
Abstract
Naringin, a flavonoid, exhibits diverse therapeutic properties and has been proven to exert cytotoxic effects on cancer cells. Nevertheless, the precise mechanism of naringin maintaining its cytotoxic effect on glioblastoma (GBM) remains unknown. Thus, the current study aimed to establish a plausible cellular mechanism for Naringin’s inhibition of GBM. We employed various system biology techniques to forecast the primary targets, including gene ontology and cluster analysis, KEGG enrichment pathway estimation, molecular docking, MD (molecular dynamic) simulation and MMPBSA analysis. Glioblastoma target sequences were obtained via DisGeNet and Therapeutic Target Prediction, aligned with naringin targets, and analyzed for gene enrichment and ontology. Gene enrichment analysis identified the top ten hub genes. Further, molecular docking was conducted on all identified targets. For molecular dynamics modelling, we selected the two complexes that exhibited the most docking affinity and the two most prominent genes of the hub identified through analysis of the enrichment of genes. The PARP1 and ALB1 signalling pathways were found to be the main regulated routes. Naringin exhibited the highest binding potential of − 12.90 kcal/mol with PARP1 (4ZZZ), followed by ABL1 (2ABL), with naringin showing a − 8.4 kcal/mol binding score, as determined by molecular docking. The molecular dynamic approach and MM-PBSA investigation along with PCA study revealed that the complex of Naringin, with 4ZZZ (PARP1) and, 2ABL (ABL1), are highly stable compared to that of imatinib and talazoparib. Analyses of the signalling pathway suggested that naringin may have anticancer effects against GBM by influencing the protein PARP and ALB1 levels. Cytotoxicity assay was performed on two different glioblastoma cell lines C6 and U87MG cells. Naringin demonstrates a higher cytotoxic potency against U87MG human glioblastoma cells compared to C6 rat glioma cells.
Similar content being viewed by others

Introduction
Glioblastoma (GBM) is a highly aggressive and benign brain tumor that has a dismal prognosis, with a median survival of only sixteen months and a five-year survival rate of fewer than ten percent1,2. Gliomas comprise approximately thirty percent of primary brain tumors and eighty percent of all malignant ones. GBM is highly vascularized, with increased expression of angiogenic factors such as vascular endothelial growth factor (VEGF), which is crucial in the process of GBM angiogenesis3. Both cancer stem cells and differentiated cells exhibit a strong invasive potential within the tumor4. Despite maximal treatment with surgery, chemotherapy, and focal radiotherapy, the median survival for patients with GBM remains low5. The current standard of care for patients who have recently been diagnosed with glioblastoma involves the administration of temozolomide and radiation simultaneously. Nevertheless, the overall survival rate of patients diagnosed with glioblastoma remains unacceptably low, highlighting the urgent requirement for innovative therapeutic alternatives6. There has been a surge in the use of immune treatments, such as immune checkpoint inhibition, to treat glioblastoma in patients. This approach has demonstrated remarkable efficacy in treating some tumor types7. The treatment of glioblastoma has recently seen the emergence of novel approaches that centre on glioma stem cells (GSCs), immunotherapies, biological targeting, and gene treatments8. There is an immediate need for innovative therapeutic techniques for GBM due to the dismal survival rate with existing approved medications.
Many phytotherapeutics have been investigated for their potential to combat cancer, specifically glioblastoma. In-silico and in-vitro evaluations have provided valuable insights into the activity of these compounds9. In our previous study, 50 phytocompounds were screened for their PARP inhibition potential in silico, and the most active compounds were shortlisted10. Among them, promising PARP inhibition was shown by naringin. This study aims to investigate the potential pharmacological impacts of naringin in relation to the management of glioblastoma.
Naringin is classified as a natural dihydroflavone compound, predominantly found in citrus fruits belonging to the Rutaceae family. It is also present in other traditional Chinese medicinal plants, including trifoliate orange, exocarpium citri grandis, rhizoma dynamite, and fingered citron11. Various research related to naringin has proven that it can inhibit the progression and metastasis of GBM cells by regulating the cyclin D1/FAK pathway and promoting programmed cell death by affecting the FAK/bads pathway12. In addition, by blocking the ERK-P38-JNK signalling pathway, naringin reduces cell metastasis and matrix metalloproteinase (MMP-2) and MMP-9 production in human glioblastoma13. Additionally, naringin suppresses angiogenesis and cell migration in GBM cancer by inhibiting MMP-2 and MMP-9 and silencing the signalling pathway-p3814. These findings collectively suggest that naringin may have a multifaceted impact on GBM, affecting various aspects of its progression and metastasis. Furthermore, naringin has been shown to restore chemo-sensitivity to TMZ (temozolomide), a chemotherapeutic DNA alkylating drug regularly used for GBM, by targeted inhibition of PARP1 (poly (ADP-ribose) polymerase-1) in glioma cell lines15. This indicates that naringin may also have the potential to enhance the efficacy of existing GBM treatments. Research findings suggest that naringin exhibits promising anti-glioblastoma properties by inhibiting cell proliferation, metastasis, and angiogenesis and potentially enhancing GBM cells’ sensitivity to chemotherapy. These findings warrant further investigation into the potential therapeutic use of naringin in treating GBM. Nevertheless, the precise mechanisms behind the anticancer properties of naringin remain incompletely elucidated, prompting substantial ongoing research efforts.
To conduct drug screening, network pharmacology and in silico molecular docking are valuable tools for discovering efficient drug molecules and forecasting drug-ligand interactions. These methods involve computational analysis, molecular modelling, and network-based approaches to understand the pharmacological effects of drugs and their interactions with biological targets. In-silico molecular docking is a highly effective method for screening chemical toxicity. It predicts toxicity by analyzing the structure of ligand-receptor complexes and evaluating their binding affinity16. It involves density functional theory (DFT) studies, molecular docking, and molecular dynamic (MD) simulations17. Network pharmacology, on the other hand, constructs drug- molecule-target-disease networks and PPI (protein–protein interaction) networks to investigate the core components and important targets of pharmacological therapy, which are then verified by molecular docking18. This approach is beneficial for investigating the therapeutic effects of herbal medicine at the molecular level.
In summary, integrating network pharmacology and In-silico molecular docking offers a robust method for drug screening, identifying potential drug candidates, understanding their mechanisms of action, and predicting their interactions with biological targets. This integrated approach, supported by computational methods and behavioural phenotyping, contributes to advancing drug discovery and identifying effective treatments for various conditions. Researchers can examine complicated interactions at several levels to understand the combined impacts and fundamental mechanisms of various drugs19. The current research employed a network pharmacology approach to investigate naringin’s pivotal targets, biological functions, and related pathways to treat GBM.
Materials and methods
Materials
The phytomedicine naringin was procured from Yarrow Chem Products, Mumbai. DMSO was procured from Sigma- Aldrich. Fetal bovine serum (FBS) was purchased from Gibco (Life Technologies AG, Basel, Switzerland). Minimum Essential Medium Eagle (MEM), Nutrient mixture F-12 Ham w/2 mM l-Glutamine and 1.5 g per litre Sodium bicarbonate, Antibiotic Antimycotic solution 100× liquid, Trypsin–EDTA solution 1×, and MTT were purchased from Himedia, India. The GBM cell lines C6 (rat glioma) and U-87MG (Human; Glioblastoma; Astrocytoma) were procured from the cell repository, National Centre for Cell Science (NCCS), Pune, India.
Methods
Identification of targets related to GBM and Naringin
The gene targets involved in the disease progression of glioblastoma was taken from DisGeNET (https://www.disgenet.org/home/) with ID number: C0017636, and the selected keyword was glioblastoma. Target proteins of Naringin were retrieved from Swiss target prediction (http://www.swisstargetprediction.ch/). In addition, commonly identified collective genes were predicted from an online tool (https://bioinfogp.cnb.csic.es/tools/venny/) with a version, Venny 2.1.0.
Network construction and analysis of protein–protein interaction (PPI network)
ALL known and expected protein linkages, including functional and physical interactions, are stored in STRING (https://string-db.org/). Database (version 11.5) was utilized in this investigation to examine the correlation between GBM-related and common Naringin targets, which were thought to be promising targets for treatment. The following criteria were used in the construction of the PPI network of possible therapeutic targets: the species was “Homo sapiens,” and the reliability was “medium confidence of ≥ 0.4”. Topology parameters were obtained by analyzing the PPI network with CytoscapeV3.10.0 software.
Gene ontology and KEGG predicted pathway analysis
The Gene Ontology (GO) and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway enrichment tools20,21,22, which are accessible through the DAVID database (https://david.ncifcrf.gov/), were utilized in order to conduct an analysis of each and every prospective therapeutic target.
This analysis aimed to elucidate the associated pathways and relevant GO terms, encompassing the categories of CC (cellular component), MF (molecular function), and BP (biological Process). Pathways and GO terms meeting the significance threshold of p < 0.05 were considered noteworthy and included in further analysis. We utilised the Bioinformatics platform (http://www.bioinformatics.com.cn/) to visually portray the results of the GO and KEGG enrichment studies. Cytoscape software version 3.10.0 was used to visualize the network consisting of active chemicals, possible targets, and signalling pathways.
Cluster analysis
The ClueGO add-on module integrated with the Cytoscape version3.9.0 was employed to conduct a comprehensive cluster analysis. This analysis focused on genes enriched in the CC, MF, and BP categories. A two-sided hypergeometric functional analysis was utilized to identify significant clusters, with a P-value restricted to less than 0.05. The Bonferroni step-down correction method was implemented to account for multiple tests and reduce the likelihood of false positives. Further refinement of the analysis involved setting the ‘GO tree interval’ parameter to cover a range of 3 to 8 pathways, ensuring a focused yet comprehensive examination of the gene ontology landscape. Moreover, the criterion for gene ontology term selection of cluster was configured to include a minimum of three genes, ensuring that the clusters identified were biologically meaningful and relevant. This meticulous approach facilitated a detailed and accurate exploration of the enriched gene clusters, contributing valuable insights into their functional roles and interactions.
Molecular docking
Both molecular docking and pharmacokinetic research were carried out with the assistance of the Schrodinger program developed by Maestro 2020-4. Subsequent to the molecular docking study, the ligand’s generation, the protein’s preparation, the glide’s formation, and the ligand’s docking were carried out. The Schrodinger application, Maestro V-12.3, was employed to estimate the drug-receptor interactions. This application was programmed on a 17-inch workstation machine manufactured by Lenovo Inc., which was powered by an Intel Core i-9 processor with a clock speed of 3.60 GHz and an octa-core processor. Additionally, the machine featured 16 GB of RAM, a 2048 GB hard drive, and an operating system (Linux-x8664-bit). For the process of protein, Maestro’s add-on protein preparation wizard was utilized. The RCSB gateway was utilized to sort out the X-ray crystallized structure of the proteins. After deducting the potential ionization for the heteroatoms, the maximum stability state was chosen, and the OPLS-3 force field was used to meet the energy reduction goal. After retrieving the ligands from PubChem, the LigPrep module was utilized to prepare the experiment. The significant binding site of the receptor grid for docking originated through utilizing the site map tool. The molecular docking process was carried out in this grid box, and the end results were portrayed using the Glide module-XP visualizer. Additionally, two-dimensional interactions were studied.
Molecular dynamics
MD simulation tests were carried out on a Dell Perc.3650 tower, equipped with 11th generation of intel core (i-9-11700, 16 MB) Cache octa-core clock speed ranging from 2.5 to 4.9 GHz, 16 GB RAM (2X16) DDR4, SSD 2TBPCIe Gen4 M2 and a 12 GB NVIDIARTX A4100 Graphic Card. MD simulation was performed for the top two docking-scored protein–ligand complexes via GROMACS 2023.223. The ligands were prepared using UCSF chimera software, and the ligands were uploaded to the swissparam.ch portal to get the other ligand format. The pdb2gmx tool via the TIP3P water model and charmm27 force field was selected to generate the protein’s topology. The water-filled complex that was neutralised by the addition of ions resulted in the formation of a Dodecahedron case approximately 10 Å in size. A verlet cut-off strategy was utilized in conjunction with the steepest descent algorithm, which was utilized for a maximum of 55,000 steps in order to minimize energy consumption. For the purpose of constraining the atoms, the LINCS algorithm was utilized, and the particle mesh Ewald (PME) algorithm was also utilized in order to equilibrate the electrostatic forces. For the purpose of maintaining the system’s pressure and temperature at 300 degrees Kelvin, the isotropic V-rescale and C-rescale algorithms were utilised here. A total of 200 ns was spent running the simulation, and for every 200 picoseconds, energy was conserved. As soon as the MD run was finished, XM grace was utilised in order to perform the analysis of the data.
Molecular mechanics Poisson–Boltzmann surface area (MMPBSA) analysis
The MM-PBSA (Molecular Mechanics Poisson-Boltzmann Surface Area) method is a computational approach that combines molecular mechanical (MM) energies with a continuum solvent model for polar solvation and a nonpolar solvation term dependent on solvent-accessible surface area. These include bonds, angles, dihedrals, Coulomb and Van der Waals interactions (MM part), solvation energy from continuum solvent calculations (PB part), and non-polar hydrophobic energy correlated with Solvent Accessible surface area (SA part). The gmx_MMPBSA module was used to analyse the energy contribution parameters. A total of 10,000 frames were used for the MMPBSA run, and each frame was separated by 100 frames. The run was completed for 100 frames. The Poisson Boltzmann calculations were carried out with the assistance of an internal PBSA solver that was placed within a sander. Additionally, the MM-PBSA_ana module was utilized in order to visualize the outcomes that were received from the gmx_MMPBSA run.
Principal component analysis (PCA)
PCA was performed using GROMACS. It is a statistical method used to reduce the dimensionality of large datasets while preserving most of the variation in the data. It identifies new uncorrelated variables called principal components, which are linear combinations of the original variables. The PCA process begins with standardizing the dataset to have a mean of zero and a variance of one, ensuring equal contribution from each variable. Following this, the covariance matrix is computed to capture the relationships between variables. Eigenvalues and eigenvectors of this matrix are then calculated, with eigenvalues indicating the magnitude of variance along the eigenvectors, which are directions of maximum variance. The eigenvalues and corresponding eigenvectors are sorted in descending order, and the top k eigenvalues and their eigenvectors are selected to form a matrix of principal components. The original data is then projected onto this new feature space, resulting in a reduced dataset with fewer dimensions.
In vitro cytotoxicity assay of naringin
The GBM cell lines C6 (rat glioma) and U-87MG (Human; Glioblastoma; Astrocytoma) were procured from the cell repository, National Centre for Cell Science (NCCS), Pune, India. Rat glioblastoma C6 cells were cultured in a Nutrient mixture of F-12 Ham supplemented with 10% FBS and 1% of the antibiotic–antimycotic solution. U-87MG (Human; glioblastoma; astrocytoma) cells were cultured in MEM media, supplemented with 10% FBS and 1% of the antibiotic–antimycotic solution. The cells were subcultured in T25 and T75 flasks and maintained at 37 °C with 5% CO2 in a humidified incubator.
For the MTT assay, the cells were seeded on 96 well plates with a seeding density of 1 × 105 cells/well-kept for 24 h incubation for attaching the cells. A stock solution of naringin was prepared by using ethanol as the solvent. Further different concentrations of naringin (2.5, 5, 10, 20, 40, 80, and 160 µg/ml) were prepared by using the media. After 24 h, the media was removed, and the cells were treated with different concentrations of naringin with media (100 µl) and incubated for 24 h. After the incubation period, media was removed, and the cells were incubated with MTT reagent (5 mg/ml) for 4 h, followed by 1-h incubation with solubilization buffer (DMSO). The optical density was determined at 570 nm with gentle shaking on a Thermo Scientific Multiskan ELISA plate reader24. Cells in media alone devoid of drug acted as positive control and were well treated with 1% of Triton-X 100 acted as negative control. Triplicate samples were analyzed for each experiment. The percentage of cytotoxicity and EC50 values were analyzed based on the viability of the cells. Naringin’s cytotoxicity dose–response curve and EC50 values were calculated by using Combenefit software25.
Results
Identification of targets
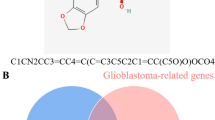
The keyword “glioblastoma” was utilized to retrieve data from multiple databases. A comprehensive search was conducted based on the data obtained from Swiss Target Prediction, and 196 genes have been identified to be associated with naringin. DisGeNET identified glioma-related targets (9039 genes). One hundred twenty-nine intersection targets were obtained from the two gene sets for further analysis. An online tool, Venny 2.1.0 (Fig. 1), was utilized to display the intersection of naringin targets and GBM-related differentially expressed genes. Categorical analysis of matching proteins between naringin and GBM indicated the top three categories to be affiliated as protease (21.0%), enzymes (17.0%), and lyase (11.0%) to be the top three classes. Figure 2 represents the pie chart of the predicted target of naringin based on the protein family.

Identification of targets related to Naringin and GBM: Venn diagram of naringin targets and GBM targets.

Pie chart of the predicted target of naringin based on protein family.
Analysis of protein–protein interaction, protein selection for molecular docking and network construction
A database called “STRING” was employed to analyze the relationship between 115 potential therapeutic targets. The Cytoscape version 3.9.0 software was employed to represent the protein–protein interaction network visually. The visualization indicates that the PPI network comprised 570 edges and 129 nodes, with an average node degree 8.84. The nodes are intended to represent the individual proteins, while the edges are meant to depict the relationships that exist between the proteins. Figure 3 represents the PPI networking of common genes from disease and naringin. An increase in the degree value indicates that the protein plays a more substantial function in the network. There were 10 hub genes found when the aforementioned criteria were used to filter the data (Fig. 4): ALB, CASP3, ESR1, PARP1, HSP90AB1, CDK2, CDK1, ABL1, CSP8, and ACE. When it comes to the PPI networks, these genes are considered to be core targets since they possess superior network topological metrics (CC, MF, and BP). A variety of biological regulating activities, including human signal transcription and protein phosphorylation, are regulated by these proteins, which are biological enzymes and cytokines. There were five nodal targets that were shown to be the most essential: ALB, CASP3, ESR1, PARP1, and HSP90AB1. These nodal targets are highly connected with other prospective pharmacological targets and have a vital role in the treatment of GBM. For the purpose of conducting a cluster analysis of protein groups, the MCODE plugin was utilized, and the result was the acquisition of six cluster modules (Fig. 5). There were sixteen significant protein targets that were grouped together in the top functional modules. These targets included MMP7, MMP12, TCF12, ADAM17, MMP3, ALB, MME, FGF2, TYR, CAB39, CXCL10, FOLH1, PON1, ACHE, SLC5A2, and MMP13.It was shown that MMP7 and BACE1 are the core nodes in each functional module, which suggests that these major targets have a considerable influence on the effects that naringin has against GBM.

PPI (protein–protein interaction) networking of common genes from disease and naringin.

Top10 proteins or genes by degree analysis from common targets.

Cluster analysis of the PPI network.
GO (gene ontology) enrichment analysis
In order to investigate the molecular mechanisms by which naringin acts against GBM, we conducted a GO enrichment analysis on 101 possible therapeutic targets of naringin that are involved in the treatment of GBM. This analysis was performed using the DAVID database. The evaluation was categorized into three distinct groups: BP (Biological Process), MF (Molecular Function), and CC (Cellular Component). We acquired 137 base pairs (BP), 10 MF, and 3 CC terms. The bar graph (Fig. 6) illustrates the top 5 Gene Ontology (GO) items. It provides the gene enrichment and corresponding p-value. A greater abundance of therapeutic genes within that GO term shows a stronger association between the detected GO term and the treatment of GBM compared to other GO terms. The findings showed that primary enriched biological process categories were the response to naringin, regulation of apoptosis, response to an organic substance, response to chemicals, and response to oxygen-containing substances. Endopeptidase activity, hydrolase activity, peptidase activity, catalytic activity, and activity acting on proteins were the primary enriched MFs. The terms with the highest enrichment in CC analysis were the extracellular exosome, extracellular space, and caspase complex.

Gene ontology analysis of the top five biological processes, molecular functions, and cellular component categories associated with naringin and GBM.
KEGG pathway analysis
The enrichment studies of the KEGG pathway were used to identify the metabolic pathways that could be possible therapeutic targets for treating GB. Utilizing the DAVID database. Table 1 highlights the signalling pathways associated with naringin and GBM, arranged from least to greatest by P value. The findings demonstrated the enrichment of the major targets, which included numerous signalling pathways associated with cancer, such as the TNF, IL-17, and p53 pathways. Supplementary sheet 1 illustrates the network connectivity between the forecasted pathways of glioblastoma and the target genes obtained from the KEGG pathway. With Cytoscape v3.10.0, we created a visual network to examine the connections between active chemicals, possible therapeutic targets, and important pathways in more detail (Supplementary sheet 2). ABL1, CSP8, ACE, CDK2, CDK1, ABL1, PARP1, HSP90AB1, and CASP3 were among the core GBM targets with which the visual network shown a strong correlation, making it the most crucial drug. Consequently, we have chosen these specific targets to be examined in more detail through molecular docking investigations.
Molecular binding analysis
Molecular binding is a fundamental technique that is essential in drug discovery processes. It involves predicting the interactions between a protein (receptor) and a small molecule (ligand) to understand binding energetics, molecular interactions, and induced conformational changes. If the configuration of the ligand and the receptor is stable, then the potential for action will increase while the binding energy will decrease simultaneously. An interaction between the ligand and the receptor that occurs spontaneously is indicated by a binding energy that is less than 0 kcal/mol, but a binding energy that is less than − 5 kcal/mol implies a snug binding. A selection of the top 10 proteins (ALB, CASP3, ESR1, PARP1, HSP90AB1, CDK2, CDK1, ABL1, CASP8, ACE) were made for conducting molecular docking studies. These proteins were then compared with the drugs the FDA approved. Evidence demonstrates that naringin had superior binding activity compared to the FDA-approved medicines talazoparib and imatinib. Table 2 depicts the docking score of naringin with its corresponding protein and the binding interaction of FDA-authorised drugs with proteins. Docking interactions of naringin with PARP-1 (4ZZZ) and ABL1 (2ABL) proteins and with their standard drugs (talazoparib and imatinib) were illustrated in Supplementary sheet 3 as well as in Fig. 7. Among the PARP-1 inhibitors approved by the FDA, Naringin demonstrated superior PARP binding activity to Talazoparib. The docking score for Naringin was − 12.90 kcal/mol, indicating that it exhibited excellent binding interactions with PARP-1 (4ZZZ). Amino acids such as ILE872,TRP861, LEU877, TYR889, ILE879, ALA880, ILE895, TYR896, LEU984, PHE897, MET890,ALA898, TYR907, CYS908, and, TYR689 showed hydrophobic interaction. It was shown that other amino acids, such as SER904, TYR907, ARG878, ASP770, ASP776, and SER864, had a role in the inhibition of PARP-1 (4ZZZ), which indicates that they interact with the ligand through hydrogen bonds and form π–π bonds with TYR 907. It was observed that the amino acid complexes of the enzyme PARP1 inhibitors (4ZZZ) exhibited polar interaction with ligands. These complexes included ASN767, SER864, HIS862, ASN868, HIE909, and SER904. Talazoparib, an FDA-approved PARP inhibitor, had a − 4.45 kcal/mol binding score, which is lower than naringin’s affinity for the 4ZZZ protein. Only one amino acid, the ASP766 amino acid of the PARP1 inhibitor protein, was involved in the formation of an H-bond interaction with this molecule. Moreover, hydrophobic interaction (TYR907, ILE895, TYR896, PHE897, VAL762, MET890, TYR889, and LEU877), polar interaction (GLN759, HIS862, SER864, SER904), pi–pi stacking (HIS862, TYR889) was observed. Naringin exhibited superior binding activity compared to FDA-approved Tyrosine kinase protein (2ABL) inhibitors (imatinib) in the instance of Protein ABL1 (2ABL) with a score of binding energy of − 8.4 kcal/mol. In comparison, imatinib showed a binding score of − 5.12 kcal/mol. Naringin having established H-bonding interaction with (GLU142, THR136, ILE135, ASN115, GLU174, ASN83) amino acid residue of 2ABL protein. We observed that lipophilic interactions (LEU141, PRO137, ILE135, TYR112, PRO150, TYR147, LEU88, PHE85, and PRO82), polar interactions (SER140, THR136, ASN133, SER132, ASN115, SER173, SER175, SER176, ASN83) and no π–π stacking involved with amino acid complex of 2ABL protein. The molecular docking interactions of imatinib with 2ABL proteins were illustrated in Supplementary sheet 3. Among the top 10 proteins screened, the top two leads with the highest binding activity than FDA-approved drugs were selected for further studies (Molecular dynamic simulation studies).

2D &3D interaction of naringin (A,E) and talazoparib (B,F) with protein 4ZZZ and naringin (C,G) and imatinib (D,H) with 2ABL.
Molecular dynamics simulation analysis
Complex of naringin with 4ZZZ
The RMSD (root mean square deviation) of the complex exhibited a peak variation of around 11.1 Å. It demonstrated deviations below 0.7 Å, showing the consistent stability of the complex for the whole duration of the experiment. After a stabilization time of 50 ns, the RMSD remained constant and showed variations of less than 0.3 Å. The root mean square fluctuation (RMSF) of the complex atoms exhibited a peak fluctuation of approximately 10.2 Å, which was the same as the residues that showed the highest variation in the naringin–4ZZZ complex (Fig. 8; Movie 1—Supplementary sheet 6). These findings suggest that the binding between naringin and PARP-1 forms a durable complex, and that this interaction occurs specifically at the active site of the protein. The Radius of Gyration (RoG) for the complex exhibited fluctuations ranging from around 20.7 Å to 21.5 Å, with slight deviations confirming the protein’s compactness throughout the entire process. The surface area of the solvent-accessible region (SASA) exhibited variability between approximately 168 nm2 and 186 nm2. Initially, during the first 25 ns of the molecular dynamics (MD) simulation, the SASA was unstable. However, subsequently, there was a progressive increase in the SASA, which might be attributed to the formation and deformation of bonds throughout the MD run. During the MD run, a maximum of 10 hydrogen bonds were observed, while a minimum of 8 bonds remained constant. However, the production and deformation of hydrogen bonds occurred intermittently throughout the run. The energy decomposition analysis per residue revealed that ARG218 and TYR247 had the lowest energy contributions of − 2.13 and − 2.54 kcal/mol, respectively. It was discovered that for the approximately 95 ns of the MD run, the ligand’s (Naringin) total energy contribution was − 34.71 kcal/mol (Fig. 9).

Molecular dynamic simulation result of PARP1-Talazoparib and PARP1-Naringin complex where: (a) Root mean square deviation (RMSD) of the backbone (black) and complex for PARP1-Talazoparib (red), and (green) backbone and complex (Blue) for PAPRP1-Naringin, (b) (root mean square fluctuations) RMSF of c-alpha atoms (black) and complex (red) for PARP1-Talazoparib and c-alpha atoms (green) and complex (blue) for 4ZZZ-Naringin, (c) Radius of gyration of complex for PARP1-Talazoparib (Black) and PARP1-Naringin (Red), (d) Number of hydrogen bonds formed for PARP1-Talazoparib (Black) and 4ZZZ-Naringin(Red), (e) SASA of complex for PARP1-Talazoparib (Black) and PARP1-Naringin (Red).

The decomposition energy of Naringin (blue) and Talazoparib (Orange) with PARP1 where (a) Total energy decomposition vs time and (b) Total energy decomposition vs residue.
Complex of talazoparib with 4ZZZ
The root mean square deviation (RMSD) of the complex exhibited a peak variation of around 9.4 Å. However, the RMSD of the complex demonstrated deviations below 0.9 Å, suggesting that the complex remained stable throughout the whole run. After a stabilisation time of 100 ns, the RMSD was steady and showed variations of less than 0.3 Å. The RMSF of the complex atoms exhibited a maximal fluctuation of approximately 12.4 Å (Fig. 8 and Movie 2—Supplementary sheet 6). The Radius of Gyration (RoG) for the complex exhibited variations between approximately 20.6 Å and 21.4 Å, with slight deviations suggesting that the protein maintained a compact structure throughout the experiment. The surface area of the SASA exhibited variability between about 169 nm2 and 190 nm2. Initially, during the first 25 ns of the molecular dynamics (MD) simulation, the SASA was unstable. However, it gradually increased subsequently. This might be attributed to the formation and deformation of bonds occurring during the MD run. Throughout the MD run, the strongest hydrogen bonds out of the three was consistently observed. However, the formation and breaking of hydrogen bonds were also observed during the run. The energy breakdown analysis per residue revealed that TYR236 and LYS243 had the lowest energy contributions of − 1.63 and − 5.51 kcal/mol, respectively. The total energy contribution of the ligand was determined to be − 19.12 kcal/mol for the MD run that lasted approximately 100 ns, as depicted in Fig. 9.
Complex of naringin with tyrosine kinase protein (2ABL)
Over the course of the 200 ns MD run, the complex’s RMSD fluctuated at a maximum of ~ 5.2 Å, falling between ~ 2.5 Å and 5.2 Å, suggesting that the complex is stable. Following an approximately 120-ns MD run, the complex’s RMSD was shown to stabilise, and it subsequently had RMSD deviations of less than 2.0 Å. A maximum fluctuation of approximately 7.4 °C was seen in the RMSF of the complex atoms (Fig. 10; Movie 3—Supplementary sheet 6). There was variation in the complex’s Radius of Gyration (RoG), which ranged from about 16.5 °C to about 18 °C. For the first twenty ns, the SASA displayed instability, fluctuating between approximately 90 nm2 and 108 nm2. After around 80 ns of simulation, there was a progressive increase in SASA with little fluctuations. A total of six H bonds were seen during the whole run. Initially, for approximately 75 ns, the bonds were not stable, but they became stable after approximately 125 ns of molecular dynamics (MD) simulation. Consistently, at least four hydrogen bonds were seen over the duration of approximately 100 ns. The analysis of energy decomposition, per residue demonstrated that PRO77 and PRO64 had the lowest energy contributions of − 1.04 and − 0.57 kcal/mol, respectively. Ligand’s total energy contribution was − 31.79 kcal/mol, after an MD run of approximately 40 ns, as depicted in Fig. 11.

Molecular dynamic simulation results of Tyrosine-protein kinase-imatinib and Tyrosine-protein kinase—Naringin complex were, (a) Root mean square deviation (RMSD of complex (red) and backbone (black) for Tyrosine-protein kinase-imatinib, and complex (blue) and backbone (green) for Tyrosine-protein kinase-Naringin, (b) RMSF of carbon-alpha atoms (black) and complex (Red) for Tyrosine-protein kinase-imatinib and c-alpha atoms (green) and complex (blue) for Tyrosine-protein kinase—Naringin, (c) RoG of complex for Tyrosine-protein kinase-Imatinib (Black) and Tyrosine-protein kinase -Naringin (Red), (d) Number of H bonds formed for Tyrosine-protein kinase-Imatinib (Black) and Tyrosine-protein kinase-Naringin (Red), (e) SASA of complex for Tyrosine-protein kinase-Imatinib (Black) and Tyrosine-protein kinase-Naringin (Red).

The energy decomposition of Naringin (blue) and Imatinib (Orange) with tyrosine kinase protein where (a) Total energy decomposition vs time and (b) Total energy decomposition vs residue.
Complex of imatinib with tyrosine kinase protein (2ABL)
During the MD run of 200 ns, RMSD of the complex exhibited a maximum fluctuation of around 9.4 Å, which fell within the range of approximately 4.2 Å to 9.48 Å. This fluctuation serves as an indication of the complex stability. The RMSD value of the complex reached a stable state after approximately 100 ns of MD simulation. Subsequently, the RMSD exhibited variations of less than 3.0 Å. The RMSF of the complex atoms exhibited a maximal fluctuation of approximately 7.0 Å (see Fig. 10; Watch Movie 4—Supplementary sheet 6). The complex and backbone exhibited a fluctuation in the Radius of Gyration (RoG) between approximately 16.5 Å and 18 Å. After 20 ns of unstable behaviour, the SASA fluctuated between ~ 94 nm2 and ~ 110 nm2, and then began to gradually increase with very slight fluctuations after ~ 125 ns of simulation. During the run time a maximum of seven H bonds were observed; the bonds were unstable for around 75 ns at first and became stable after about 125 ns of MD run. Consistently, three H bonds were observed for a duration of approximately 100 ns. PRO09 and THR124 were found to have the lowest energy contributions of − 2.21 and − 0.65 kcal/mol, respectively, according to the total energy decomposition per residue analysis. The ligand’s overall energy contribution was determined to be − 39.95 kcal/mol during a ~ 70 ns MD simulation. According to the results of the total energy decomposition for each residue, PRO09 and THR124 are more likely to interact with one another. They have the lowest energy contribution, which is − 2.21 and − 0.65 kcal/mol, respectively (Fig. 11).
Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) analysis of naringin and talazoparib with PARP1 (4ZZZ)
Based on the MMPBSA analysis, which was presented in Supplementary sheet 4, it was found that the naringin-PARP1 complex exhibited the lowest values of VDWAALS, EPB, and GSOLV, with respective values of − 60.37 ± 0.42, 101.20 ± 1.02, and 95.20 ± 1.01 kcal/mol. Similarly, the conventional Talazoparib provided a total binding energy of − 31.80 ± 0.45 kcal/mol. This value is lesser than the naringin–4ZZZ complex, which reveals that the complex is highly stable and has a higher affinity for binding with protein.
MM-PBSA analysis of naringin and imatinib with ABL-1 (2ABL)
The MMPBSA analysis, which was presented in Supplementary sheet 5, demonstrated that the Naringin-Tyrosine kinase protein complex exhibited the lowest values of VDWAALS, EPB, and GSOLV, with respective values of − 31.18 ± 0.62, 46.80 ± 1.63, and 43 ± 1.60 kcal/mol. In the same vein, the complex exhibited the least amount of electrostatic and non-polar contributions from solute–solvent interactions to the solvation energy and total gas phase molecular mechanics energy. The energy decomposition for the complex was − 3.81 ± 0.06, and − 59.34 ± 1.87 kcal/mol, respectively. FDA-approved drug Imatinib exhibited a total relative binding energy of − 27.05 ± 0.58 kcal/mol, which is lower than that of the naringin-tyrosine kinase protein complex.
Principal component analysis
The principal component analysis (PCA) study helps in examining the important coordinated movements that occur during ligand binding. Only the first few eigenvectors are known to define the overall motion of the protein. In this study, the eigenvectors are obtained by diagonalizing the matrix. This study used the top 40 and 200 eigenvectors to estimate coordinated movements. Figure 12a,b show the eigenvalues generated from the diagonalization of the covariance matrix of atomic fluctuations versus the appropriate eigenvector in decreasing order. Out of the 40 eigenvectors, it can be safely inferred that both of the studied complexes had lesser movements and formed a stable association with naringin than imatinib and talazoparib. This could only be possible if the ligand association has altered protein structure and dynamics. Using PCA to construct 2D projection plots is another approach for obtaining the dynamics of protein–ligand complexes. Figure 12b,d depict a two-dimensional projection of the trajectories in phase space for the first two main components, eigenvector1 and eigenvector2, for both complexes. In general, the complexes occupying a compact phase space along with a stable cluster are considered more stable, whereas those complexes taking greater space with unstable clusters are considered less stable. As they took up minimal area in the phase space, all of the complexes were found to be very stable. The above PCA and other MD simulation findings were also in accordance with the 2D PCA reports.

Principal component analysis. The plot of the eigenvalues vs first 40 eigenvectors (a,b), the first two eigenvectors showing the protein motion in phase space for naringin (black) and Imatinib (red), (c,d) naringin (black) and Talazoparib (red).
In vitro anticancer effect of Naringin
The cytotoxic effects of Naringin on C6 rat glioma cells and U-87MG human glioblastoma cells were assessed using the MTT assay. Figure 13 demonstrated a dose-dependent reduction in cell viability for both cell lines upon treatment with naringin. The EC50 value, representing the concentration of naringin required to inhibit cell viability by 50%, was determined to be 34.1 µg/ml. This suggests that C6 cells exhibit moderate sensitivity to Naringin treatment. A more pronounced cytotoxic effect was observed in U-87MG cells, with an EC50 value of 10 µg/ml. This indicates that U-87MG cells are significantly more sensitive to Naringin compared to C6 cells.

In vitro cytotoxicity assay of naringin against (A) C6 cells and (B) U87MG cells.
Discussion
Glioma, a subtype of brain tumor, presents a substantial therapeutic obstacle owing to its invasive characteristics and resistance to standard therapies. Finding possible therapeutic targets and processes for glioma treatment through network pharmacology has become a very promising approach. Several investigations have employed network pharmacology to examine the impact of different compounds on glioma cells and to clarify the molecular mechanisms at stake. Nevertheless, the exact mechanism that underpins its anticancer activity against GBM is still unknown. We employed network pharmacology in the current study to reveal the mechanism of action that underlies the therapeutic effect of naringin in the treatment of GBM. Naringin, a flavonoid glycoside extracted from citrus fruits, has been investigated for its potential therapeutic applications in a variety of conditions, such as cancer. From a public database, we predicted the anticancer activity of naringin and GBM-related targets in naringin. Our research revealed that naringin has 129 shared targets with GBM, which directly impact apoptosis and cell growth. We discovered that these targets were grouped together as protein modules by analyzing the connections between possible therapeutic targets and a variety of biological processes. Because of their strong relationships with one another, these 129 prospective therapeutic targets serve as “hubs” for the entire therapeutic network.
Protein–protein interaction analysis revealed ten crucial proteins that play an additive role in different mechanisms such as apoptosis, metastasis, angiogenesis, and therapeutic resistance of GBM cells. In this case, ALB, CASP3, ESR1, PARP1, HSP90AB1, CDK2, CDK1, ABL1, CSP8, and ACE are crucial in the pathogenesis of GBM. The ALB gene has been implicated in various cancer-related studies, including gliomas. While the direct role of ALB in gliomas is not extensively discussed anywhere, its involvement in cancer biology and potential implications for GBM research can be inferred. Huang et al. identified ALB as one of the core targets of luteolin against glioma, indicating its potential relevance in anti-glioma mechanisms26. Caspase-3 (casp3) is a key mediator of apoptosis in glioblastoma, playing a crucial role in inducing programmed cell death in these aggressive brain tumors. Various treatments for glioblastoma have been linked to caspase-3 activation. For example, the compound 5-demethylnobiletin triggered programmed cell death in glioblastoma cells by increasing the production of cleaved caspase-327. The ESR-1 gene, also known as Estrogen Receptor 1, plays a major part in glioblastoma through its involvement in several molecular mechanisms. Sharanek et al. identified the LGALS1 gene, which encodes the galectin1 protein, as a critical regulator of Brain Tumor Stem- Cell (BTSC) fate in glioblastoma tumors with the EGFRvIII mutation. This highlights the transcriptional control of genes like LGALS1 in influencing the behaviour of cancer stem cells within glioblastoma28. HSP90 proteins act as molecular chaperones for client proteins, essential for cell survival. In glioma, HSP90AB1 has been associated with promoting autophagy and cell death by inducing ER stress29. Upregulation of HSP90AB1 has been connected to enhanced autophagy in glioma cells, leading to increased susceptibility to TGF-β1-triggered epithelial–mesenchymal transition30. HSP90AB1 emerges as a critical player in glioma pathogenesis, impacting various cellular processes such as autophagy, ER stress induction, and susceptibility to treatment-induced transitions. CDK2 plays a vital role in GBM progression through multiple mechanisms. According to studies, glioma cell growth is inhibited by CDK20 knockdown, which results in cell cycle arrest of the G1 phase and leads to decreasing CDK2 phosphorylation31. Long noncoding RNA LINC00958 has been identified as an oncogenic gene in glioma genesis through miR-203-CDK2 regulation, shedding light on a novel aspect of glioma tumorigenesis32. Furthermore, glioma cells undergo cell cycle arrest in the G0/G1 phase due to the downregulation of nuclear protein-1 via P27, indicating a potential regulatory pathway involving CDK2 in glioma cell cycle progression33. Studies have shown that CDK1 is essential for mitosis initiation and progression in glioma cells34. Inhibition of CDK1 expression can impact the proliferation of glioma cells by affecting the mitotic cell cycle. CDK1 is involved in cell cycle arrest and programmed cell death in glioma cells, further underlining its role in GBM progression and potential therapeutic targeting35. CSP8, a complex network of molecular signals, is pivotal in regulating various cellular processes implicated in cancer progression. CSP8 is intricately linked to the resistance of glioblastoma cells to current standard treatments, including chemotherapy and radiation therapy. Activating specific signaling pathways within CSP8 confers resistance mechanisms, rendering these therapies less effective and limiting patient treatment options. Targeting CSP8 pathways presents a promising strategy for combating glioblastoma. By disrupting the signaling cascades involved in tumor growth, invasion, and therapy resistance, novel therapeutic agents can be developed to target GBM cells while sparing healthy brain tissue specifically. The complexity of CSP8 signaling networks and the heterogeneity of glioblastoma tumors underscores the need for comprehensive research efforts and interdisciplinary collaborations to unravel the intricacies of CSP8 in glioblastoma biology. Angiotensin-converting enzyme (ACE) has been implicated in glioma. In anti-glioma therapy, the tumor microenvironment and angiogenic blood vessels are targeted. The direct mechanism of ACE’s action in glioma may not be fully understood, but its genetic variants and interactions with other molecules play a role in glioma susceptibility, progression, and patient outcomes.
The PARP pathway is crucial in glioma, particularly in DNA repair mechanisms and cell death processes. Studies have shown that inhibiting the DNA-PARP repair pathway can sensitize IDH1-mutated glioma cells to TMZ (temozolomide), a standard chemotherapy drug used in glioma treatment36. Additionally, PARP inhibitors have been found to enhance the cytotoxic effects of TMZ against glioma cells, suggesting a potential therapeutic strategy for improving treatment outcomes37. Furthermore, the PARP pathway has been implicated in glioma cell apoptosis, with evidence showing that PARP cleavage and activation of caspases are involved in inducing cell death in response to various treatments38. Targeting this pathway with PARP inhibitors shows promise as a therapeutic strategy, particularly in sensitizing IDH1-mutated gliomas to chemotherapy and enhancing treatment outcomes.
One of the genes implicated in glioblastoma is ABL1 (Abelson tyrosine-protein kinase1), which belongs to the family of non-receptor tyrosine kinases. Initially identified for its association with leukemia, ABL1 has since been implicated in various solid tumors, including glioblastoma. Its physiological functions span a broad spectrum, including regulation of cell proliferation, survival, adhesion, migration, and DNA repair39. In the context of targeted cancer therapy, kinase inhibitors have shown promise in treating various cancers. For instance, imatinib has successfully induced remission in leukemia patients with BCR-ABL1 fusions. Additionally, ABL1 is one of the genes encoding protein kinases that are affected by glioblastoma mutations, highlighting its significance in the pathogenesis of this cancer. Furthermore, the upregulation of ABL1 at the mRNA level has been observed in multiple cancers, including glioblastoma40,41. The journey to deciphering the role of ABL1 in glioblastoma is far from over. While significant strides have been made in elucidating its molecular mechanisms and therapeutic potential, many questions remain unanswered. Future research endeavors will undoubtedly illuminate the intricate interplay between ABL1 and the myriad factors governing glioblastoma pathogenesis. With this knowledge, scientists and clinicians stand poised to usher in a new era of precision medicine, offering hope to patients battling this formidable foe.
The gene ontology study identified several significantly enriched biological process categories, including the response to naringin, regulation of the apoptotic process, response to an organic substance, response to chemicals, and response to oxygen-containing compounds. Naringin has shown promise in inhibiting glioblastoma cell metastasis by targeting multiple aspects of the MAPK signalling pathways, such as p38, ERK, and JNK, and downregulating MMP-9 and MMP-2 expression and activities42. Moreover, naringin has been found to restore chemo-sensitivity to temozolomide in glioblastoma by targeting PARP1, which not only enhances chemo-sensitivity but also induces anti-angiogenic effects that may impact tumor growth15. Additionally, naringin has been reported to inhibit apoptosis induced by cyclic stretch in rat annular cells, potentially delaying intervertebral disc degeneration by inhibiting the ROS/NF-κB pathway43. Furthermore, naringin has been shown to inhibit glioblastoma cancer neo-angiogenesis, exerting anti-cancer effects and slowing cancer progression44. These results imply that naringin may have a complex mechanism of action against gliomas, including anti-angiogenic, anti-metastatic, and chemo-sensitizing qualities. To develop potentially new therapeutic approaches, it would be necessary to do additional studies to thoroughly understand the precise molecular targets and pathways that naringin uses to affect gliomas. The KEGG enrichment analysis revealed several important signaling pathways in naringin’s mechanism against glioblastoma. Most of the gene targets were enriched in the IL-17, P53, and TNF pathways, indicating that these pathways are crucial.
Molecular docking is a fundamental technique in network pharmacology that evaluates the binding affinity between molecular ligands and protein receptors, thereby validating the outcomes of network pharmacology analyses. Our findings indicate a direct interaction between the model features and the essential amino acids that hinder the activity of PARP-1 and the protein complex including Tyrosine kinase (ABL1). Therefore, these features can be considered essential chemical attributes in the quest for novel inhibitors of PARP1 and ABL1. Furthermore, GO analysis and network pharmacology prediction research identified PARP1 and ABL1 as the genes with the highest edge count. These particular genes were considered as hub genes for MD simulation. According to MD simulation, the naringin-PARP1 complex was shown to be the most stable, with a total energy of 18.42 ± 0.78 kcal/mol. Similarly, it was demonstrated that the ABL1 complex remained stable when combined with naringin, resulting in a total energy contribution of − 16.34 ± 0.56 kcal/mol. The stability characteristics demonstrate that the ligand naringin’s association with 4ZZZ and 2ABL remained stable for the whole MD simulation, hence confirming the accuracy of the molecular docking studies. The PCA study revealed that naringin forms more stable protein–ligand complexes compared to imatinib and talazoparib. By analyzing the top 40 and 200 eigenvectors, we observed that naringin complexes showed lesser movements, indicating greater stability. The 2D projection plots of the first two main components further confirmed this, as all complexes occupied minimal area in phase space, suggesting high stability. These findings align with other MD simulation results, highlighting naringin’s potential to induce favourable structural and dynamic changes in proteins. This suggests that naringin could enhance protein stability, making it a promising candidate for drug design. The MTT assay results indicate that naringin exerts a cytotoxic effect on both C6 and U87MG glioma cells, with U87MG cells being more sensitive. The lower EC50 value observed in U87MG cells suggests a higher potency of naringin against human glioblastoma cells compared to rat glioma cells. This differential sensitivity may be attributed to differences in cell origin, genetic background, and the expression of molecular targets relevant to naringin’s mechanism of action. U87MG cells might possess specific pathways that make them more susceptible to naringin’s induced cytotoxicity. These findings highlight naringin’s potential as a therapeutic agent for glioblastoma, particularly in tumors resembling the U87MG cell line. Further studies are necessary to elucidate the underlying mechanisms and to explore the potential of Naringin in combination with other therapies to enhance its efficacy in glioblastoma treatment.
Conclusion
Based on our findings from computational studies, we found that naringin-mediated therapy of GBM involved several targets and pathways. Naringin controls important targets involved in apoptosis, necrosis, angiogenesis, metastasis, and chemotherapy resistance, including those relevant in treating GBM via the TNF, P53, and IL-17 signaling pathways. These targets include ABL1, CSP8, ACE, CDK2, CDK1, ABL1, CASP3, ESR1, PARP1, HSP90AB1, and CDK2. Naringin holds significant promise as an anticancer agent against GBM, but realizing its full potential requires extensive and multidisciplinary research. Naringin demonstrates a higher cytotoxic potency against U-87MG human glioblastoma cells compared to C6 rat glioma cells, indicating its potential as a therapeutic agent for glioblastoma. The differential sensitivity suggests the need for further investigation into the specific mechanisms and potential combinatory strategies to enhance naringin’s efficacy in glioblastoma treatment. By exploring unexplored mechanisms, conducting rigorous preclinical and clinical studies, developing advanced delivery systems, and integrating personalized approaches, naringin could become a valuable addition to the arsenal of anticancer therapies.
Data availability
All data generated or analysed during this study are included in this published article as supplementary data.
References
Vollmann-Zwerenz, A., Leidgens, V., Feliciello, G., Klein, C. A. & Hau, P. Tumor cell invasion in glioblastoma. Int. J. Mol. Sci. 21(6), 1932. https://doi.org/10.3390/IJMS21061932 (2020).
Seystahl, K., Gramatzki, D., Roth, P. & Weller, M. Pharmacotherapies for the treatment of glioblastoma—Current evidence and perspectives. Expert Opin. Pharmacother. 17(9), 1259–1270. https://doi.org/10.1080/14656566.2016.1176146 (2016).
Xiao, B. et al. MicroRNA-566 modulates vascular endothelial growth factor by targeting Von Hippel-Landau in human glioblastoma in vitro and in vivo. Mol. Med. Rep. 13, 379–385 (2016).
Vasileva, N. et al. Double recombinant vaccinia virus: A candidate drug against human glioblastoma. Life 11(10), 1084. https://doi.org/10.3390/LIFE11101084 (2021).
Chamberlain, M. C. Treatment options for glioblastoma. Neurosurg. Focus 20(4), E19. https://doi.org/10.3171/FOC.2006.20.4.12 (2006).
Saki, M. et al. Combined treatment with dopamine receptor antagonists and radiation creates a metabolic vulnerability in mouse models of glioblastoma. bioRxiv https://doi.org/10.1101/2020.01.13.905380 (2020).
Ricklefs, F. L. et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. https://doi.org/10.1126/SCIADV.AAR2766/SUPPL_FILE/AAR2766_SM.PDF (2018).
Xi, Z. et al. Overexpression of miR-29a reduces the oncogenic properties of glioblastoma stem cells by downregulating Quaking gene isoform 6. Oncotarget 8(15), 24949–24963. https://doi.org/10.18632/ONCOTARGET.15327 (2017).
Shah, F. H. et al. Current progress of phytomedicine in glioblastoma therapy. Curr. Med. Sci. 40(6), 1067–1074. https://doi.org/10.1007/S11596-020-2288-8 (2020).
Tharamelveliyil Rajendran, A. et al. Selection of potential natural compounds for poly-ADP-ribose polymerase (PARP) inhibition in glioblastoma therapy by in silico screening methods. Saudi J. Biol. Sci. 30(7), 103698. https://doi.org/10.1016/J.SJBS.2023.103698 (2023).
Stabrauskiene, J., Kopustinskiene, D. M., Lazauskas, R. & Bernatoniene, J. Naringin and naringenin: Their mechanisms of action and the potential anticancer activities. Biomedicines https://doi.org/10.3390/BIOMEDICINES10071686 (2022).
Cirmi, S. et al. Chemopreventive agents and inhibitors of cancer hallmarks: May citrus offer new perspectives?. Nutrients 8(11), 698. https://doi.org/10.3390/NU8110698 (2016).
Aroui, S. et al. Naringin suppresses cell metastasis and the expression of matrix metalloproteinases (MMP-2 and MMP-9) via the inhibition of ERK-P38-JNK signaling pathway in human glioblastoma. Chem. Biol. Interact. 244, 195–203. https://doi.org/10.1016/J.CBI.2015.12.011 (2016).
Ding, S. et al. The new exploration of pure total flavonoids extracted from Citrus maxima (Burm.) Merr. as a new therapeutic agent to bring health benefits for people. Front. Nutr. 9, 958329. https://doi.org/10.3389/FNUT.2022.958329/BIBTEX (2022).
Rao, V. et al. Restoring chemo-sensitivity to temozolomide via targeted inhibition of poly (ADP-ribose) polymerase-1 by naringin in glioblastoma. Chem. Pap. 75(9), 4861–4871. https://doi.org/10.1007/S11696-021-01700-0/FIGURES/6 (2021).
Jeong, J., Kim, H. & Choi, J. In silico molecular docking and in vivo validation with Caenorhabditis elegans to discover molecular initiating events in adverse outcome pathway framework: Case study on endocrine-disrupting chemicals with estrogen and androgen receptors. Int. J. Mol. Sci. 20(5), 1209. https://doi.org/10.3390/IJMS20051209 (2019).
Ahmed, A. et al. Design, synthesis, kinetic analysis and pharmacophore-directed discovery of 3-ethylaniline hybrid imino-thiazolidinone as potential inhibitor of carbonic anhydrase II: An emerging biological target for treatment of cancer. Biomolecules 12(11), 1696. https://doi.org/10.3390/BIOM12111696/S1 (2022).
Zhang, K. et al. Mechanism study of Cinnamomi ramulus and Paris polyphylla Sm. drug pair in the treatment of adenomyosis by network pharmacology and experimental validation. Evid. Based Complement Alternat. Med. https://doi.org/10.1155/2022/2624434 (2022).
Hu, M. et al. Use of network pharmacology and molecular docking to explore the mechanism of action of curcuma in the treatment of osteosarcoma. Sci. Rep. 13(1), 1–13. https://doi.org/10.1038/s41598-023-36687-z (2023).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51(D1), D587–D592. https://doi.org/10.1093/NAR/GKAC963 (2023).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28(11), 1947–1951. https://doi.org/10.1002/PRO.3715 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30. https://doi.org/10.1093/NAR/28.1.27 (2000).
Van Der Spoel, D. et al. GROMACS: Fast, flexible, and free. J. Comput. Chem. https://doi.org/10.1002/jcc.20291 (2005).
Shirvalilou, S. et al. Enhancement radiation-induced apoptosis in C6 glioma tumor-bearing rats via pH-responsive magnetic graphene oxide nanocarrier. J. Photochem. Photobiol. B Biol. 205, 111827. https://doi.org/10.1016/J.JPHOTOBIOL.2020.111827 (2020).
Di Veroli, G. Y. et al. Combenefit: An interactive platform for the analysis and visualization of drug combinations. Bioinformatics 32(18), 2866–2868. https://doi.org/10.1093/BIOINFORMATICS/BTW230 (2016).
Huang, R. et al. Exploring the antiglioma mechanisms of luteolin based on network pharmacology and experimental verification. Evid. Based Complement Alternat. Med. https://doi.org/10.1155/2021/7765658 (2021).
Zhang, X. et al. 5-Demethylnobiletin mediates cell cycle arrest and apoptosis via the ERK1/2/AKT/STAT3 signaling pathways in glioblastoma cells. Front. Oncol. 13, 1143664. https://doi.org/10.3389/FONC.2023.1143664/BIBTEX (2023).
Sharanek, A. et al. Transcriptional control of brain tumour stem cells by a carbohydrate binding protein. bioRxiv https://doi.org/10.1101/2021.04.14.439704 (2021).
Salazar, M. et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Investig. 119(5), 1359–1372. https://doi.org/10.1172/JCI37948 (2009).
Zou, M. et al. AEG-1/MTDH-activated autophagy enhances human malignant glioma susceptibility to TGF-β1-triggered epithelial–mesenchymal transition. Oncotarget 7(11), 13122. https://doi.org/10.18632/ONCOTARGET.7536 (2016).
Lai, L., Shin, G. Y. & Qiu, H. The role of cell cycle regulators in cell survival—Dual functions of cyclin-dependent kinase 20 and p21Cip1/Waf1. Int. J. Mol. Sci. 21(22), 1–14. https://doi.org/10.3390/IJMS21228504 (2020).
Guo, E. et al. Long noncoding RNA LINC00958 accelerates gliomagenesis through regulating miR-203/CDK2. DNA Cell Biol. 37(5), 465–472. https://doi.org/10.1089/DNA.2018.4163 (2018).
Li, J. et al. Downregulation of nuclear protein-1 induces cell cycle arrest in G0/G1 phase in glioma cells in vivo and in vitro via P27. Neoplasma 67(4), 843–850. https://doi.org/10.4149/NEO_2020_190814N759 (2020).
Wang, Y. et al. SYST-23 CDK1 as an independent prognostic indicator and potential therapeutic target in glioma. Neuro-Oncol. Adv. 5(Suppl 3), iii32. https://doi.org/10.1093/NOAJNL/VDAD070.125 (2023).
Ferreira, W. A. S. et al. Pisosterol induces G2/M cell cycle arrest and apoptosis via the ATM/ATR signaling pathway in human glioma cells. Anticancer Agents Med. Chem. 20(6), 734–750. https://doi.org/10.2174/1871520620666200203160117 (2020).
Lu, Y. et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. 77(7), 1709–1718. https://doi.org/10.1158/0008-5472.CAN-16-2773 (2017).
Ortiz, R. et al. Temozolomide: An updated overview of resistance mechanisms, nanotechnology advances and clinical applications. Curr. Neuropharmacol. 19(4), 513–537. https://doi.org/10.2174/1570159X18666200626204005 (2021).
Chang, C. Y. et al. Endoplasmic reticulum stress contributes to indomethacin-induced glioma apoptosis. Int. J. Mol. Sci. https://doi.org/10.3390/IJMS21020557 (2020).
Tripathi, R., Liu, Z. & Plattner, R. EnABLing tumor growth and progression: Recent progress in unraveling the functions of ABL kinases in solid tumor cells. Curr. Pharmacol. Rep. 4(5), 367–379. https://doi.org/10.1007/S40495-018-0149-Y (2018).
Gross, S., Rahal, R., Stransky, N., Lengauer, C. & Hoeflich, K. P. Targeting cancer with kinase inhibitors. J. Clin. Investig. 125(5), 1780–1789. https://doi.org/10.1172/JCI76094 (2015).
Varmus, H., Unni, A. M. & Lockwood, W. W. How cancer genomics drives cancer biology: Does synthetic lethality explain mutually exclusive oncogenic mutations?. Cold Spring Harb. Symp. Quant. Biol. 81(1), 247–255. https://doi.org/10.1101/SQB.2016.81.030866 (2016).
Ghanbari-Movahed, M., Jackson, G., Farzaei, M. H. & Bishayee, A. A systematic review of the preventive and therapeutic effects of naringin against human malignancies. Front. Pharmacol. https://doi.org/10.3389/FPHAR.2021.639840/FULL (2021).
Zhang, Y. H. et al. Naringin inhibits apoptosis induced by cyclic stretch in rat annular cells and partially attenuates disc degeneration by inhibiting the ROS/NF-κB pathway. Oxid. Med. Cell Longev. https://doi.org/10.1155/2022/6179444 (2022).
Aroui, S., Fetoui, H. & Kenani, A. Natural dietary compound naringin inhibits glioblastoma cancer neoangiogenesis. BMC Pharmacol. Toxicol. 21(1), 1–10. https://doi.org/10.1186/S40360-020-00426-1/FIGURES/5 (2020).
Acknowledgements
We acknowledge NITTE (Deemed to be University), Mangaluru for providing software and technical support for the smooth conduction of the research work.
Author information
Authors and Affiliations
Contributions
A.T.R: manuscript writing, methodology, and data analysis; G.D.R: methodology and data analysis; H.A and A.S: methodology; P.K: Study plan, supervision and proofreading; AN V: conceptualization, study plan, and supervision. P.K and A.N.V contributed equally to this work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tharamelveliyil Rajendran, A., Dheeraj Rajesh, G., Ashtekar, H. et al. Uncovering naringin’s anticancer mechanisms in glioblastoma via molecular docking and network pharmacology approaches. Sci Rep 14, 21486 (2024). https://doi.org/10.1038/s41598-024-72475-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-72475-z
- Springer Nature Limited