conditions as evidenced by Sirius red staining, hydroxyproline and fibrotic marker expression, including αSMA. These outcomes contrast with earlier findings where complete OASL1 depletion in a chronic inflammation model aggravated plaque formation in atherosclerosis patients38. Nonetheless, our findings align with research showing improved antitumor immunity due to increased type I IFN production resulting from OASL1 deficiency39.
Macrophage polarization by hepatocytes may lead to changes in the local hepatic immune microenvironment and thus affect the immune state40. As a result, we investigated how OASL1 might affect the production of type I IFN in acute liver injury. In vitro, hepatocytes treated with 0.2% CCl4 treatment (Supplementary Fig. S5) showed a considerable increase in IFN-α and IFN-β mRNA levels in cells with high OASL1 concentrations, regulated possibly by IRF3. Nevertheless, these cells leaked significantly more lactate dehydrogenase (LDH) than the OASL1-deficient cells, suggesting that even though hepatocytes released type I IFN more in WT-CCl4 treatment, they were more vulnerable to membrane damage (Supplementary Fig. S5C). These contradicting consequences of the involvement of OASL1 in vitro in hepatocytes inspired an additional acute in vivo study. Interestingly, 24 h treatment of CCl4 revealed that ALT levels and pro-inflammatory cytokines like TNF-α and IL-1β were lower in OASL1−/− mice acutely challenged with CCl4, and this was consistent with the cholestatic and hepatotoxin-induced fibrosis models. When OASL1−/− mice were acutely stimulated with CCl4 in vivo, their IRF7-upregulated levels of IFN-α and IFN-β mRNA while downregulating proinflammatory cytokines TNF-α and IL-1β, this, in turn, established an environment to ameliorate acute liver injury (Fig. 8). It is possible that in the OASL1−/− mice's liver, another type of cell contributed significantly to the total synthesis of type I IFN, as this phenomenon was previously alluded10. Therefore, we turned to the macrophage population, and surprisingly, even though not significantly appreciated, immunofluorescence (red) targeting of F4/80+ macrophages in untreated OASL1−/− mice livers revealed slightly more positive stained cells than WT-negative control livers. Upon 24 h of CCl4 treatment, OASL1−/− mice had significantly higher expression of F4/80+ macrophages. F4/80 is usually used to mark macrophages41,42. Most studies have revealed how OASL1 is mainly related to dendritic cells and macrophages37,43and thus we further investigated the direct knockdown of OASL1 in RAW macrophages, and upon stimulation with CCl4 we observed that the knockdown of OASL1 in RAW macrophages did significantly release type I IFN related genes. Indicating that there is a strong synergy in the effect of OASL1 deficiency on the macrophage and production of large amounts of IFN-I this function maybe strongly dependent on the macrophage/Kupffer cell niche and thus this synergy would contribute to therapeutics involving liver injury. Because OASL1 may act differently depending on the condition and type of cell, this must be taken into account in setting up future treatment modalities.

Proposed mechanism for the effects of OASL1 on biliary and hepatotoxin-induced liver injury in mice. Liver injury leads to the upregulation of OASL1, inhibiting IRF7 expression thus negatively regulating IFN-α and IFN-β production thereby enhancing proinflammatory cytokines TNF-α, IL-6 and IL-1β, via NF-κB. The figure was created with BioRender (biorender.com).
The TGF-β protein crucially influences liver fibrosis progression, activating fibroblasts that may lead to cirrhosis and cancer44. In our study, 24 h in vivo treatment of CCl4 showed significant TGF-β level differences between WT and OASL1−/− mice. Intriguingly, consistent in cases of chronic injury, OASL1−/− mice exhibited notably lower TGF-β levels in CCl4-treated livers. Consequently, we investigated how OASL1 knockdown directly impacts primary HSCs, finding a consistent and significant reduction in TGF-β expression, suggesting OASL1’s pivotal role in chronic liver fibrosis progression45. Beyond TGF-β, the Ras-MAPK pathway collaborates in regulating tumor development46, and enhanced OASL expression activates retinoic acid-inducible gene I signaling, leading to IRF3 dimerization and NF-κB-regulated gene induction7. Our findings demonstrated that OASL1−/− mice with chronic liver injury showed no significant changes in ERK and p38 activation, with the most notable impact being on NF-κB activation, likely due to OASL1’s regulatory effects on inflammation, TGF-β levels, and fibrosis in chronic liver injury.
In conclusion, this study reveals that OASL1 deficiency promotes type I IFN synthesis via IRF7 causing amelioration of liver injury. Accordingly, leveraging OASL1 as a therapeutic target requires an in-depth investigation into its expression dynamics concerning type I IFN production as the data indicates that multiple factors may involve the action of OASL1 on individual cells.
Materials and methods
Animals and induction of murine fibrosis and acute liver injury
The study was conducted using eight-week-old male WT C57BL/6 mice (supplied by Taconic Farms, Inc., Samtako Bio Korea, O-San, Korea) and OASL1−/−. PCR genotyping confirmed the OASL1−/− status of the animals (see Supplementary Fig. S1). The mice were provided ad libitum access to food and water, and maintained in an environment with controlled temperature (23 ± 2 °C) and humidity (50 ± 10%), under a 12 h light–dark cycle. Following a period of 4 weeks, two distinctive models were employed to induce liver fibrosis. In the initial phase of 4 weeks, both WT and OASL1−/− mice were administered a DDC (obtained from Saeronbio, Inc., Uiwang, Korea) diet to incite biliary fibrosis, or a standard chow diet, hereafter denoted as NC. CCl4; acquired from Sigma‒Aldrich, MO, USA, and diluted to 2:5 v/v in corn oil) was utilized over another period of up to 4 weeks to elicit hepatotoxin-mediated liver fibrosis, replicating established protocols47. To induce ALI in mice, a single dose of CCl4 or an equal volume of corn oil as control following 16 h starvation was administered by intraperitoneal injections at a dose of 5 mL/kg of BW (2.64 g/kg) as previously48. Preceding necropsy, the mice underwent an 8 h fasting period and were then anaesthetized with Zoletil® (Tiletamine-zolazepam, Virbac, South Korea) at 0.2 ml/kg BW administered intramuscularly. Post which blood was collected via cardiac puncture, tissues were harvested, fixed in formalin, and enrobed in paraffin.
Ethics approval
The ethics and care of all animal experiments received approval from the Animal Care and Ethics Committees of Jeonbuk National University and the Institutional Animal Care and Use Committee (IACUC) of Korea and all methods were in accordance the relevant protocols, guidelines and regulations. This was approved by the Korean Council on Animal Care and the Korean Animal Protection Law, 2007; Article 13 (Animal Experiments). All animal studies adhered to the ARRIVE 2.0 requirements, which included study design, animal numbers, randomization, and statistical methods.
ELISA
The quantification of proinflammatory cytokines, including tumor TNF-α, IL-6, IFN-α, and IL-1β, was accomplished with ELISA kits (Thermo Fisher Scientific Inc., MA, USA) and an EMax precision microplate reader spectrophotometer (Molecular Devices, CA, USA).
Serum bilirubin and transaminase assays
The quantification of bilirubin was conducted using a serum bilirubin ELISA kit (BioVision Inc., CA, USA), with the optical density readings obtained at 450 nm employing a precision EMax microplate reader spectrophotometer (Molecular Devices). AST levels were determined according to methodologies previously outlined49. Serum ALT was assayed using an ALT/SGPT activity kit (BioVision Inc., CA, USA; catalog # K752-100).
Assay for hydroxyproline
Using a hydroxyproline colorimetric assay kit (Biovision Inc., Milpitas, USA; Catalog # K555-100) and individual liver weights (10 mg tissue), the total hydroxyproline was calculated per the manufacturer's instructions. Samples of liver tissue were hydrolyzed in 12N HCl and then placed in a drying oven (Vision Bionex, VS-1202D3, Gyeonggi-do Bucheon, South Korea) for 3 h at 120 °C. In a microplate reader, optical density (OD) was measured at a wavelength of 560 nm.
RNA extraction, reverse transcription, and quantitative RT-qPCR
Total RNA was isolated from liver tissues or cells utilizing a GeneAll RNA Extraction Kit (GeneAll Biotechnology, Seoul, Korea), with RNA purity subsequently assessed through a Thermo Fisher Scientific NanoDrop 2000 (Thermo Fisher Scientific Inc.). The ReverTra Ace® qPCR RT Master Mix with gDNA Remover (Toyobo, Japan) facilitated the reverse transcription of RNA into cDNA. The RT–qPCR analysis was performed using a CFX96TM RT–qPCR System (Bio-Rad, CA, USA) and 2 × qPCR BIOB SyGreen (PCR Biosystems, London, UK). A melting curve analysis verified the specificity of the PCR products post-amplification. Gene expression quantification hinged on comparing the Ct value (cycle threshold) of each sample to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Table 1 delineates the sequences for the RT–qPCR primers employed.
Microscopic observation of tissue sections and basic staining
Tissues were embedded in paraffin using a Sakura Tissue-Tek embedding station (Sakura Finetek, Tokyo, Japan) subsequent to fixation in 10% neutral buffered formalin, employing a Myr Spin Tissue Processor-STP 120 (Especialidades Médicas Myr, S.L., Catalonia, Spain). The embedded tissues were sectioned into 5 μm thick slices and mounted on glass slides. These slides were then processed through an automated staining sequence with H&E using the Myr Eva slide strainer SS-30 (Especialidades Médicas Myr, S.L.). For the examination of all liver sections derived from each specimen, a light microscope (BX-51; Olympus Corp., Tokyo, Japan) was utilized.
Western blot analysis
Liver tissues were homogenized on ice using extraction buffer (T-PER, Thermo Fisher Scientific, Inc.). Following centrifugation at 12,000×g for 15 min at 4 °C, the protein concentration in the supernatant was determined with a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific Inc.) in accordance with the manufacturer’s instructions. Subsequently, proteins underwent electrophoretic separation as previously outlined50. Overnight incubation at 4 °C was then performed with primary antibodies, each diluted to 1:1000 in blocking buffer. Utilized antibodies included: rabbit αSMA (Abcam, Cambridge, UK), rabbit anti-ERK, rabbit anti-phospho-ERK (Thr202/Tyr204), rabbit anti-p38, and rabbit anti-phospho-p38 (Thr180/Tyr182) all from Cell Signaling Technology, MA, USA; mouse anti-β-actin and anti-β-actin from Santa Cruz Biotechnology; and rabbit anti-NF-κB p65 and rabbit anti-phospho-NF-κB p65 (Ser536) from Cell Signaling Technology; rabbit anti-IRF7 from Cell Signaling Technology. Detecting antigen–antibody complexes involved the use of peroxidase-conjugated secondary antibodies from Santa Cruz Biotechnology, Inc., diluted to 1:2000 in blocking buffer and incubated at room temperature for 1 h. Protein bands were visualized utilizing an enhanced chemiluminescence system with an ImageQuant™ LAS 500 (GE Healthcare Life Sciences, IL, USA). Protein expression levels were subsequently quantified using ImageQuant™ TL software.
In vitro CCl4 treatment and transfection
RAW 264.7 macrophages were cultured and maintained in DMEM (Welgene, Gyeongsan, South Korea) containing 10% fetal bovine serum (Young in frontier, Seoul, South Korea) at 37 °C in a 5% CO2 incubator (Visionbionex VS-9170CT, Gyeonggi-do, South Korea). For in vitro treatments, RAW macrophages were seeded in 12-well plates and, on the next day, transfected with OASL1-siRNA (Thermo Fisher Scientific Inc., Catalogue (Cat) number (#) AM16708) or with a control silencer (Thermo Fisher Scientific Inc, Cat # AM4611) for 4 h, and were then treated either with 0.2% CCl4 for 12 h.
Primary HSCs from mice was conducted as previously described47,51. Initially, the liver was subjected to two-step digestion using collagenase and pronase, followed by the isolation of the hepatocyte pellet through centrifugation at 50×g. To further separate the HSCs, a gradient centrifugation method employing OptiPrep (Sigma-Aldrich) at 60% and 11.5% underlaid with Gey’s Balanced Salt Solution at 0%, was utilized; this mixture was then centrifuged for 30 min at 1800 rcf (g). After washing, the cells were plated in six-well plates that lacked any coating. On the fourth day of culture, primary mouse HSCs underwent transfection with OASL1-siRNA or with a control silencer.
LDH
The assessment of LDH release from the cytosol into the culture medium was conducted utilizing a cytotoxicity detection kit (Sigma-Aldrich), following the manufacturer’s instructions. The absorbance of the sample at 490 nm was quantified with an EMax spectrophotometer (Molecular Devices).
Sirius red and TUNEL staining
The degree of fibrosis was quantified by Sirius red staining, which employs an aqueous solution comprising picric acid and Direct Red 80 (Sigma-Aldrich), and the fibrotic regions were quantified as a percentage of the total area using digital imaging software (Analysis TS, Olympus Corp., Tokyo, Japan). To identify cellular apoptosis within the liver, TUNEL staining was performed on paraffin-embedded tissue sections employing an in situ apoptosis detection kit, ApopTag Peroxidase (EMD Millipore, CA, USA) kit that also detects DNA fragments utilizing terminal deoxynucleotidyl transferase (TdT) for detection by staining with 3,3-diaminobenzidine (DAB), adhering to the manufacturers recommendations. All liver slices were inspected using a light microscope (BX-51, Olympus Corp., Tokyo, Japan).
Immunofluorescence
The process of staining with immunofluorescence was conducted using an antibody against rabbit αSMA obtained from Abcam, mouse GFAP (2E1)-sc-33673; Santa Cruz Biotechnology. This procedure involved incubating liver sections at 4 °C overnight, followed by additional incubation in a dark room with fluorescein isothiocyanate (FITC)-conjugated secondary antibodies, either goat anti-mouse IgG or goat anti-rabbit IgG, sourced from Thermo Fisher Scientific Inc., for a duration of 45 min. Subsequent to a thorough rinse with phosphate-buffered saline, the slides were prepared with an antifade mounting medium that included 4′,6-diamidino-2-phenylindole (labeled as DAPI; Vector Laboratories, CA, USA). Finally, these slides were scrutinized employing fluorescence microscopy software (BX-51, Olympus Corp.).
IHC
Liver sections, embedded in paraffin, were initially fixed in 10% phosphate-buffered formalin prior to undergoing IHC staining procedures. Liver paraffin sections, measuring 4 µm in thickness, underwent a deparaffinization process, were rehydrated, and subsequently submerged in antigen retrieval solution (Dako, Jena, Germany), followed by treatment with sodium citrate buffer (pH 6.0) for 30 min, facilitating antigen retrieval. Following the inhibition of endogenous peroxidase activity and non-specific binding, the sections received an overnight treatment at 4 °C with a primary rabbit αSMA antibody (Abcam), and mouse CK19 (sc-376126; Santa Cruz Biotechnology). Subsequently, a horseradish peroxidase -conjugated secondary antibody (Vector Laboratories) was applied to the sections for 45 min. Detection was ultimately achieved by counterstaining the slides with hematoxylin (ScyTek Laboratories) before applying a DAB substrate for staining (Vector Laboratories).
Statistical analysis
The data in this study are presented as means ± SD. Differences between two groups were assessed using two-sided unpaired Student’s t tests, with significance levels denoted as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (GraphPad Prism 8.0.1, CA, USA).
Data availability
The datasets utilized or analyzed over the course of this investigation are available from the corresponding author upon reasonable request.
References
Marcellin, P. & Kutala, B. K. (2018) Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. 38, 2–6. https://doi.org/10.1111/liv.13682.
He, J., Bai, K., Hong, B., Zhang, F. & Zheng, S. Docosahexaenoic acid attenuates carbon tetrachloride-induced hepatic fibrosis in rats. Int. Immunopharmacol. 53, 56–62. https://doi.org/10.1016/j.intimp.2017.09.013 (2017).
Roh, Y. S. et al. Toll-like receptor 7-mediated type I interferon signaling prevents cholestasis- and hepatotoxin-induced liver fibrosis. 60, 237–249, https://doi.org/10.1002/hep.26981 (2014).
Roehlen, N., Crouchet, E. & Baumert, T. F. Liver fibrosis: mechanistic concepts and therapeutic perspectives. 9, 875. https://doi.org/10.3390/cells9040875 (2020).
Sadler, A. J. & Williams, B. R. J. N. r. i. Interferon-inducible antiviral effectors. 8, 559–568. https://doi.org/10.1038/nri2314. (2008).
Liao, X. et al. 2ʹ,5ʹ-Oligoadenylate synthetase 2 (OAS2) inhibits zika virus replication through activation of type Ι IFN signaling pathway. Viruses. https://doi.org/10.3390/v12040418 (2020).
Zhu, J. et al. Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity 40, 936–948. https://doi.org/10.1016/j.immuni.2014.05.007 (2014).
Eskildsen, S., Justesen, J., Schierup, M. H. & Hartmann, R. J. N. A. R. (2003) Characterization of the 2′–5′‐oligoadenylate synthetase ubiquitin‐like family. 31, 3166–3173. https://doi.org/10.1093/nar/gkg427.
Choi, U. Y., Kang, J.-S., Hwang, Y. S. & Kim, Y.-J. Oligoadenylate synthase-like (OASL) proteins: Dual functions and associations with diseases. Exp. Mol. Med. 47, e144. https://doi.org/10.1038/emm.2014.110 (2015).
Lee, M. S., Kim, B., Oh, G. T. & Kim, Y.-J. OASL1 inhibits translation of the type I interferon–regulating transcription factor IRF7. Nat. Immunol. 14, 346–355. https://doi.org/10.1038/ni.2535 (2013).
Ghosh, A. et al. (2019) Oligoadenylate-synthetase-family protein OASL inhibits activity of the DNA sensor cGAS during DNA virus infection to limit interferon production. 50, 51–63.e55. https://doi.org/10.1016/j.immuni.2018.12.013.
Zhan, Y. et al. Novel role of macrophage TXNIP-mediated CYLD-NRF2-OASL1 axis in stress-induced liver inflammation and cell death. JHEP Rep. Innovation Hepatol. 4, 100532. https://doi.org/10.1016/j.jhepr.2022.100532 (2022).
Sim, C. K. et al. 2′–5′ Oligoadenylate synthetase-like 1 (OASL1) deficiency in mice promotes an effective anti-tumor immune response by enhancing the production of type I interferons. Cancer Immunol. Immunotherapy 65, 663–675. https://doi.org/10.1007/s00262-016-1830-9 (2016).
Choi, B. Y. et al. 2′-5′ Oligoadenylate synthetase-like 1 (OASL1) deficiency suppresses central nervous system damage in a murine MOG-induced multiple sclerosis model. Neurosci. Lett. 628, 78–84. https://doi.org/10.1016/j.neulet.2016.06.026 (2016).
Oh, J. E., Lee, M. S., Kim, Y. J. & Lee, H. K. OASL1 deficiency promotes antiviral protection against genital herpes simplex virus type 2 infection by enhancing type I interferon production. Sci. Rep. 6, 19089. https://doi.org/10.1038/srep19089 (2016).
Guarda, G. et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34, 213–223. https://doi.org/10.1016/j.immuni.2011.02.006 (2011).
Baghdasaryan, A. et al. Inhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitis. 64, 674–681. https://doi.org/10.1016/j.jhep.2015.10.024 (2016).
Hassan, S., Syed, S. & Kehar, S. I. Glial fibrillary acidic protein (GFAP) as a mesenchymal marker of early hepatic stellate cells activation in liver fibrosis in chronic hepatitis C infection. Pak. J. Med. Sci. 30, 1027–1032. https://doi.org/10.12669/pjms.305.5534 (2014).
Best, J. et al. Macrophage depletion attenuates extracellular matrix deposition and ductular reaction in a mouse model of chronic cholangiopathies. PloS One 11, e0162286. https://doi.org/10.1371/journal.pone.0162286 (2016).
Assy, N., Jacob, G., Spira, G. & Edoute, Y. Diagnostic approach to patients with cholestatic jaundice. World J. Gastroenterol. 5, 252–262. https://doi.org/10.3748/wjg.v5.i3.252 (1999).
Kumar, S., Wang, J., Shanmukhappa, S. K. & Gandhi, C. R. J. T. A. J. O. P. Toll-like receptor 4–independent carbon tetrachloride–induced fibrosis and lipopolysaccharide-induced acute liver injury in mice: role of hepatic stellate cells. 187, 1356–1367. https://doi.org/10.1016/j.ajpath.2017.01.021 (2017).
Sayed, E. A. et al. Induction of liver fibrosis by CCl4 mediates pathological alterations in the spleen and lymph nodes: The potential therapeutic role of propolis. Saudi J. Biol. Sci. 28, 1272–1282. https://doi.org/10.1016/j.sjbs.2020.11.068 (2021).
Knittel, T. et al. Expression patterns of matrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells of rat liver: Regulation by TNF-α and TGF-β1. 30, 48–60. https://doi.org/10.1016/s0168-8278(99)80007-5 (1999).
Hafez, M. M. et al. Effect of ginseng extract on the TGF-β1 signaling pathway in CCl(4)-induced liver fibrosis in rats. BMC Complement. Alternative Med. 17, 45. https://doi.org/10.1186/s12906-016-1507-0 (2017).
Qing, F. & Liu, Z. Interferon regulatory factor 7 in inflammation, cancer and infection. Front. Immunol. 14, 1190841. https://doi.org/10.3389/fimmu.2023.1190841 (2023).
Sunami, Y. et al. Hepatic activation of IKK/NFκB signaling induces liver fibrosis via macrophage-mediated chronic inflammation. Hepatology (Baltimore, Md.) 56, 1117–1128. https://doi.org/10.1002/hep.25711 (2012).
Yu, Q., Cheng, P., Wu, J., Guo, C. J. PPARγ/NF‐κB and TGF‐β1/Smad pathway are involved in the anti‐fibrotic effects of levo‐tetrahydropalmatine on liver fibrosis. 25, 1645–1660. https://doi.org/10.1111/jcmm.16267 (2021).
Song, Q. et al. Type I interferon signaling facilitates resolution of acute liver injury by priming macrophage polarization. Cellular Mol. Immunol. 20, 143–157. https://doi.org/10.1038/s41423-022-00966-y (2023).
Ivashkiv, L. B. & Donlin, L. T. J. N. R. I. Regulation of type I interferon responses. 14, 36–49. https://doi.org/10.1038/nri3581 (2014).
Li, W., Chang, N. & Li, L. Heterogeneity and function of kupffer cells in liver injury. Front. Immunol. 13, 940867. https://doi.org/10.3389/fimmu.2022.940867 (2022).
Chen, S. et al. Macrophages in immunoregulation and therapeutics. Signal Transduct. Targeted Therapy 8, 207. https://doi.org/10.1038/s41392-023-01452-1 (2023).
Friedman, S. L. J. J. o. h. Liver fibrosis—From bench to bedside. 38, 38–53 (2003).
Bataller, R. & Brenner, D. A. Liver fibrosis. J. Clin. Investig. 115, 209–218. https://doi.org/10.1172/jci24282 (2005).
Honda, M. et al. Hepatic ISG expression is associated with genetic variation in interleukin 28B and the outcome of IFN therapy for chronic hepatitis C. Gastroenterology 139, 499–509. https://doi.org/10.1053/j.gastro.2010.04.049 (2010).
Sim, C. K., Lee, J. H., Baek, I.-J., Lee, S.-W. & Lee, M. S. J. J. o. I. R. Enhanced antitumor immune response in 2–5 oligoadenylate synthetase-Like 1-(OASL1-) deficient mice upon cisplatin chemotherapy and radiotherapy. 2019 https://doi.org/10.1155/2019/7596786 (2019).
Chang, Y. et al. OASL1-mediated inhibition of type I IFN reduces influenza A infection-induced airway inflammation by regulating ILC2s. Allergy Asthma Immunol. Res. 14, 99–116. https://doi.org/10.4168/aair.2022.14.1.99 (2022).
Lee, M. S., Park, C. H., Jeong, Y. H., Kim, Y.-J. & Ha, S.-J. J. P. p. Negative regulation of type I IFN expression by OASL1 permits chronic viral infection and CD8+ T-cell exhaustion. 9, e1003478. https://doi.org/10.1371/journal.ppat.1003478 (2013).
Kim, T. K. et al. 2ʹ–5ʹ Oligoadenylate synthetase-like 1 (OASL1) protects against atherosclerosis by maintaining endothelial nitric oxide synthase mRNA stability. Nat. Commun. 13, 6647. https://doi.org/10.1038/s41467-022-34433-z (2022).
Sim, C. K. et al. 2ʹ–5ʹ Oligoadenylate synthetase-like 1 (OASL1) deficiency in mice promotes an effective anti-tumor immune response by enhancing the production of type I interferons. Cancer Immunol. Immunother. CII 65, 663–675. https://doi.org/10.1007/s00262-016-1830-9 (2016).
Xie, D. & Ouyang, S. The role and mechanisms of macrophage polarization and hepatocyte pyroptosis in acute liver failure. 14, https://doi.org/10.3389/fimmu.2023.1279264 (2023).
Nguyen-Lefebvre, A. T. & Horuzsko, A. Kupffer cell metabolism and function. J. Enzymol. Metab. 1 (2015).
Li, P.-Z., Li, J.-Z., Li, M., Gong, J.-P. & He, K. An efficient method to isolate and culture mouse Kupffer cells. Immunol. Lett. 158, 52–56. https://doi.org/10.1016/j.imlet.2013.12.002 (2014).
Reheman, A. et al. Involvement of 2′-5′ oligoadenylate synthetase-like protein in the survival of Mycobacterium tuberculosis avirulent strain in macrophages. Animal Diseases 3, 5. https://doi.org/10.1186/s44149-023-00068-w (2023).
Peng, D., Fu, M., Wang, M., Wei, Y. & Wei, X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol. Cancer 21, 104. https://doi.org/10.1186/s12943-022-01569-x (2022).
Fabregat, I. et al. TGF‐β signalling and liver disease. 283, 2219–2232. https://doi.org/10.1111/febs.13665 (2016).
Chapnick, D. A., Warner, L., Bernet, J., Rao, T. & Liu, X. Partners in crime: The TGFβ and MAPK pathways in cancer progression. Cell Biosci. 1, 42. https://doi.org/10.1186/2045-3701-1-42 (2011).
Zhou, Z. et al. Dual TBK1/IKKɛ inhibitor amlexanox attenuates the severity of hepatotoxin-induced liver fibrosis and biliary fibrosis in mice. J. Cell. Mol. Med. 24, 1383–1398. https://doi.org/10.1111/jcmm.14817 (2020).
Zhou, Z. et al. AK-1, a Sirt2 inhibitor, alleviates carbon tetrachloride-induced hepatotoxicity in vivo and in vitro. 30, 324–335. https://doi.org/10.1080/15376516.2020.1729915 (2020).
Yang, D. et al. Oral administration of Jinan Red Ginseng and licorice extract mixtures ameliorates nonalcoholic steatohepatitis by modulating lipogenesis. 46, 126–137. https://doi.org/10.1016/j.jgr.2021.05.006. (2022).
Qi, J., Kim, J.-W., Zhou, Z., Lim, C.-W. & Kim, B. J. T. A. J. o. P. Ferroptosis affects the progression of nonalcoholic steatohepatitis via the modulation of lipid peroxidation–mediated cell death in mice. 190, 68–81. https://doi.org/10.1016/j.ajpath.2019.09.011 (2020).
Kim, J.-W. et al. Spliceosome-associated protein 130 exacerbates alcohol-induced liver injury by inducing NLRP3 inflammasome-mediated IL-1β in mice. Am. J. Pathol. 188, 967–980. https://doi.org/10.1016/j.ajpath.2017.12.010 (2018).
Funding
This research was supported by Basic Science Research Program through the National Research Foundation (NRF) funded by the Ministry of Science and ICT (2020R1A2C1007178), and by the Ministry of Education (2019R1A6A1A03033084), Republic of Korea.
Author information
Authors and Affiliations
Contributions
Methodology, Formal Analysis, Investigation, Data Curation, Writing – Review & Editing were conducted by M.L., D.Y., T.H.T.N.B., and H.J.; Writing – Original Draft Preparation, Visualization, M.L; Supervision, Validation, Visualization S.O, and J.H.; Conceptualization, Resources, Validation, Writing – Review & Editing were provided by J.W.K.; Resources, Project Administration, Supervision, Validation, Visualization, Funding Acquisition, and Writing – Review & Editing were performed by B.K.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Leya, M., Yang, D., Bao, T.H.T.N. et al. The role of 2ʹ–5ʹ-oligoadenylate synthase-like protein (OASL1) in biliary and hepatotoxin-induced liver injury in mice. Sci Rep 14, 21873 (2024). https://doi.org/10.1038/s41598-024-72465-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-72465-1
- Springer Nature Limited