
Abstract
Heme serves as a prosthetic group in hemoproteins, including subunits of the mammalian mitochondrial electron transfer chain. The first enzyme in vertebrate heme biosynthesis, 5-aminolevulinic acid synthase 1 (ALAS1), is ubiquitously expressed and essential for producing 5-aminolevulinic acid (ALA). We previously showed that Alas1 heterozygous mice at 20–35 weeks (aged-A1+/−s) manifested impaired glucose metabolism, mitochondrial malformation in skeletal muscle, and reduced exercise tolerance, potentially linked to autophagy dysfunction. In this study, we investigated autophagy in A1+/−s and a sarcopenic phenotype in A1+/−s at 75–95 weeks (senile-A1+/−s). Senile-A1+/−s exhibited significantly reduced body and gastrocnemius muscle weight, and muscle strength, indicating an accelerated sarcopenic phenotype. Decreases in total LC3 and LC3-II protein and Map1lc3a mRNA levels were observed in aged-A1+/−s under fasting conditions and in Alas1 knockdown myocyte-differentiated C2C12 cells (A1KD-C2C12s) cultured in high- or low-glucose medium. ALA treatment largely reversed these declines. Reduced AMP-activated protein kinase (AMPK) signaling was associated with decreased autophagy in aged-A1+/−s and A1KD-C2C12s. AMPK modulation using AICAR (activator) and dorsomorphin (inhibitor) affected LC3 protein levels in an AMPK-dependent manner. Our findings suggest that heme deficiency contributes to accelerated sarcopenia-like defects and reduced autophagy in skeletal muscle, primarily due to decreased AMPK signaling.
Similar content being viewed by others
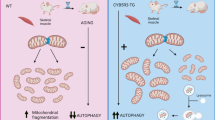


Introduction
Heme, a crucial biomolecule, serves as the prosthetic group for hemoproteins like hemoglobin and myoglobin and is a component of several complexes in the mammal mitochondrial electron transfer chain (ETC)1,2. It also acts as a ligand for heme-binding transcription factors such as Bach1/23. Recent research has highlighted heme’s emerging role as a signaling molecule in diverse cellular processe4.
5-Aminolevulinic acid (ALA), the first heme biosynthetic precursor in the vertebrate, is synthesized from succinyl CoA and glycine in the mitochondrial matrix, which is a rate-limiting step of heme biosynthesis and catalyzed by 5-aminolevulinic acid synthase (ALAS). When administered, ALA is rapidly metabolized to heme in normal cells and has been used as a supplement for improving glucose metabolism5.
Ubiquitous heme production, inducing mitochondrial ETC complexes, is regulated by a ubiquitous isozyme of ALAS known as ALAS16. This is consistent with our previous findings that Alas1-null mice were early embryonic lethal7, whereas knockout mice for the erythroid-selective isozyme Alas2 gene exhibited later embryonic lethality compared to Alas1-null mice due to severe anemia from heme deficiency in hemoglobin8,9,10. Despite no apparent abnormalities in heterozygous animals (hereafter designated as A1+/−s)7, we found that A1+/−s more than 20 weeks after birth (designated as aged-A1+/−s below) suffered from glucose intolerance and insulin resistance, accompanying atrophied mitochondria in skeletal muscle (SM)11. Administration of ALA improved the defects of glucose metabolism, exercise endurance, and mitochondria in aged-A1+/−s, suggesting that heme deficiency causes those abnormalities11. In many tissues, a significant part of ATP is produced in mitochondrial ETC complexes, and an adequate supply of heme is essential for normal ATP synthesis. We previously reported an age-dependent reduction in free heme levels and Alas1 mRNA expression in SM in mice12, indicating that age-dependent mitochondrial dysfunction could be attributed partially to age-dependent heme deficiency in SM.
Autophagy is a catabolic mechanism that degrades cytoplasm components in lysosomes and makes its products available for reuse13. Autophagy works in the removal of abnormal intracellular organelles, including mitochondria. In muscle diseases characterized by abnormal mitochondrial accumulation and vacuole formation, autophagy is inhibited or altered14, contributing to the degeneration and weakening of myofibers and progressively worsening in aging muscles. Sarcopenia, the age-related degenerative loss of SM mass and strength due to aging, is mainly responsible for functional decline and frailty in older adults15. Previous studies have indicated that a reduction in autophagic capacity contributes to sarcopenia16,17.
Several signaling pathways regulate autophagic activity. AMP-activated protein kinase (AMPK) is a crucial regulator of cellular energy homeostasis and is known to play a critical role in the regulation of autophagy18. AMPK signaling is activated by various stimuli, such as low energy levels, oxidative stress, and hypoxia, which trigger an increase in the AMP/ATP ratio, resulting in the activation of AMPK. Thr172-phosphorylated AMPKα (p-AMPKα) can phosphorylate the ULK1 complex responsible for initiating the autophagosome formation18. AMPK also inhibits mTORC1, a negative regulator of autophagy19, thereby activating autophagy through ULK1 phosphorylation18. The eukaryotic initiation factor 2α (eIF2α) plays a crucial role in autophagy regulation. eIF2α is phosphorylated at Ser51 in response to various cellular stresses by protein kinases, such as heme-regulated inhibitor (HRI), decreasing global protein translation. While global protein synthesis is reduced, the translation of some specific mRNAs, such as those encoding the transcription factor activating transcription factor 4 (ATF4), is enhanced. ATF4 promotes the expression of various autophagy-related genes, such as microtubule-associated protein 1A/1B-light chain 3 (Map1lc3a/b) encoding LC3 protein, leading to the induction of autophagy20. p62 also promotes autophagy by binding to ubiquitinated proteins and targeting them to autophagosomes and inhibits degradation of Nrf2 by Keap1, which enhances the formation of autophagosomes (p62-Kelch-like ECH-associated protein 1 (Keap1)- Nrf2 pathway)21.
Our previous electron microscopic examinations observed that aged-A1+/−s manifesting reduced exercise endurance showed atrophied mitochondria and vacuoles more frequently11. Abnormal autophagy in SM may contribute to those defects of A1+/−s. Reduction in autophagy was reported in SM of insulin-resistant patients with type 2 diabetes22. Mitofusion2 (Mfn2) deficiency in mouse SM caused reduced autophagy and impaired mitochondrial quality23. These facts prompted us to investigate autophagy in SM of aged-A1+/−s suffering from insulin resistance.
This study confirmed the accelerated sarcopenia-like phenotype, that is, a decline in body weight, muscle weight, and muscle strength, in A1+/−s between 75–90 weeks of age, henceforth designated as senile-A1+/−s. Then, we hypothesized that those phenotypes like sarcopenia and an age-dependent mitochondrial abnormality in SM of A1+/−s11 can be attributed to abnormal autophagy. We also analyzed autophagic activity in SM of aged-A1+/−s, revealing reduced autophagy. The reduced autophagic activity might be attributed to the AMP-activated kinase (AMPK) signaling reduction in SM of aged-A1+/−s.
Results
Accelerated sarcopenia-like defects in senile Alas1+/− mice
Previously, we reported no significant difference in body weight between WTs and A1+/−s at approximately 25-weeks11. Previous studies24,25 suggested that grip strength, exercise endurance, muscle volume, and muscle mass of 75-week‐old mice were significantly lower than those of 10‐week‐old C57BL/6 J mice, indicating that at least 75-week‐old mice present sarcopenia. Then, older A1+/−s (45–60 and 75–90 weeks) exhibited significant reductions in body weight compared to WTs (p < 0.05) (Fig. 1a). Fast-twitch fibers are characterized by their ability to generate rapid, powerful contractions compared to slow-twitch fibers. Sarcopenia is characterized by a selectively reduced size and greater atrophy of type 2 fibers26,27. Therefore, we analyzed the mass of the gastrocnemius and triceps brachii muscles, both of the fast-twitch fiber-rich muscles28. Similarly, the gastrocnemius and triceps brachii muscle weights in A1+/−s at 90 weeks were significantly lower than those in WTs (p < 0.05) (Fig. 1b,d). This trend was also observed in both muscle weights when normalized to tibia or humeral length (Figs. 1c,e) at 90 weeks. The muscle strength of WTs and A1+/−s at 15 weeks old, 26–35 weeks old, and 75–90 weeks old, designated as young-, aged-, and senile-WTs /-A1 ± s, respectively, was assessed using the hanging wire test (Fig. 1f) and the grip strength test (Fig. 1g). There were significant reductions in both the holding impulse (HI), the product of duration time of hanging and body weight, and grip strength, between senile groups and young or aged groups of WTs and -A1 ± s, respectively (Fig. 1f,g), supporting the sarcopenia-like phenotypes in both senile groups. Senile-A1+/−s showed a significant decrease in HI and grip strength compared to senile-WTs (p < 0.05) (Fig. 1d,e). At 90-weeks, A1+/−s showed an accelerated loss of body weight and SM mass, along with a decline in muscle strength, indicating exacerbated senile sarcopenia-like defects in these mice.

Senile Alas1+/− mice exhibit a decline in body weight, muscle weight, and muscle strength. (a) Body weight of wild-type (WTs) and Alas1+/− mice (A1+/−s) at 45–60 weeks (WTs: n = 16, A1+/−s: n = 21) and at 75–90 weeks of age (n = 9 per group). (b–e) Gastrocnemius muscle weight (b) and gastrocnemius muscle weight standardized by tibia length (c), and triceps brachii muscle weight (d) and triceps brachii muscle weight standardized by humeral length (e) in senile mice (75–90 weeks; n = 4 per group). (f) Holding impulse (HI) (body weight [g] x hanging time [sec]) from the hanging wire test assessing muscle strength) at 15 and 26–34 weeks (n = 6 per group), and at 75–90 weeks (WT: n = 9; A1+/−s: n = 8) of age. (g) Grip strength (grams) at 15 weeks (WT: n = 20; A1+/−s: n = 16), at 30–35 weeks (WT: n = 15; A1+/−s: n = 13), and at 90 weeks (n = 10 per group) of age. WTs represent wild-type littermates, and A1+/−s represents Alas1 heterozygous mice. Results are means ± standard error of the mean. * p < 0.05 indicates a significant difference between groups.
Decreased autophagy in skeletal muscle of aged Alas1+/− mice and Alas1-knockdown or heme-deficient C2C12 myocytes
Since autophagic activities under normally fed conditions are generally lower, we examined them under fed and fasting conditions. Alas1 mRNA levels in SM of aged-A1+/−s were confirmed to be significantly lower than aged-WTs under fasting conditions like under fed conditions, while were not significantly augmented by fasting, unlike in aged-WTs (Fig. 2a).

Reduction in Alas1 mRNA, map1lc3a mRNA (encoding LC3), and LC3 protein levels in skeletal muscle of aged Alas1+/− mice under the fed and fasting conditions. (a,b) Levels of Alas1 mRNA (a) and map1lc3a mRNA (b) in skeletal muscle of aged mice (20–35 weeks old; n = 6 per group). (c,d) Total LC3 protein and LC3-II protein levels under fed (c) and fasting (d) conditions. (c n = 5 per group; d WTs: n = 5, A1+/−s: n = 4, ALA-administered A1+/−s (ALA-adm-A1+/−s): n = 3). (e) Total LC3 protein and LC3-II protein in skeletal muscle of young mice (10 weeks old; WTs n = 6; A1+/−s n = 5) under the fasting conditions. ALA-adm-A1+/−s represents A1+/−s orally administered with ALA. The data presented are representative of results obtained from multiple independent experiments, each performed with biological replicates and technical replicates.. Results are means ± standard error of the mean.* p < 0.05, ** p < 0.01 indicate significant differences between groups.
LC3 protein is one of the components of autophagosome. LC3-I, cytosolic intact form, is converted to LC3-II by adding phosphatidylethanolamine (PE) anchored in the autophagosome membrane. LC3-II, localized on autophagosomes, is ultimately degraded during the autophagy process29,30. Decreases in the protein levels of total LC3 and LC3-II suggest reduced autophagic activity. Aged-A1+/−s showed significantly decreased levels of Map1lc3a mRNA encoding LC3 protein in SM compared to controls under fed and fasting conditions (p < 0.01 and p < 0.05, respectively) (Fig. 2b). Aged ALA-administered A1+/−s, designated aged-ALA-adm-A1+/−s, recovered from decreased Map1lc3a mRNA expression only under fed conditions (p < 0.01, Fig. 2b). In each group, increases in Map1lc3a mRNA expression by fasting were observed (p < 0.01, respectively) (Fig. 2b).
Protein levels of LC3 in SM under fed conditions showed no significant difference between aged-WTs and aged-A1+/−s (Fig. 2c). However, protein levels of total LC3 and LC3-II in aged-A1+/−s were significantly lower than those in WTs under fasting conditions (p < 0.01, respectively) (Fig. 2d), while administration of ALA for aged-A1+/−s significantly increased protein levels of total LC3 and LC3-II (p < 0.01, respectively) (Fig. 2d). No significant differences were observed in the ratios of LC3-I to LC3-II among aged-WTs, aged-A1+/−s, and ALA-administered aged-A1+/−s, as well as in the C2C12 cell experiments below (data not shown).
We previously reported that approximately 10-weeks-old A1+/−s (young-A1+/−s) did not manifest phenotypes such as glucose intolerance, insulin resistance, or mitochondrial abnormalities in SM11, and free heme deficiency12. Protein levels of LC3-II of young-A1+/−s were not significantly less than those of young-WTs, but those of total LC3 of young-A1+/−s were significantly less (Fig. 2e).
Mouse Alas1-knockdown C2C12 myocytes (A1KD-C2C12s), which we had previously established as a model of SM cells of A1+/−s11, cultured in high-glucose medium, corresponding to mice under fed conditions, showed no significant decrease in the protein levels of both total LC3 and LC3-II compared to control C2C12 myocytes (CT-C2C12s) (Fig. 3a). However, when those in high-glucose medium were treated with chloroquine (CQ), which is an inhibitor of autophagosome degradation, protein levels of total LC3 and LC3-II significantly decreased in A1KD-C2C12s in comparison with CT-C2C12s (p < 0.05, respectively) (Fig. 3b). When low-glucose culture, corresponding to mice under fasting conditions, protein levels of LC3-II in A1KD-C2C12s were significantly lower than CT-C2C12s (p < 0.01) with or without treatment of CQ (Figs. 3c,d), and significant reductions in protein levels of total LC3 were found in A1KD-C2C12s cultured only with the treatment of CQ. The reduced protein levels of LC3-II in A1KD-C2C12s were recovered by ALA treatment (p < 0.05) (Figs. 3b,d).

Decreased autophagic activity in Alas1-knockdown and heme-deficient C2C12 myocytes. (a,b) Total LC-3 protein and LC3-II protein levels in C2C12 myocytes cultured in high-glucose medium without (a) or with (b) chloroquine (CQ) (control C2C12 myocytes: CT, n = 4; Alas1-knockdown C2C12 myocytes: A1KD, n = 4). (c,d) Levels in myocytes cultured in low-glucose medium without (c) or with CQ (d) (n = 4 for each condition) (e) Levels in CQ-treated myocytes cultured in low-glucose medium with or without succinylacetone (SA), a heme biosynthesis inhibitor (n = 6 for each condition). CT represents control C2C12 myocytes, and A1KD represents Alas1-knockdown C2C12 myocytes. A1KD + ALA represents Alas1-knockdown C2C12 myocytes treated with ALA. The presented data are representative of results obtained from multiple independent experiments, each performed with biological replicates and technical replicates. Results are means ± standard error of the mean. * p < 0.05, ** p < 0.01 indicate significant differences between groups.
To determine whether the deficiency of ALA or heme causes decreased autophagy in SM cells, we examined the effect of succinylacetone (SA) on LC3 protein expression in C2C12 myocytes (Fig. 3e). SA is an inhibitor of 5-aminolevulinate dehydratase (ALAD), the next biosynthetic enzyme to ALAS in vertebrate heme biosynthetic pathway, and can induce cellular heme deficiency accompanying surplus rather than deficiency of ALA. SA-treated C2C12 myocytes also showed reductions in protein levels of total LC3 and LC3-II (Fig. 3e).
These data suggest that a deficiency of heme rather than ALA can decrease autophagic activity in SM and myocytes.
Association of decreased AMPK activation with reduced autophagic activity in heme-deficient skeletal muscle and myocytes
We first investigated major signaling pathways for autophagy regulation in SM of aged mice and C2C12 myocytes under autophagy-stimulating conditions: starvation and culture in low-glucose medium, respectively. Under fasting conditions, there was a significant decrease in the protein levels of p-AMPKα and in the ratio of p-AMPKα to total AMPKα (p-AMPKα/AMPKα) in aged-A1+/−s compared to aged-WTs (p < 0.05, respectively) (Fig. 4a), while those in aged-ALA-adm-A1+/−s significantly increased in comparison to aged-A1+/−s (p < 0.05, respectively) (Fig. 4a).


Proteins Expression in autophagy regulatory pathways in skeletal muscle of aged Alas1+/− mice under fasting conditions. (a) Levels of Thr172-phosphorylated AMPKα (p-AMPKα), total AMPKα, Thr389-phosphorylated p70S6K (p-p70S6K), and total p70S6K in skeletal muscle of aged mice (20–35 weeks; WT: n = 5, A1+/−s: n = 5, ALA-adm-A1+/−s: n = 3). Below are quantifications of p-AMPK, the p-AMPKα/AMPKα ratio, p-p70S6K, and the p-p70S6K/p70S6K ratio. (b) Levels of Ser51-phosphorylatd eIF2α (p-eIF2α), total eIF2α, and ATF4 (WT: n = 4, A1+/−s: n = 4, ALA-adm-A1+/−s: n = 4) with quantifications below. (c) Ulk1 mRNA levels (WT: n = 5, A1+/−s: n = 5, ALA-adm-A1+/−s: n = 3). (d) p62 and Nrf2 protein levels (WT: n = 5, A1+/−s: n = 4) with quantifications below. (e) p-AMPKα and total AMPKα levels in young mice (10–15 weeks; WT: n = 5, A1+/−s: n = 5) with quantifications below. The presented data are representative of results obtained from multiple independent experiments, each performed with biological replicates and technical replicates. Results are means ± standard error of the mean. * p < 0.05, ** p < 0.01 indicate significant differences between groups.
Activation of mTORC1 leads to the inhibition of autophagy19. p70S6K is phosphorylated by active mTORC1, making the protein levels of the phosphorylated p-70S6K (p-p70S6K) a marker of the mTORC activation. The protein levels of p-p70S6K in A1+/−s compared were significantly greater than WTs (p < 0.05) (Fig. 4a). Protein levels of p-p70S6K expression in aged-ALA-adm-A1+/−s decreased compared to aged-A1+/−s (Fig. 4a). There was no difference in the p-p70S6K/p70S6K ratio between groups (Fig. 4a).
Eukaryotic initiation factor2α (eIF2α), a translation factor, can regulate autophagy through ATF4, a transcription factor whose translation is activated by phosphorylation of eIF2α20. Both protein levels of p-eIF2α and p-eIF2α/eIF2α ratio significantly decreased in aged-A1+/−s under fasting conditions compared to aged-WTs (p < 0.05, respectively) (Fig. 4b). Protein levels of p-eIF2α and p-eIF2α/eIF2α ratio (p-eIF2α/eIF2α) in aged-ALA-adm-A1+/−s significantly increased compared to aged-A1+/−s (p < 0.05, respectively) (Fig. 4b). Protein levels of ATF4 also significantly decreased in aged-A1+/−s compared to aged-WTs (p < 0.05) (Fig. 4b). Protein levels of ATF4 in aged-ALA-adm-A1+/−s significantly increased in contrast to A1+/−s (p < 0.05) (Fig. 4b). Consistently above, mRNA expression levels of UlK1, whose transcription is activated by ATF4, decreased significantly in aged-A1+/−s in comparison to aged-WTs (p < 0.05) (Fig. 4c). Ulk1 mRNA levels in aged-ALA-adm-A1+/−s significantly increased higher than those of aged-A1+/−s (p < 0.05) (Fig. 4c).
p62 and Nrf2, regulators of autophagy21, exhibited similar protein levels in aged-WTs and aged-A1+/−s (Fig. 4d), indicating that heme deficiency-related autophagy decrease in SM is not associated with Nrf2 modulation.
In Alas1 knockdown C2C12 myocytes (A1KD-C2C12s) cultured in low-glucose medium, we noted a significant decrease in the phosphorylation levels of AMPKα and eIF2α (p < 0.05 and p < 0.01, respectively). This pattern mirrors that seen in the skeletal muscle (SM) of aged Alas1 heterozygous mice (aged-A1 ± s). Furthermore, treatment with 5-aminolevulinic acid (ALA) in A1KD-C2C12s led to a significant increase in both the phosphorylation levels and ratios of p-AMPKα/AMPKα and p-eIF2α/eIF2α (p < 0.01 for both) (Fig. 5), indicating that ALA treatment effectively enhances the phosphorylation of these proteins.

Expression of AMPKα and eIF2α in C2C12 myocytes cultured in low glucose medium. Levels of Thr172-phosphorylated AMPKα (p-AMPKα), total AMPKα, Ser51-phosphorylatd eIF2α (p-eIF2α), and total eIF2α in control C2C12 (CT), Alas1 knockdown C2C12 (A1KD), and ALA-treated Alas1 knockdown C2C12 (A1KD + ALA) myocytes (CT, A1KD, A1KD + ALA: n = 4 for each condition). Quantifications are shown below. The presented data are representative of results obtained from multiple independent experiments, each performed with biological replicates and technical replicates. Results are means ± standard error of the mean. * p < 0.05, ** p < 0.01 indicate significant differences between groups.
Similarly, in A1KD-C2C12s cultured in low-glucose medium, we observed a significant decrease in the phosphorylation of AMPKα and eIF2α (p < 0.05 and p < 0.01, respectively). This was also observed in the ratios of p-AMPKα/AMPKα and p-eIF2α/eIF2α (p < 0.05 and p < 0.01, respectively), when compared to CT-C2C12s (Fig. 5), like in SM of aged-A1+/−s. ALA treatment on A1KD-C2C12s led to a significant increase in both the phosphorylation levels and ratios of AMPKα and eIF2α (p < 0.01 for both) (Fig. 5), indicating that ALA treatment effectively enhances the phosphorylation of these proteins.
In young-A1+/−s, there were reductions in the mean protein levels of p-AMPKα and the mean p-AMPKα/AMPKα ratio in SM, although these reductions were not significant (Fig. 4e). These results suggest that decreased autophagic activities in heme-deficient SM and myocytes under fasting/low-glucose conditions are associated with decreases in AMPK activation and/or eIF2α phosphorylation.
Comparative analysis of AMPK and eIF2α signaling in autophagy regulation in skeletal muscle of aged Alas1+/− mice under fed and fasting conditions
Under fed conditions, we observed no significant decreases in the levels of LC3 protein in SM of aged-A1+/−s (Fig. 2c), unlike in chloroquine-treated A1KD-C2C12s cultured in high-glucose medium (Fig. 3b). We speculate that the lack of decrease in LC3 protein expression in SM of aged-A1+/−s under fed conditions could be attributed to the relatively low expression of LC3 protein, suggesting that autophagy may be decreased under both fasting and fed conditions in SM of aged-A1+/−s. Given that nutritional conditions can modulate the signaling pathways of AMPK and eIF2α18,19,20, we compared these pathways in SM of A1+/−s between under fed and fasting conditions.
The expression levels of p-AMPK and p-AMPKα/AMPKα ratio were significantly decreased in aged-A1+/−s compared to aged-WTs under both fed and fasting conditions (p < 0.01, respectively) (Fig. 6). Conversely, increased protein levels of p-eIF2α were found in aged-A1+/−s under fed conditions compared to WTs (p < 0.05) (Fig. 6), which contrasts with lower levels in aged-A1+/−s under fasting conditions compared to WTs (p < 0.01) (Figs. 4b and 6). Increased levels of p-eIF2α are supposed to activate autophagy, suggesting that a reduction in AMPK signaling may be more relevant to the decreased autophagy than changes in eIF2α signaling in SM of aged-A1+/−s.

AMPK and eIF2α signaling under fed and fasting conditions in aged Alas1+/− mice. Protein levels of Thr172-phosphorylated AMPKα (p-AMPKα), total AMPKα, Ser51-phosphorylatd eIF2α (p-eIF2α), and total eIF2α in skeletal muscle of aged-WTs and A1+/−s (n = 3 for each condition). Quantifications shown below. The presented data are representative of results obtained from multiple independent experiments, each performed with biological replicates and technical replicates. Results are means ± standard error of the mean. * p < 0.05, ** p < 0.01 indicate significant differences between groups.
Impact of modulation of AMPK and eIF2α signaling on reduced autophagy in alas1-knockdown C2C12 myocytes
To evaluate the impact of AMPK and eIF2α signaling on autophagy in A1KD-C2C12s, we analyzed the protein levels of LC3 in CQ-treated A1KD-C2C12s following treatment with AICAR, (an AMPK activator), sarubrinal (a phosphatase inhibitor that increases phosphorylated eIF2α), or dorsomorphin (an AMPK inhibitor).
AICAR treatment in A1KD-C2C12s significantly increased the protein levels of total LC3 and LC3-II compared to untreated A1KD-C2C12s (p < 0.05, respectively), while salubrinal treatment did not have this effect (Fig. 7a). Additionally, dorsomorphin treatment suppressed the elevation of total LC3 and LC3-II in ALA-treated A1KD-C2C12s (Fig. 7b). This data suggests that the decreased autophagy observed in A1KD-C2C12s, induced by heme deficiency in SM and myocytes, might be attributed to reduced AMPK signaling.

Impact of AMPK or eIF2α signaling modulation on autophagy in Alas1-knockdown C2C12 myocytes cultured in low-glucose medium. (a,b) LC3-I/II protein levels in chloroquine (CQ)-treated control (CT) and Alas1-knockdown C2C12 (A1KD) myocytes under low-glucose conditions treated with ALA, AICAR (an AMPK activator), or salubrinal (an eIF2α dephosphorylation inhibitor) (a), or treated with ALA, dorsomorphin (an AMPK inhibitor), or both (b) (n = 3 for each condition). Quantifications shown below. The presented data are representative of results obtained from multiple independent experiments, each performed with biological replicates and technical replicates. Results are means ± standard error of the mean. * p < 0.05, ** p < 0.01 indicate significant differences between groups.
Discussion
In this study, senile A1+/−s (75–90 weeks old) exhibited decreases in the whole body and muscle weights, along with weakened muscle strength compared to senile wildtype controls (Fig. 1). The senile wildtype controls showed lower muscle strength than young controls (15 weeks old) (Fig. 1f,g), suggesting that the senile wildtype controls sponteneously manifest sarcopnia-like defects. These data indicate that ALA deficiency might accelerate defects similar to ‘sarcopenia’, characterized by an age-dependent decrease in muscle mass and strength.
We did not measure the skeletal muscle mass of ALA-administered A1+/−s. However, Fujii et al. have already reported that ALA administration increases the skeletal muscle weight of 100-week-old mice31, suggesting that ALA administration at a pharmacological dose increases skeletal muscle mass. We consider that the effect of ALA administration in A1+/−s is substantially similar to that in wild-type controls. Therefore, our focus was on the muscle weight of A1 ± mice, as the effect of heme deficiency on skeletal muscle weight had not been previously investigated.
Reduced protein expression of LC3-II and total LC3 was observed in SM of aged-A1+/-s (~ 30 weeks-old) under fasting and in A1KD-C2C12s cultured in low-glucose medium, which was reversed following ALA treatment (Figs. 2,3). Aging appears to exacerbate the reduction in autophagy in SM of A1+/−s, as young-A1+/−s (~ 10 weeks-old) did not show a significant decrease in LC3-II protein expression in SM (Fig. 2e). Chloroquine (CQ)-treated A1KD-C2C12s demonstrated lower autophagy levels, even when cultured even in high-glucose medium (Figs. 3a–d). Additionally, decreased autophagy was also observed in succinylacetone (SA)-treated C2C12 myocytes cultured in low-glucose medium (Fig. 3e). These results indicate that heme deficiency, rather than a deficit in ALA, is primarily responsible for the suppression of autophagic processes in SM and myocytes.
We previously reported that aged-A1+/−s exhibited myopathy accompanied by mitochondrial abnormalities in SM, which improved following ALA administration11. Several studies have suggested that age-dependent decreases in autophagy contribute to sarcopenia16. Indeed, we observed that senile-A1+/−s showed an accelerated sarcopenic phenotype (Fig. 1). Amanta et al. have argued that symptoms induced by heme deficiency are similar to those observed in aging32. Given our previous findings of substantial decreases in free heme, which is weekly protein-bound cellular signaling heme, in SM of aged-A1+/−s12, it is plausible that age-related heme deficiency may significantly contribute to the development of sarcopenia-like defects. Therefore, the administration of ALA could represent a viable pharmacological strategy to mitigate the progression of sarcopenia-like defects.
Heme, a small molecule, plays a pivotal role in cellular bioenergetics as a prosthetic group of several subunits within mitochondrial ETC2. Consequently, heme deficiency could lead to decreased energy production. Supporting this, our previous study revealed that aged-A1+/−s experienced glucose intolerance and insulin resistance, which is attributable to reduced insulin-stimulated glucose uptake in SM11. This finding aligns with the notion that heme deficiency can inhibit cellular processes that promote energy production. Furthermore, autophagy itself enhances cellular energy production through its catabolic activities. Therefore, it is plausible that heme deficiency in SM could lead to reduced autophagy, particularly from the perspective of cellular bioenergetics. Ikeda et al. have reported that heme enhances autophagy by upregulating GATA-1 in erythroid cells33. However, this mechanism is unlikely to play a significant role in the reduced autophagy observed in SM under heme-depleted conditions, given the scant expression of GATA-1 in SM.
The ratio of LC3-II protein to LC3-I protein is used as an index for evaluating autophagic activity30. However, we observed decreases in the protein levels of total LC3 and LC3-II in SM of aged-A1+/−s and A1KD-C2C12s (Figs. 2,3). The LC3-II/LC3-I ratios were similar between aged-WTs and A1+/−s, as well as CT- and A1KD-C2C12s (data not shown). This suggests that heme deficiency may not significantly affect the conversion of LC3-I to LC3-II. However, the use of CQ, an inhibitor of the fusion of autophagosome to lysosome, appeared to reduce the inhibition of autophagy in A1KD-C2C12s (Figs. 3c,d). Hence, it’s likely that heme deficiency affects both autophagosome-lysosome fusion and LC3 protein and mRNA expression (Fig. 2b).
The amounts and ratio of p-AMPK and p-eIF2α positively correlate with autophagy activation18,20,, while those of p-70S6K correlate negatively19. The protein expression levels of AMPK, p70S6K, and eIF2α in SM of aged-A1+/−s under fasting conditions (Figs. 4a,b) correlate with the observed reduction in autophagic activity. However, this diminished autophagy in aged-A1+/−s appears more likely due to increased mTORC1 signaling, as indicated by the increase in phosphorylated p70S6K (Fig. 4a). This is potentially mediated by reduced phosphorylation of mTORC1’s regulatory subunit Raptor by AMPK, rather than being directly influenced by nutritional status. This theory is bolstered by the fact that aged-A1+/−s were subjected to fasting conditions, wherein nutritional suppression of autophagy would be less probable. Moreover, the protein expression profiles of AMPK and eIF2α in A1KD-C2C12s cultured in low-glucose medium mirrored those in SM of aged-A1+/−s under fasting conditions (Fig. 5). A comparative analysis between fed and fasting conditions (Fig. 6) implies that the reduced autophagy in aged-A1+/−s results from decreased AMPK signaling rather than eIF2α signaling. This inference is drawn from the decrease in Map1lc3a mRNA levels, encoding for LC3 protein, in SM of aged-A1+/−s under fed conditions (Fig. 2b), coupled with diminished autophagy in A1KD-C2C12s cultured in high-glucose medium (Fig. 3b).
Additionally, the effects of AICAR (an AMPK activator), Sarubrinal (a phosphatase inhibitor increasing phosphorylated eIF2α), or dorsomorphin (an AMPK inhibitor) on CT- and A1KD-C2C12s (Fig. 7) align with the notion that decreased AMPK signaling is a crucial contributor to reduced autophagy under heme deficiency. Our findings showed that AICAR treatment enhances autophagic activities in A1KD-C2C12s, even in heme depletion (Fig. 7a). Conversely, dorsomorphin treatment inhibits autophagy in CT-C2C12s and ALA-treated A1KD-C2C1s, irrespective of heme repletion (Fig. 7b). These results suggest that heme deficiency is not necessary for decreased autophagic activity. Instead, they indicate that heme depletion may reduce AMPK signaling, resulting in diminished autophagy.
Contrary to expectations, our findings suggest that heme deficiency leads to a decrease in AMPK signaling. This outcome appears paradoxical since heme deficiency, potentially leading to ATP shortage in the mitochondrial ETC, would logically be expected to upregulate AMPK signaling. AMPK is activated by increasing the AMP/ATP ratio and ADP/ATP ratio during exercise or fasting18. Additionally, enzymes like Ca2+/calmodulin-dependent protein kinase kinase-β (CaMKK) -involved in intracellular calcium signaling phosphorylation cascade—and TAK1, which is activated by factors such as TGF-β, IL-1, and TNF-α, have been reported to phosphorylate and activate AMPK directly34. Our following study aims to analyze the cause of decreased AMPK activity and to elucidate the potential impact of heme deficiency on SM pathology. The precise mechanisms underlying the reduction of AMPK signaling due to heme deficiency in SM are yet to be fully understood and will be the focus of our future research. Interestingly, only under fasting conditions did eIF2α signaling seem relevant to decreased autophagy in heme-deficient SM (Fig. 4b). However, this signaling pathway is not essential to autophagy reduction, as it appears to increase autophagy under fed conditions (Fig. 6). Nonetheless, the perturbation of eIF2α signaling in heme-deficient SM is noteworthy. The heme-regulated inhibitor, a member of the eIF2α kinase family, is unlikely to be responsible for the observed eIF2α signaling perturbations due to differing responses under fed and fasting conditions (Fig. 6). Therefore, GCN2, which senses amino acid availability35, could be a candidate for mediating these eIF2α signaling perturbations.
The correlation between autophagy dysfunction and SM disorders has been previously suggested14,36. It was initially hypothesized that inhibiting autophagy might mitigate sarcopenia, considering that increased autophagy could trigger muscle fiber atrophy36. However, recent studies have revised this view, showing that autophagy plays a critical role in maintaining muscle mass and that its age-related dysfunction leads to maladaptive stress responses, resulting in muscle weakness and atrophy14,36. In our previous study11, A1+/−s demonstrated an age-dependent decline in physical endurance, as assessed through treadmill tests, which was more pronounced than changes in body weight. In this study, senile-A1+/−s (75–90 weeks-old) exhibited reductions in body weight, SM mass, and muscle strength (Fig. 1). These findings suggest an increased susceptibility of A1+/−s to sarcopenia-like phenotypes compared to wild-type controls. Hence, heme deficiency could be considered a potential causative or exacerbating factor in sarcopenia-like defects. Additionally, 5-aminolevulinic acid (ALA) supplementation has been shown to ameliorate sarcopenia-like defects in humans37. We have demonstrated that ALA administration increases heme levels in SM12, and one of the mechanisms through which ALA may exert its therapeutic effect could be by enhancing autophagy via increased heme levels in SM.
In this study, our analysis of Alas1 knockout heterozygous mice and Alas1 knockdown C2C12 myocytes revealed that heme deficiency accelerates sarcopenia-like defects and reduces autophagy within skeletal muscle. We identified a decrease in AMPK signaling as the primary cause of this autophagic dysfunction. Furthermore, our findings suggest that 5-aminolevulinic acid (ALA) may enhance autophagy by improving AMPK signaling. Given the challenges of an aging society, ALA administration could potentially be a viable treatment approach for mitigating age-related changes such as sarcopenia.
Matrials and methods
Mice
As previously described, A1+/−s were generated on a mixed background, 129 Sv/C57BL/67. These animals were maintained through crossbreeding with BDF1 (F1 hybrid of C57BL/6 and DBA2), using male A1+/−s and wild-type littermates (WTs) for experiments. Mice were kept under a 14 h-10 h light–dark cycle and allowed access to a regular chow diet and water ad libitum. Three groups of mice were studied: WTs group, A1+/−s group, and A1+/−s treated with ALA (ALA-adm-A1+/−s). 5-Aminolevulinic acid hydrochloride (ALA), procured from KYAN PHARMA, was administered orally at a 2 mg/ml concentration by allowing the animals to drink freely11. Mice typically drink 6 ml of water daily, amounting to 12 mg of ALA daily, 40 mg/kg in a 30 g mouse. Mice treated with ALA for more than two weeks were used for the analysis. Since it has been reported that sarcopenia is more strongly induced in fast-twitch muscle fibers38, we used the quadriceps or gastrocnemius muscles, predominantly fast-twitch muscles39, for the analysis. All animal studies are conducted following the "The Regulation of Animal Experiments in Yamagata University " and are approved by the Institutional Animal Care and Use Committee of Yamagata University (Approval No. R2-131). Mice were deeply anesthetized via isoflurane inhalation in a glass desiccator and euthanized by cervical dislocation. This study is reported in accordance with ARRIVE guidelines (https://arriveguidelines.org).
Phenotyping
Mice aged 75–90 weeks were analyzed for sarcopenic phenotypes. Previous studies have shown that mice aged at least 18 months (75 weeks) naturally exhibit sarcopenia24,25. Therefore, 75–90 weeks-old mice are suitable for assessing accelerated sarcopenic defects. Hanging wire tests and grip strength tests were performed at 15, and 26–35, and 75–90 weeks of age to access muscle strength during aging. For the hanging wire test, we used the holding impulse (HI) calculated as body weight [g] x hanging time [sec], following previous reports40,41. This index accounts for the effect of body weight on the test. For the hanging wire test method, each mouse was placed by a wire mesh of about 2 mm vertically on the ground, and the time until it fell was examined. The wire mesh was about 30 cm to 45 cm above the ground, and a cage lined with wood shavings was placed underneath to ensure safety. The test was conducted three times a day, and the average value was measured as a result for that day. The recovery time between each time was measured as 30 s, and the cutoff for the maximum hanging time was 180 s. After two days of practice, the test was conducted for three days, and the average value was measured. The gastrocnemius muscle weight of each mouse was measured at 90–100 weeks of age. For the grip strength tests, we performed basically according to the method described in ref42. Briefly, the test subject, a mouse, was placed on the exposed section of the cage lid and elevated vertically by grasping the tail. The negative force generated by the animal as it resisted removal from the grip platform (the cage lid) was measured. For each trial, three consecutive measurements were recorded per mouse.
Gene and protein expression analysis
Quantitative RT-PCR and Western blot analyses were performed as previously described12. The quadriceps muscle of 25–35 weeks of age was used for the study. Autophagy was induced in mice through a 24-h fasting period, during which they had free access to drinking water, prior to sample collection. Each reaction in Q-PCR was performed in duplicate using the specific primers below. Map1lc3a (Microtubule Associated Protein 1 Light Chain 3 Alpha): Forward Primer 5’-TACATGGTCTACGCCTCCCA-3’; Reverse Primer 5’-GCCTAATCCACTGGGGACTG-3’, Ulk1 (unc-51 like kinase 1): Forward Primer 5’-CTCACCTAAGCTGCCTGACT-3’; Reverse Primer 5’-TAGCCAACAGGGTCAGCAAA-3’. Primary antibody reactions were performed using antibodies against the following proteins: AMPKα, p-AMPKα (Thr172), Light Chain 3B (LC3), α-tubulin, p-p70S6K (Thr389) (Cell Signaling Technology), and p70S6K, ATF4 (Santa Cruz), and eIF2α, p-eIF2α (Ser51) (Abcam). p62, Nrf2 (MBL). All experiments were conducted with biological replicates, with each biological replicate comprising at least three technical replicates. The data presented are representative of results obtained from multiple independent experiments. The original blot images for Figs. 2b–e, 3a–e, 4a–b,d–e, 5, 6, and 7a–b are provided in the Supplementary figures.
Cell culture
The Alas1 knockdown C2C12 cells (A1KD-C2C12s) and control C2C12 cells (CT-C2C12s) were established as previously described11. C2C12 cells were maintained in high-glucose (4500 mg/L) DMEM with 10% fetal bovine serum at 37 °C with 5% CO2 and induced to differentiate into myocytes by culturing in DMEM with 2% horse serum for four days11. For autophagy induction under low-glucose conditions, C2C12 cells were initially cultured in high-glucose DMEM with 2% horse serum for three days, followed by culturing in low-glucose (500 mg/L) DMEM with 2% horse serum for an additional day. Chloroquine diphosphate (Tokyo Chemical Industry), succinylacetone (Sigma-Aldrich), AICAR (FUJIFILM-Wako), and salubrinal (Cayman Chemical) were treated for one day on the final day of myocyte differentiation induction. The final concentration of each drug was 50 μM for CQ, 10 μM for SA, 2 mM for AICAR, and 15 μM for SB.
Statistical analysis
When equal variances were assumed in the F-test, the Student’s t-test was used. When equal variances were not assumed, Welch’s t-test was employed for analysis except data on hanging wire tests and grip strength tests. For data on hanging wire tests and grip strength tests, one-way ANOVA with Bonferroni post hoc test was used to evaluate differences in age between subjects from different clusters. EZR was used for the statistical analysis of this study.
Data availability
All data supporting the findings of this study are available within the paper.
References
Ilka, U. H., Martina, J. & Dieter, J. The biochemistry of heme biosynthesis. Arch. Biochem. Biophys.474, 238–251 (2008).
Kim, H. J., Khalimonchuk, O., Smith, P. M. & Winge, D. R. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim. Biophys. Acta Mol. Cell Res.1823, 1604–1616 (2012).
Igarashi, K. & Watanabe-Matsui, M. Wearing red for signaling: the heme-bach axis in heme metabolism, oxidative stress response and iron immunology. Tohoku J. Exp. Med.232, 229–253 (2014).
Liao, R. & Bresnick, E. H. Heme as a differentiation-regulatory transcriptional cofactor. Int. J. Hematol.116, 174–218 (2022).
Higashikawa, F., Noda, M., Awaya, T., Tanaka, T. & Sugiyama, M. 5-Aminolevulinic acid, a precursor of heme, reduces both fasting and postprandial glucose levels in mildly hyperglycemic subjects. Nutritionhttps://doi.org/10.1016/j.nut.2013.02.008 (2013).
Yomogida, K., Yamamoto, M., Yamagami, T., Fujita, H. & Hayashi, N. Structure and expression of the gene encoding rat nonspecific form δ-aminolevulinate synthase. J. Biochem.113, 364–371 (1993).
Okano, S. et al. Indispensable function for embryogenesis, expression and regulation of the nonspecific form of the 5-aminolevulinate synthase gene in mouse. Genes Cells15, 77–89 (2010).
Nakajima, O. et al. Heme deficiency in erythroid lineage causes differentiation arrest and cytoplasmic iron overload. EMBO J.18, 6282–6289 (1999).
Nakajima, O. et al. Transgenic rescue of erythroid 5-aminolevulinate synthase-deficient mice results in the formation of ring sideroblasts and siderocytes. Genes Cells11, 685–700 (2006).
Yamamoto, M. & Nakajima, O. Animal models for X-linked sideroblastic anemia. Int. J. Hematol.72, 157–164 (2000).
Saitoh, S. et al. 5-aminolevulinic acid (ALA) deficiency causes impaired glucose tolerance and insulin resistance coincident with an attenuation of mitochondrial function in aged mice. PLoS One13, 1–20 (2018).
Koen, V. W. et al. Heterozygous disruption of ALAS1 in mice causes an accelerated age-dependent reduction in free heme, but not total heme, in skeletal muscle and liver. Arch. Biochem. Biophys.697, 1–7 (2021).
Mizushima, N. & Komatsu, M. Autophagy: Renovation of cells and tissues. Cell147, 728–741 (2011).
Masiero, E. et al. Autophagy is required to maintain muscle mass. Cell Metab.10, 507–515 (2009).
Sayer, A. A. & Cruz-Jentoft, A. Sarcopenia definition, diagnosis and treatment: consensus is growing. Age Ageinghttps://doi.org/10.1093/ageing/afac220 (2022).
Fan, J. et al. Autophagy as a Potential Target for Sarcopenia. J. Cell. Physiol.231, 1450–1459 (2016).
Nichenko, A. S. et al. Lifelong Ulk1-mediated autophagy deficiency in muscle induces mitochondrial dysfunction and contractile weakness. Int. J. Mol. Sci.22, 1937 (2020).
Saikia, R. & Joseph, J. AMPK: A key regulator of energy stress and calcium-induced autophagy. J. Mol. Med.99, 1539–1551 (2021).
Kim, J., Kundu, M., Viollet, B. & Guan, K. L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol.13, 132–141 (2011).
Pike, L. R. G. et al. Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem. J.449, 389–400 (2013).
Ichimura, Y. et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell.51, 618–631 (2013).
Hye, S. J. & Myung, S. L. Role of autophagy in diabetes and mitochondria. Ann. N. Y. Acad. Sci.1201, 79–83 (2010).
Sebastián, D. et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J.35, 1677–1693 (2016).
Kim, C. & Hwang, J. K. The 5,7-dimethoxyflavone suppresses sarcopenia by regulating protein turnover and mitochondria biogenesis-related pathways. Nutrients12, 1079 (2020).
Xie, W. Q. et al. Mouse models of sarcopenia: Classification and evaluation. J. Cachexia Sarcopenia Muscle.12, 538–554 (2021).
Lexell, J. Human aging, muscle mass, and fiber type composition. J. Gerontol. A Biol. Sci. Med. Sci.50, 11–16 (1995).
Nilwik, R. et al. The decline in skeletal muscle mass with aging is mainly attributed to a reduction in type II muscle fiber size. Exp. Gerontol.48, 492–498 (2013).
Zhang, B. et al. Histochemical characteristics of soleus muscle in angiotensin-converting enzyme gene knockout mice. Hypertens. Res.28, 681–688 (2005).
Kabeya, Y. et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J.19, 5720–5728 (2000).
Mizushima, N. & Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy3, 542–545 (2007).
Fujii, C. et al. Treatment of sarcopenia and glucose intolerance through mitochondrial activation by 5-aminolevulinic acid. Sci. Rep.7, 4013 (2017).
Atamna, H. Heme, iron, and the mitochondrial decay of ageing. Ageing Res. Rev.3, 303–318 (2004).
Ikeda, M. et al. Heme-dependent induction of mitophagy program during differentiation of murine erythroid cells. Exp. Hematol.118, 21–30 (2023).
Jens, F. H., Stine, R., Maja, M. N., Peter, O. & Henriette, P. PGC-1α promotes exercise-induced autophagy in mouse skeletal muscle. Physiol. Rep.4, e12698 (2016).
Maurin, A. C. et al. GCN2 upregulates autophagy in response to short-term deprivation of a single essential amino acid. Autophagy Rep.1, 119–142 (2022).
Jiao, J. & Demontis, F. Skeletal muscle autophagy and its role in sarcopenia and organismal aging. Curr. Opin. Pharmacol.34, 1–6 (2017).
Tamura, Y. et al. Efficacy and safety of 5-aminolevulinic acid combined with iron on skeletal muscle mass index and physical performance of patients with sarcopenia: A multicenter, double-blinded, randomized-controlled Trial (ALADDIN Study). Nutrientshttps://doi.org/10.3390/nu15132866 (2023).
Lexell, J., Taylor, C. C. & Sjöström, M. What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J. Neurol. Sci.84, 275–294 (1988).
Kuznetsov, A. V. et al. Striking differences between the kinetics of regulation of respiration by ADP in slow-twitch and fast-twitch muscles in vivo. Eur. J. Biochem.241, 909–915 (1996).
Hosaka, Y. et al. Alpha1-syntrophin-deficient skeletal muscle exhibits hypertrophy and aberrant formation of neuromuscular junctions during regeneration. J. Cell Biol.158, 1097–1107 (2002).
Zhou, H. et al. Myostatin inhibition in combination with antisense oligonucleotide therapy improves outcomes in spinal muscular atrophy. J. Cachexia Sarcopenia Muscle.11, 768–782 (2020).
Homolak, J. et al. A hacked kitchen scale-based system for quantification of grip strength in rodents. Comput. Biol. Med.144, 105391 (2022).
Acknowledgements
We thank Dr A. Tanaka, Yamagata University School of Medicine, for his technical advice. Mrs. S. Takanashi, A. Kikawa, and K. Kato for experimental support.
Funding
This study was supported by JSPS KAKENHI (Grant-in-Aid for Scientific Research(C)) Grant Number 18K06949 to O.N. and was a cooperative research effort with SBI Pharmaceuticals and KIYAN PHARM, who provided financial support for the work.
Author information
Authors and Affiliations
Contributions
Study concept and design: O.N.; Investigation: T.A, H.S., K.W., S.S., T.K., and S.O.; Data curation: K.K., K.W., M.N., T.T., M.T., and O.N.; Funding acquisition: M.N., T.T., and O.N.; Resources: K.K., K.W., M.N., T.T., M.T., and O.N.; Writing the manuscript: T.A. and O.N.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Akabane, T., Sagae, H., van Wijk, K. et al. Heme deficiency in skeletal muscle exacerbates sarcopenia and impairs autophagy by reducing AMPK signaling. Sci Rep 14, 22147 (2024). https://doi.org/10.1038/s41598-024-73049-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-73049-9
- Springer Nature Limited