
Abstract
Patients with advanced idiopathic pulmonary fibrosis (IPF), a complex and incurable lung disease with an elusive pathology, are nearly exclusive candidates for lung transplantation. Improved identification of patient subtypes can enhance early diagnosis and intervention, ultimately leading to better prognostic outcomes for patients. The goal of this study is to identify new immune patterns and biomarkers in patients. Immune subtypes in IPF patients were identified using single-sample gene set enrichment analysis, and immune subtype-related genes were explored using the weighted correlation network analysis algorithm. A machine learning integration framework was used to establish the optimal prognostic model, known as the immune-related risk score (IRS). Single-cell sequencing was conducted to investigate the major role of macrophage-derived PLA2G7 in the immune microenvironment. We assessed the stability of celecoxib in targeting PLA2G7 through molecular docking and surface plasmon resonance. IPF patients present two distinct immune subtypes, one characterized by immune activation and inflammation, and the other by immune suppression. IRS can predict the immune status and prognosis of IPF patients. Furthermore, multi-cohort analysis and single-cell sequencing analysis demonstrated the diagnostic and prognostic value of PLA2G7 derived from macrophages and its role in shaping the inflammatory immune microenvironment in IPF patients. Celecoxib could effectively and stably bind with PLA2G7. PLA2G7, as identified through IRS, demonstrates marked stability in diagnosing and predicting the prognosis of IPF patients as well as predicting their immune status. It can serve as a novel biomarker for IPF patients.
Similar content being viewed by others


Introduction
Idiopathic pulmonary fibrosis (IPF) is a complex and incurable pulmonary disease with an elusive etiology1. Its prevalence varies, affecting 20–80 individuals per 100,000, with a notable predominance in males with a history of cigarette smoking2. A hallmark feature of IPF is the progressive interstitial fibrosis in lung parenchyma, resulting in a gradual decline in respiratory function. This decline often culminates in irreversible conditions and, ultimately, respiratory system failure and mortality3,4,5. Unfortunately, the treatment options for IPF remain highly limited, with only pirfenidone6 and nintedanib7 currently approved for IPF treatment. Late-stage IPF patients face limited options, typically lung transplantation or palliative care. Thus, early intervention and diagnosis are crucial for enhancing patient survival. Furthermore, the significant heterogeneity among IPF patients underscores the need to identify distinct patient subtypes, each with varying risk profiles, to enable more precise therapeutic approaches and management8.
The pathogenesis of IPF is exceedingly intricate, involving a complex interplay of various environmental risk factors9 and genetic elements10,11. These factors and genetic variations collectively trigger epithelial cell damage and apoptosis, leading to the massive recruitment of immune cells to the affected areas12. Subsequently, these activated immune cells stimulate fibroblasts to secrete extracellular matrix, thereby initiating the process of lung tissue repair and regeneration. This pathological process encompasses multiple domains, including genetics, environment, and cellular biology, all of which are crucial for a profound understanding of IPF pathophysiology. Although the exact etiology of IPF remains unclear, numerous studies emphasize the pivotal role of the patient’s immune system, particularly immune characteristics associated with recurrent inflammation and immune system dysregulation13,14. Nevertheless, our understanding of the precise roles and immune characteristics of the immune system in IPF patients remains limited.
In this context, machine learning has emerged as a powerful tool to assist in identifying patient characteristics and biomarkers15. However, traditional single-machine learning approaches may have limitations. Hence, integrated frameworks utilizing multiple machine learning methods have been widely applied in lung cancer16, gastric cancer17, pancreatic cancer18, and sepsis19. In our study, we employed integrated machine learning approach, initially conducting parallel analyses across multiple cohorts to reveal two stable immune subtypes present in IPF patients. One subtype displayed significant immune cell infiltration and inflammation, whereas the other exhibited lower immune cell infiltration and immune-suppressive features. Building upon these immune subtypes, we developed a robust immune-related risk score (IRS) and successfully identified a new biomarker, PLA2G7. Furthermore, through drug sensitivity analysis and molecular docking techniques, we identified the potential of celecoxib to target PLA2G7. This discovery opens up the prospect of inhibiting inflammation and pain, presenting a novel avenue for IPF treatment. Our study provides robust support for a deeper understanding of IPF pathogenesis and the development of therapeutic approaches.
Materials and methods
Bulk data download and processing
We obtained the GSE7086620, GSE11014721, and GSE1066722 datasets from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). Each dataset was matched with its corresponding sequencing platform (GSE70866: GPL17077 and GPL14550, GSE110147: GPL6244, GSE10667: GPL4133). The GSE70866 dataset included lung lavage fluid samples from 20 healthy individuals and 176 IPF patients, with recorded survival time and status. The GSE110147 dataset included lung tissue samples from 11 healthy individuals and 22 IPF patients. The GSE10667 dataset comprised lung tissue samples from 15 healthy individuals and 31 IPF patients. The available clinical characteristics of all patients can be seen in Table S1. Subsequently, the data underwent a correction in R (version 4.3.1) using the “normalizeBetweenArrays” function from the “limma” package23.
Single-sample gene set enrichment analysis (ssGSEA)
We used the ssGSEA algorithm24 to investigate the enrichment of immune-related gene sets within the IPF samples. ssGSEA is a computational method utilized for the analysis of gene set enrichment in individual samples, providing insights into the activity levels of immune-related genes across different samples. Immune-related gene sets were derived from research conducted by the Alexander Bagaev team25. We used the ‘gsva()’ function from the “GSVA” package in R to perform ssGSEA. Here, the method parameters were set to “ssgsea” and Gaussian kernel density estimation (kcdf = "Gaussian”) was employed for enrichment analysis. Additionally, we retained the absolute ranking information (abs.ranking = TRUE). To normalize the ssGSEA results, the ssGSEA score values were transformed into a range of 0–1, facilitating improved visualization and comparison.
Unsupervised clustering
To elucidate the patterns of immune cell infiltration in patients, the was used for sample clustering. Clustering was based on the immune cell enrichment scores ConsensusClusterPlus algorithm26 obtained through ssGSEA. Specifically, we used the “ConsensusClusterPlus” package in R to conduct sample clustering analysis. We set the maximum number of clusters (K value) to 9, with intervals of 1, to assess the clustering outcomes under different K values. To ensure robust clustering results, we repeated the clustering analysis 1000 times. pltem serves as a feature retention threshold, which determines the number of features (genes) retained for clustering analysis in each bootstrap iteration. In our parameter settings, pltem was set to 0.8, indicating that 80% of features would be retained in each bootstrap iteration. The parameter pFeature represents the probability of feature retention, determining whether each feature is retained in each bootstrap iteration. In this context, pFeature was set to 1, indicating that every feature would be retained and no feature selection would occur. This setting is suitable for scenarios where all features are desired to be considered or when dealing with a relatively small number of features. The clustering process utilized the k-means clustering algorithm (clusterAlg = "km”) and Euclidean distance (distance="Euclidean”). Finally, t-SNE dimensionality reduction analysis was conducted using the “Rtsne” package (perplexity = 10, max_iter = 500). Visualization of the results was performed using the ggplot2 package to assess the presence of algorithmic bias in the ConsensusClusterPlus clustering method.
Survival analysis
We conducted survival analysis using the “survival” and “survminer” packages in R to generate Kaplan–Meier survival curves. Survival differential analysis was performed using the “survdiff” function, comparing survival time and event status between different groups (experimental and control groups). The chi-square statistic was calculated and the significance P-value was obtained through this function. The “survfit” function was used to calculate and plot Kaplan–Meier survival curves, with each curve representing the survival status for different groups and displaying the corresponding significance P-value. Finally, the “ggsurvplot” function was employed to create survival curve plots.
Enrichment analysis
We conducted enrichment analysis of differential genes using the “GSEABase,” “ClusterProfiler”27 and “org.Hs.eg.db” packages. The database used for the enrichment analysis was sourced from the Gene Ontology (GO) database (http://geneontology.org/)28 and Kyoto Encyclopedia of Genes and Genomes (KEGG) database29. The enrichment analysis was conducted using the “EnrichGO” function. Pathways with a P-value < 0.05 were considered significantly enriched. The “ggplot2” and “ggpubr” packages were used for visualization.
Prognostic model construction using machine learning
We employed the following ten machine learning algorithms in R: random survival forest (RSF), elastic network (Enet), Lasso, Ridge, stepwise Cox, CoxBoost, partial least squares regression for Cox (plsRcox), supervised principal components (SuperPC), generalized boosted regression (GBM), and survival support vector machine (survival-SVM). In the process, one algorithm was used to filter the variables, and another algorithm was used to construct the prognostic signature. When the final prognostic signature contained fewer than two genes, it was considered invalid. Subsequently, we calculated Harrell’s concordance index (C-index) for each signature. After calculating the risk score for each patient using the “predict” function, the optimal cutoff value for the risk score was determined using the “surv_cutpoint” function in the “srvminer” package. We distinguished patients into high and low risk groups in the training set based on the best cutoff value. Afterwards, the validation set and the total cohort are grouped based on the best cut-off value in the training set.
Acquisition and pre-processing of single-cell transcriptome data
Single-cell transcriptome data were obtained from the GEO database (GEO registration number: GSE128033; https://www.ncbi.nlm.nih.gov/geo/) and Li Wu et al.(https://ngdc.cncb.ac.cn/gsa/browse/CRA011039)30. Quality control was performed in R using standard single-cell processing procedures. The count matrix was read using the “Read10X” function from the Seurat31 package (version 4.0.4), and it was further converted to the dgCMatrix format. The merge function was used to integrate all individual objects into an aggregate object, and the “RenameCells” function was used to ensure the uniqueness of all cell labels. We filtered out low-quality cells using the following filtering criteria: genes expressed in fewer than three cells were deleted, and cells expressing fewer than 200 genes were removed. A global-scaling normalization method (“LogNormalize”) was used to equalize the total gene expression in each cell, with a scale factor set to 10,000. The top 2000 variably expressed genes were selected for downstream analysis using the “FindVariableFeatures” function. The “ScaleData” function, with the “vars.to.regress” option set to UMI, and percent mitochondrial content were used to eliminate unwanted sources of variation. Principal component analysis (PCA) incorporating highly variable features reduced the dimensionality of this dataset, and the first 30 PCs were selected for analysis. The Harmony method32 was used to remove batch effects between samples. Cells were down-dimensioned using the UMAP method. Clustering analysis was performed based on the edge weights between any two cells. A shared nearest-neighbor graph was generated using the Louvain algorithm, which was implemented in the “FindNeighbors” and “FindClusters” functions. The parameter of resolution in the “FindClusters” function was tested repeatedly between 0.1 and 1. Cell clustering trees at different resolutions were observed using the “clustree” function, and the results indicated that the most distinct clustering outcomes were obtained at a resolution of 0.5. The scrublet al.gorithm was used to remove potential doublets.To annotate the cell clusters, we identified differentially expressed markers within the resulting clusters using the “FindAllMarkers” function. This was accomplished by using the default nonparametric Wilcoxon rank sum test with Bonferroni correction. All cells were annotated based on cell surface markers and annotated genes from the relevant literature and the cell taxonomy database (https://ngdc.cncb.ac.cn/celltaxonomy/).
Single-cell differential analysis
To mitigate the bias introduced by the sparsity of single-cell sequencing data in differential expression analysis, we used the advanced Libra package for conducting differential expression analysis. Libra33 is an R package designed for the analysis of single-cell RNA sequencing (scRNA-seq) data, providing extensive functionality for differential expression analysis, cell subpopulation analysis, and other tasks related to single-cell data analysis (https://github.com/neurorestore/Libra).
Molecular docking
In molecular docking, our objective was to predict the binding mode and affinity of a ligand molecule with a target protein (PLA2G7). First, the 3D structure of molecular was obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) and optimized using the MM2 force field in chemBio3D. The optimized structure was saved in PDB format. The protein structure information was obtained from the AlphaFold Protein Structure database (https://alphafold.ebi.ac.uk/entry/Q13093) and saved in PDB format. Molecular docking was then conducted using the AutoDock Vina program, which predicts the binding conformation of celecoxib to the target protein based on the optimization of a scoring function. This scoring function estimates the binding energy of the protein–ligand complex, which is a measure of the affinity of the ligand to the protein. After the docking was completed, the results were visualized using PyMOL software, which allowed us to observe the binding pose of the ligand in the active site of the protein.
Establishment of bleomycin-challenged mouse model
The experimental model was established using 8-week-old wild-type C57BL/6 male mice, weighing between 20 and 25 g, obtained from Zhuhai Bestway Biotechnology Company. All procedures were ethically reviewed and approved by the Institutional Animal Care and Use Committee of Zhongshan Institute for Drug Discovery. Intratracheal injections were administered using bleomycin (MCE, Cat No. HY-17565) at a dosage of 5 mg/kg, while the control group received physiological saline. Processing time is 21 days34,35,36.
Tissue processing, staining
Mice are anesthetized with isoflurane and then subjected to cervical dislocation sacrifice. The chest cavity was opened to expose the lungs. Perfusion through the heart with PBS (BBI, Cat No. B640011-0010) ensured thorough lung rinsing to remove blood. Instillation of 4% PFA (Absin, Cat No. abs9179) into the lungs via the trachea fixed the tissue. Residual thymus, muscles, and extraneous tissues were removed. Lung samples were placed in pre-chilled 4% PFA for fixation. Subsequently, lung tissues were fixed in 4% PFA for 1 h, washed in PBS for 30 min (four times), immersed overnight in a solution comprising 30% sucrose (Absin, Cat No. abs42027828) followed by a 1:1 mixture of 30% sucrose and OCT (Sakura, Cat No. 4583). This process occurred on a shaker in a 4 °C refrigerator. The tissues were then embedded in OCT, frozen at -80 °C, and sectioned at 8 μm thickness using a Leica cryostat. Subsequent steps involved re-fixing the sections in 4% PFA for 10 min, washing them in PBS for 10 min, and finally staining them using an H&E staining kit (Absin, Cat No. abs9217). Following the previous steps, sections underwent 1-hour re-warming, 1-hour fixation in 4% PFA, 1-hour Bouin’s solution (Labcoms, Cat No. LA0976) treatment at 60 °C, and Masson’s Trichrome staining using a respective staining kit (Solarbio, Cat No. G1340).
Immunofluorescence and enzyme-linked immunosorbent assay (ELISA)
Mice with bleomycin and saline were euthanised on day 21. Lung tissues were processed similarly, fixed in 4% PFA for 10 min, permeabilized in 0.2% TritonX-100 (Macklin, Cat No. 9002-93-1) in PBS for 10 min, blocked with 5% goat serum for 1 h, and incubated with CD68 (Thermo Fisher, Cat No. 14-0681-82) at 1:200 and Pla2g7 antibodies (Proteintech, Cat No. 15526-1-AP) at 1:100 overnight at 4 °C. Subsequent steps involved incubation with secondary antibodies, DAPI staining, and mounting using Fluoromount-G™. Pla2g7 levels were quantified using the mouse Lp-PLA2 enzyme immunoassay kit (MEIMIAN, Cat No. MM-46736M1) per the manufacturer’s instructions.
Surface plasmon resonance analysis
Surface plasmon resonance (SPR) analysis was conducted using the Biacore T200 system (Cytiva) by ZIDD (Guangdong, China). The SPR technique can be used to investigate and to monitor molecular interactions in real time37.The equilibrium-binding constant (KD) for the interaction between the celecoxib compound (Selleck, Cat No. S1261) and human PLA2G7 protein (MCE, Cat No. HY-P70993) was determined at 25 °C with a flow rate of 30 µl/min. PLA2G7 protein, serving as the ligand, was immobilized onto Series S CM5 sensor chips (Cytiva, Cat No. BR100530) using an amine-coupling chemistry kit (Cytiva, Cat No. BR100050) to achieve an immobilization level of 10,000 response units (RU). Celecoxib, the analyte, was flowed over the chip surface in pH 7.2–7.4 buffer (1X PBS-P+, comprising 20 mM phosphate buffer, 2.7 mM KCl, 137 mM NaCl, 0.05% Tween 20, and 5% DMSO). Celecoxib was diluted to various concentrations (0.19 µM, 0.39 µM, 0.78 µM, 1.56 µM, 3.13 µM, 6.25 µM) using the running buffer and sequentially injected onto the chip from the lowest to the highest concentration. For each cycle, a 200 µl sample at the specified concentrations was prepared by dilution into the running buffer, with a contact time of 60 s and a dissociation time of 120 s. Data were analyzed using the Biacore Insight Evaluation Software Version 3.0.12, applying the steady-state affinity 1:1 binding model.
Statistical analysis
All statistical analyses were performed using R, with the corresponding statistical methods set up in the R software. Image analysis for immunofluorescence utilized OLYMPUS FV3000 2.6.1.243 software, while H&E and Masson’s Trichrome staining analysis employed OLYMPUS VS200 software. Cell counting and statistics were performed using Image J software. Statistical significance was set at P < 0.05. Significance levels were denoted as follows: *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
Association between abundant immune cell infiltration, key gene modules, and adverse prognosis in IPF patients: unsupervised clustering analysis across multiple cohorts
Figure 1 illustrates the key workflows in our study. In total, three bulk transcriptome datasets, GSE7086620, GSE11014721, and GSE1066722 were used. The GSE70866 dataset included lung lavage fluid samples from 20 healthy individuals and 176 IPF patients, with recorded survival time and status. The GSE110147 dataset included lung tissue samples from 11 healthy individuals and 22 IPF patients. The GSE10667 dataset comprised lung tissue samples from 15 healthy individuals and 31 IPF patients. We commenced our investigation by exploring the existence of distinct immune subtypes within IPF patients. To achieve this, we employed an immune cell signature gene set as previously reported38 and calculated immune cell enrichment scores for each patient in the GSE70866 dataset using the ssGSEA method. Subsequently, we conducted an unsupervised clustering analysis26. The results uncovered two distinct subtypes (Fig. S1A,B) designated as Subtypes A and B among all patients in the GSE70866 dataset (Fig. 2A). This differentiation was further validated through t-SNE analysis, demonstrating a substantial separation between Subtypes A and B based on the reduced-dimension distribution of patients (Fig. 2A). Notably, Subtype B patients exhibited an overall higher degree of immune cell enrichment than Subtype A patients (Fig. 2B). We repeated these analyses on the GSE10667 (Fig. 2C,D) and GSE110147 datasets (Fig. 2E,F). The results validated the presence of two stable subtypes among IPF patients based on immune cell enrichment scores. Kaplan–Meier analysis in the GSE70866 dataset revealed that overall survival was significantly reduced for Subtype B patients than for Subtype A (Fig. 2G). This outcome underscores the substantial association between abundant immune cell infiltration and adverse prognosis in IPF patients.

Workflow illustrating the comprehensive process employed in this study.

Unsupervised Clustering Analysis Across Multiple Cohorts Reveals a Strong Association Between Abundant Immune Cell Infiltration Regulated by Key Gene Modules and Adverse Prognosis in IPF Patients. (A) Unsupervised clustering and t-SNE dimensionality reduction analyses were performed on IPF patients from the GSE70866 dataset based on immune cell infiltration scores. (B) Heatmaps displaying immune cell infiltration scores for Subtypes A and B in the GSE70866 dataset. (C) Unsupervised clustering and t-SNE dimensionality reduction analyses were conducted on IPF patients from the GSE10667 dataset based on immune cell infiltration scores. (D) Heatmaps displaying immune cell infiltration scores for Subtypes A and B in the GSE10667 dataset. (E) Unsupervised clustering and t-SNE dimensionality reduction analyses were performed on IPF patients from the GSE110147 dataset based on immune cell infiltration scores. (F) Heatmaps displaying immune cell infiltration scores for Subtypes A and B in the GSE110147 dataset. (G) Survival analysis of patients in Subtypes A and B in the GSE70866 dataset. (H) Soft threshold determination. (I) Identification of gene clustering modules. (J) Correlation analysis between gene modules and phenotypes, with pink modules exhibiting a high correlation with subtypes. (K) Correlation coefficient analysis between gene significance (GS) and module membership (MM). (L) KEGG enrichment analysis. (M) GO enrichment analysis. (N) KEGG enrichment analysis.
To identify genes highly correlated with these two subtypes, we conducted a weighted gene co-expression network analysis (WGCNA) within the GSE70866 dataset. The soft threshold (β) was set to 5 (Fig. 2H), providing an appropriate power value for constructing a co-expression network. In total, 27 gene modules, each represented by a different color, were identified (Fig. 2I). We evaluated the correlation between each module and the clinical traits of patients, including age, gender, and the subtypes clustered based on the ssGSEA method. The pink module exhibited the highest correlation with the subtype (correlation coefficient = 0.71; Fig. 2J). The correlation coefficient between gene significance (GS) and module membership (MM) reached 0.88 (Fig. 2K), indicating the superior construction quality of the pink module, which included 351 genes. To identify hub genes that influence the formation of immune subtypes, we applied filtering criteria (GS > 0.5 and MM > 0.6) to these genes. Ultimately, we obtained a set of 122 hub genes. To further validate the relevance of these 122 hub genes to the immune system, we conducted enrichment analysis from various perspectives. KEGG enrichment analysis (Fig. 2L) revealed significant enrichment of several immune-related pathways, including Th1 and Th2 cell differentiation; cytokine–cytokine receptor interaction; NF-kappa B, JAK-STAT, and TNF signaling pathways; and antigen processing and presentation. GO enrichment analysis (Fig. 2M) yielded consistent results, with pathways like regulation of type II interferon production, leukocyte activation involved in immune response, regulation of leukocyte differentiation, and lymphocyte-mediated immunity exhibiting significant enrichment. Disease Ontology (DO) enrichment analysis (Fig. 2N) highlighted the significant relevance of these genes to various immune system-related disorders, including primary immunodeficiency disease, acquired immunodeficiency syndrome, autoimmune diseases of the endocrine system, human immunodeficiency virus infectious disease, and COVID-19. Collectively, we identified two entirely distinct immune subtypes within IPF patients. Subtype A patients displayed lower levels of immune cell enrichment and longer overall survival, whereas Subtype B patients exhibited higher immune cell enrichment and shorter overall survival. Furthermore, we identified 122 hub genes highly correlated with these two immune subtypes.
Development of an optimal immune-related prognostic model using a multi-machine learning integration framework
Before constructing the prognostic model, we conducted a univariate Cox regression analysis on these 122 genes based on survival time and status, thereby identifying 60 prognosis-related genes (Fig. 3A). All of these genes were risk genes significantly associated with poor patient outcomes. Subsequently, to establish training and validation sets, we randomly divided all patients in the GSE70866 dataset into two groups at a 7:3 ratio. Statistical analyses showed no significant difference between the two groups in terms of gender and age (Table 1). The 60 prognosis-related genes were then subjected to a multi-machine learning integration framework. In the training set, we trained the prognostic model, which was subsequently validated in the validation set. Within this multi-machine learning integration framework, ten different machine learning algorithms were employed, namely RSF, Enet, Lasso, Ridge, stepwise Cox, CoxBoost, plsRcox, SuperPC, GBM, and survival-SVM. Additionally, these machine learning algorithms were subjected to combinatorial analysis. Specifically, the first machine learning algorithm was used for gene selection, and the second algorithm was employed for building the prognostic model. During this process, to enhance the model’s robustness across other cohorts, we considered the prognostic model ineffective if the number of genes included was fewer than two. Consequently, a total of 91 prognostic models were constructed, and C-index was calculated for each signature (Fig. 3B). After ranking the average C-index values calculated from the training and validation sets, we identified the top ten prognostic models (Fig. 3C). Among these models, the StepCox [both] + GBM model combination exhibited the highest C-index value in the validation set, reaching 0.681. This result demonstrated that this model combination displayed superior robustness and generalizability, rendering it the best immune-related risk model. Comprising 20 genes (CSGALNACT1, TNFSF14, PTPN7, RGL4, DPP4, CDK6, GZMB, ABLIM1, PBX4, ADA, HOPX, EIF4E3, ITM2C, GNG2, RHOH, MIAT, P2RY8, CD8A, ADAM19, and HMHA1), this model enabled us to calculate an immune-related risk score (IRS) for each patient. The optimal IRS cutoff value for patient stratification was determined using the “survminer” package. Survival analysis results indicated that patients in the high-IRS group had significantly reduced overall survival compared with those in the low-IRS group in both the training and validation sets (Fig. 3D). Furthermore, given that the GSE70866 dataset was originally derived from the merging of three center cohorts (Freiburg, LEUVEN, and SIENA)20, we conducted a concurrent analysis of the prognostic performance of IRS in different center cohorts. Consistent with previous findings, when data was re-stratified into the Freiburg, LEUVEN, and SIENA cohorts, patients in the high-IRS group continued to exhibit significantly reduced overall survival compared with those in the low-IRS group (Fig. 3D). This outcome further validated the robustness of the IRS. Receiver operating characteristic (ROC) curve analysis revealed that in the training cohort, the area under the curve (AUC) values were 0.933, 0.953, and 0.927 at 1, 2, and 3 years, respectively (Fig. 3E). In the validation cohort, the AUC values were 0.773, 0.677, and 0.596 at 1, 2, and 3 years, respectively (Fig. 3E). Across the Freiburg, LEUVEN, and SIENA cohorts, AUC values remained above 0.8 at 1, 2, and 3 years (Fig. 3E). Collectively, we developed the IRS and demonstrated its robust effectiveness, stability, and reliability.

Development of an optimal immune-related prognostic model using a multi-machine learning integration framework. (A) Univariate cox regression analysis was employed to select prognostically relevant genes based on survival time and status in the GSE70866 dataset. (B) Presentation of 91 prognostic models integrated through machine learning, along with their corresponding C-index values. (C) Showing the top ten models with the highest average C-index values, StepCox [backward] + GBM was selected as the best model in the validation cohort. (D) Survival analysis of high- and low-IRS patients in different cohorts. (E) ROC analysis of high- and low-IRS patients in different cohorts.
Inflammatory activation phenotype and metabolic disruptions in High-IRS patients
We conducted a comprehensive analysis of the relationship between IRS and patient phenotypes and observed a significant positive correlation between IRS and various immune cell enrichment indices. Patients with high IRS exhibited elevated levels of immune cell enrichment, especially in macrophages, neutrophils, mast cells, and regulatory T cells (Fig. 4A). Additionally, a strong positive correlation was noted among several immune cell enrichment indices, suggesting that high-IRS patients might experience acute and intense immune system hyperactivation and immune cell infiltration (Fig. 4B). Moreover, the clustering of samples from the Freiburg, LEUVEN, and SIENA centers in the heatmap revealed uniform distribution patterns as IRS values changed (Fig. 4A). This observation implies the broad applicability of IRS across different centers. Gene set enrichment analysis (GSEA) results indicated that high-IRS patients displayed activation in adaptive immune responses (Fig. 4C,D), inflammatory reactions (Fig. 4E), and white blood cell chemotaxis (Fig. 4F). In terms of metabolic activity (Fig. 4G), IRS showed a significant positive correlation with amino acid metabolism, glycosaminoglycan biosynthesis, and arachidonic acid metabolism. This suggests disruptions in protein synthesis, energy metabolism, extracellular matrix composition or structure, lipid metabolism, and inflammation in high-IRS patients. Conversely, IRS exhibited significant negative correlations with biotin, glyoxylate, dicarboxylate, and butanoate metabolisms.Biotin is a vital coenzyme involved in various enzymatic reactions, and the negative correlation may indicate abnormalities in biotin metabolism in patients with high-IRS scores, potentially affecting metabolism and cellular functions. Glyoxylate and dicarboxylate metabolism, along with butanoate metabolism, are associated with organic acid metabolism, which is crucial for maintaining cellular metabolic balance. The negative correlation suggests metabolic abnormalities in organic acid metabolism among high-IRS patients. Collectively, our results demonstrate that high-IRS patients exhibit intense immune system activation responses and an inflammatory microenvironment. Furthermore, the IRS plays a role in disrupting multiple metabolic pathways.

High-IRS Patients Exhibit an Inflammatory Activation Phenotype and Various Metabolic Disruptions. (A) Heatmap illustrating the relationship between IRS expression and immune cell infiltration in the GSE70866 dataset. (B) Correlation analysis between IRS expression and immune cell infiltration in the GSE70866 dataset. (C–F) GSEA. (G) Correlation analysis between IRS and different metabolic pathways in the GSE70866 dataset.
Identification of a robust prognostic and diagnostic biomarker, PLA2G7, using IRS
To further assess the utility of IRS, we conducted a differential expression analysis between high- and low-IRS patients, identifying differentially expressed genes (|log2FC| > 1, P-value < 0.05; Fig. 5A). Our analysis recapitulated the expression of well-established genes associated with adverse IPF prognoses, such as SPP139–41 and MMP742,43, thus validating the effectiveness of the IRS. To identify novel biomarkers, we focused on upregulated differentially expressed genes. Initially, 128 differentially expressed genes associated with prognosis, all categorized as risk genes, were identified through univariate Cox regression analysis (Fig. 5B). Subsequently, we calculated the AUC values for these genes in distinguishing healthy samples from IPF samples in the GSE70866, GSE10667, and GSE110147 datasets (Fig. 5C). The top three genes with the highest average AUC values were SPP1, MMP7, and PLA2G7. The value of PLA2G7 in IPF was previously unknown. PLA2G7 exhibited AUC values exceeding 0.8 in all three datasets, with an average AUC of 0.889. Furthermore, the expression of PLA2G7 was significantly elevated in IPF patients compared with that in healthy samples in all three datasets (Fig. 5D–F). Survival analysis in the GSE70866 dataset revealed that patients with high PLA2G7 expression experienced a significant reduction in overall survival (Fig. 5G). Additionally, PLA2G7 expression displayed a significant positive correlation with various immune cell enrichment indices (Fig. 5H), such as macrophages, neutrophils, regulatory T cells, and mast cells, consistent with the results of IRS. Collectively, based on the IRS, we successfully identified PLA2G7 as a novel biomarker. PLA2G7 demonstrated high value for both prognosis and diagnosis, making it a novel candidate in the field of biomarker genes.

Identification of a Robust Prognostic and Diagnostic Biomarker, PLA2G7, Using IRS. (A) Identification of differentially expressed genes in high-IRS patients. (B) Univariate cox regression analysis of differentially expressed genes to select prognostically relevant genes. (C) Calculation of ROC diagnostic capabilities for each gene in distinguishing IPF samples from healthy samples in different cohorts. (D) Differential expression of SPP1, MMP7, and PLA2G7 in healthy and IPF samples in the GSE70866 dataset. (E) Differential expression of SPP1, MMP7, and PLA2G7 in healthy and IPF samples in the GSE110147 dataset. (F) Differential expression of SPP1, MMP7, and PLA2G7 in healthy and IPF samples in the GSE10667 dataset. (G) Survival analysis of IPF patients with high and low PLA2G7 expression in the GSE70866 dataset. (H) The association between PLA2G7 gene expression levels and the abundance of immune cells.
Validation of PLA2G7-positive macrophages driving inflammatory activation in IPF through single-cell sequencing analysis
We obtained single-cell sequencing data from the GSE128033 dataset in the GEO database44. This dataset comprised single-cell sequencing data from lung tissue samples of eight healthy individuals and eight IPF patients, fresh bronchoalveolar lavage fluid samples from a healthy individual, and frozen bronchoalveolar lavage fluid samples from another healthy individual. To avoid potential bias introduced by sample freezing, the frozen bronchoalveolar lavage fluid sample was excluded. Following quality control, we analyzed a total of 68,064 cells, including 22,121 cells from healthy individuals and 34,943 cells from IPF patients. Using common cell markers (Fig. 6A), we performed dimensionality reduction, clustering, and annotation of these cells, including macrophages, endothelial, T cells, smooth muscle cell (SMC), dendritic cell (DC), B cells, monocytes, epithelial cells, fibroblasts, and mast cells (Fig. 6B). We found that PLA2G7 expression was primarily localized to macrophages, with a significant increase in its expression in IPF patients (Fig. 6C). Subsequently, we conducted a differential analysis of all cell types between IPF and healthy samples (Fig. 6D). Although we observed varying gene expression profiles in different cell types within IPF, all cells exhibited elevated expression of genes associated with inflammatory phenotypes. For instance, macrophages displayed increased expression of SPP1, while endothelial cells exhibited increased expression of CXCL11 and CXCL10. T cells showed elevated expression of CCL4 and IFNG, monocytes and DCs displayed high expression levels of HLA family genes, and epithelial cells exhibited increased expression of MMP7 and S100A2. The expression of these genes was significantly correlated with inflammatory activation pathways. To further validate our results, we processed single-cell sequencing data from Li Wu et al.30 (https://ngdc.cncb.ac.cn/gsa/browse/CRA011039). In this dataset, the lung Cd45+ immune cells were isolated using fluorescence-activated cell sorting from naïve mice (day 0) and at various stages after bleomycin administration: acute inflammatory (D7), profibrotic (D14), and later fibrotic (D21) phases. After dimensionality reduction and cell annotation (Fig. 6E–G), we isolated macrophages (Fig. 6H) and examined the expression level of Pla2g7 (Fig. 6I). As the treatment time extended, the expression level of Pla2g7 gradually increased, and the number of Pla2g7-positive macrophages increased gradually (Fig. 6J). Additionally, we observed a gradual increase in Spp1 levels within macrophages (Fig. 6I), which is consistent with our previous results, further validating the high reliability of our findings.

Validation of PLA2G7-positive macrophages driving inflammatory activation in IPF through single-cell sequencing analysis. (A) Heatmap depicting marker gene expression for cell annotation in different cell types. (B) t-SNE dimensionality reduction landscape of single-cell sequencing data. (C) Differential expression of PLA2G7 in IPF and healthy samples. (D) Differential analysis of different cell types in IPF and healthy samples. (E) t-SNE dimensionality reduction landscape of single-cell sequencing data samples from mice. (F) t-SNE dimensionality reduction landscape of cell types in the single-cell sequencing data from mice. (G) Heatmap displaying the marker gene expression in different cell types. (H) t-SNE dimensionality reduction landscape after re-extraction of macrophages in the single-cell sequencing data from mice. (I) Bubble plot exhibiting the expression levels of PLA2G7 and SPP1 in different samples. (J) t-SNE plot displaying the expression levels of PLA2G7 in different mouse samples. (K) In human single-cell sequencing data, violin plot depicting PLA2G7 expression in PLA2G7-positive and PLA2G7-negative macrophages. (L) In human single-cell sequencing data, differential analysis of PLA2G7-positive and PLA2G7-negative macrophages. (M) In human single-cell sequencing data, GSEA of PLA2G7-positive and PLA2G7-negative macrophages. (N) H&E, Masson’s trichrome staining and Multiplex immunofluorescence in a mouse model. (O) Counting of Cd68 + Pla2g7 + cells in a mouse model. (P) ELISA for Pla2g7 expression levels in alveolar lavage fluid and lung tissues of the mouse model.
To delve deeper into the impact of PLA2G7 on macrophage function, we extracted macrophages from GSE128033 data for further analysis (Fig. S1C). Increased expression levels of PLA2G7 in macrophages from IPF patients are demonstrated in a violin plot (Fig. S1D). Based on whether macrophages expressed PLA2G7, we classified them into PLA2G7-positive and PLA2G7-negative cells (Figs. 6K, S1E). Subsequent differential analysis (Fig. 6L) revealed that PLA2G7-positive macrophages exhibited high expression of HAMP and CCR5, indicating the involvement of PLA2G7 in inflammation regulation. Elevated expression of KLK4 and TIMP3 in PLA2G7-positive macrophages suggested a correlation with cell proliferation and apoptosis regulation. The high expression of CCR5 and SLAMF7 indicated the involvement of PLA2G7 in promoting immune responses. We further conducted GSEA using the HALLMARK database of pathway gene sets for these two groups of cells (Fig. 6M). The results showed that PLA2G7-positive macrophages exhibited activation in apoptosis, DNA repair, and STAT3-related pathways. We established a mouse model with lung fibrosis by treating them with bleomycin. Using H&E staining and Masson’s trichrome staining, we observed damage in the mouse lung tissues (Fig. 6N, Figure S1F). In comparison to healthy mice, we observed a significant infiltration of macrophages in the lung tissues of bleomycin-challenged mice in this model, along with a notable increase in Pla2g7-positive macrophages (Fig. 6N,O). Furthermore, ELISA analysis of mouse bronchoalveolar lavage fluid and lung tissue samples (Fig. 6P) revealed a significant increase in Pla2g7 concentration compared to healthy samples in bleomycin-challenged mice. These findings strongly support the establishment of the bleomycin-challenged model and suggest a potential role for Pla2g7 in the development of bleomycin-challenged. In summary, through single-cell analysis, we identified a group of PLA2G7-positive macrophages that increased in number in IPF patients and exhibited an inflammatory activation state. This state is significantly associated with the inflammatory characteristics observed in these patients.
Celecoxib exerts anti-inflammatory effects by Targeting PLA2G7
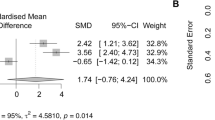
To enhance the clinical relevance of our study, we conducted a small molecule sensitivity analysis using the Enrichr database (https://maayanlab.cloud/Enrichr/). We identified the top five drugs sensitive to PLA2G7, ranked by P-values. Celecoxib emerged as the most sensitive drug (Table 2). As a nonsteroidal anti-inflammatory drug, celecoxib is primarily used to treat pain and inflammation-related conditions, alleviating pain and swelling during the inflammatory process45. Furthermore, we employed computer simulation techniques for molecular docking, considering PLA2G7 as the protein receptor and celecoxib as the small molecule ligand. Our results demonstrated that celecoxib could stably bind within the protein pocket of PLA2G7 (Fig. 7A) with an affinity of − 9.0 kcal/mol (Fig. 7B). It formed hydrogen bonds with the Gly-154 (3.3 Å) and Gln-352 (2.3 Å) residues in the PLA2G7 protein (Fig. 7A). Surface plasmon resonance analysis revealed that celecoxib could bind to PLA2G7 in a dose-dependent manner (Fig. 7C). The KD value of celecoxib binding to PLA2G7 was 2.607 µM, indicating that this binding is very stable. Additionally, in molecular docking, we also analyzed the affinity between PLA2G7 and four other small molecules as well as the PLA2G7-specific inhibitor darapladib. Our results showed that celecoxib had the best affinity with PLA2G7, even better than darapladib. In addition, we note that both celecoxib and darapladib can bind to GLN-352, and thus their binding to PLA2G7 may be competitive with each other. Collectively, our findings substantiate the potential of celecoxib to exert anti-inflammatory effects by targeting PLA2G7.

Celecoxib exerts anti-inflammatory effects by targeting PLA2G7. (A) Schematic representation of the binding mode of molecules with PLA2G7. (B) Affinity between small molecules and PLA2G7. (C) Surface plasmon resonance analysis of celecoxib and PLA2G7. Celecoxib, as the ligand, was dissolved and injected at concentrations of 0 µM, 0.19 µM (purple), 0.39 µM (black), 0.78 µM (blue), 1.56 µM (orange), 3.13 µM (green), and 6.25 µM (red).
Discussion
IPF, a complex and deadly progressive interstitial lung disease affecting millions worldwide46, continues to present treatment challenges despite numerous therapeutic approaches. Early diagnosis and intervention are pivotal for improving patient prognosis. Immune dysregulation is a hallmark of IPF, yet much remains unknown about the immunophenotype of IPF patients. In our study, we initiated unsupervised clustering and parallel analyses across multiple cohorts, identifying two distinct immune phenotypes among IPF patients: an inflammation-activated immune cell infiltration phenotype and an immune-suppressive phenotype. Notably, this subtype distinction was reproduced in both alveolar lavage fluid and lung tissue, demonstrating that this immune signature is widespread in the lung microenvironment and that there is a partial similarity in gene expression alterations between alveolar lavage fluid samples and lung tissue samples. This divergence may account for the longer overall survival observed in subtype A patients compared to the shorter overall survival observed in subtype B patients. This finding holds considerable clinical and biological significance, highlighting that immune system characteristics may offer new insights into patient prognosis.
Subsequently, we identified key gene modules regulating the formation of these two phenotypes through WGCNA. Enrichment analysis reaffirmed the close association of these genes with the immune system. KEGG enrichment analysis highlighted the enrichment of crucial immune pathways, including Th1 and Th2 cell differentiation, cytokine–cytokine receptor interaction, and NF-kappa B, JAK-STAT, and TNF signaling pathways, demonstrating the critical role of these genes in regulating immune responses47,48. GO enrichment analysis results emphasized the involvement of these genes in regulating several biological processes, such as immune cell activation, cell differentiation, and lymphocyte-mediated immune responses. The DO enrichment analysis results demonstrated the strong correlation of these genes with various immunological disorders, including primary immunodeficiency diseases, acquired immunodeficiency syndromes, autoimmune diseases of the endocrine system, human immunodeficiency virus infection, and COVID-19. These findings highlight that these genes not only play a critical role in IPF but may also have an impact on various other immune-related diseases. This discovery suggests that these genes, besides being instrumental in IPF, may have been involved in multiple other immunological disorders, providing valuable clues for exploring common mechanisms underlying immune-related diseases and developing new therapeutic strategies.
In our machine learning integration framework, we utilized ten different machine learning algorithms, taking a comprehensive approach aimed at enhancing model performance and robustness. We employed these key genes to establish 91 prognostic models and identified the IRS. The stability and effectiveness of the IRS in the training group, validation group, and cohorts from different medical centers (Freiburg, LEUVEN, and SIENA) were validated through clustering analysis. Our analysis revealed a uniform distribution of IRS in samples from different medical centers, affirming the widespread applicability of IRS as an assessment tool across different locations. This validation further strengthens the case for incorporating IRS into clinical practice. Moreover, we associated the immune and metabolic systems with IRS, underscoring the intense immune system activation in high-IRS patients, marked by the enrichment of multiple immune cell types, including macrophages, neutrophils, mast cells, and regulatory T cells. This result suggests that high-IRS patients exhibit a more complex and active pulmonary inflammatory microenvironment. Through GSEA, we found that high-IRS patients displayed activated adaptive immune responses and inflammation reactions. This could lead to immune system overactivation, ultimately damaging lung tissue. Perturbations associated with metabolic pathways correlated with high-IRS patients, indicating potential issues in amino acid metabolism49 and lipid metabolism50, among others. Of particular significance is the negative correlation with biotin metabolism, implying that patients with higher risk scores may have abnormalities in this regard, potentially affecting overall health and disease progression negatively. Altogether, our results demonstrate that IRS can be used to determine the risk level of IPF patients, guiding treatment choices more effectively to restore immune system balance and repair disrupted metabolic pathways. Physicians can adjust treatment plans in advance based on a patient’s IRS, improving treatment effectiveness, extending survival, alleviating symptoms, and even reducing treatment-related adverse events.
In this study, we did not directly analyze the 20 genes used to construct the IRS. This is because while these genes directly contribute to the construction of the IRS, we believe the best biomarkers may involve the synergistic action of multiple genes, not limited to those used for IRS construction. As anticipated, with the aid of the IRS, we successfully identified a new biomarker: PLA2G7. It belongs to the phospholipase A2 (PLA2) enzyme family and plays a vital biological role in the body. This enzyme is primarily responsible for hydrolyzing ester bonds in phospholipid molecules, releasing free fatty and phosphoric acids51. We found that high PLA2G7 expression was significantly associated with a reduced overall survival time in IPF patients. Furthermore, in multiple datasets, the average AUC value for PLA2G7 exceeded 0.8, indicating its exceptional performance in distinguishing healthy samples from IPF samples. Consistent with the IRS results, PLA2G7 expression exhibited a significant positive correlation with the infiltration of various immune cells. This observation suggests that PLA2G7 is closely related to immune system activation and inflammation response, reinforcing the rationale for PLA2G7 as an IPF biomarker. Thus, the discovery of PLA2G7 has significant clinical potential, assisting in improving the prognosis assessment and early diagnosis of IPF patients. It provides a new biomarker for IPF research and underscores the value of IRS in the fields of prognosis and diagnosis, offering new possibilities for better understanding and treating IPF.
Through single-cell sequencing analysis, we revealed that PLA2G7 is primarily concentrated in macrophages in IPF patients, with significant upregulation in these cells. The results were replicated in the single-cell sequencing data of the mouse IPF model. Similarly, studies on pulmonary fibrosis52 and fibrotic hypersensitivity pneumonitis53 have shown that the PLA2G7-positive macrophage subset is associated with disease status, which is consistent with our research findings. These PLA2G7-positive macrophages displayed significantly increased expression of inflammation-related genes, underscoring the potential role of PLA2G7 in regulating the inflammatory response. Additionally, elevated expression of inflammation-related genes was observed in various cell types, including endothelial cells, T cells, monocytes, and epithelial cells. This observation indicates an overall immune activation characteristic in IPF patients, transcending individual cell types. Further analysis of PLA2G7-positive and PLA2G7-negative cell groups within macrophages revealed the involvement of PLA2G7 in immune regulation, cell proliferation, and apoptosis regulation. High expression of genes, such as HAMP and CCR5, in PLA2G7-positive macrophages suggests the role of PLA2G7 in regulating inflammation and cell proliferation. Lastly, we screened a series of potential drugs that could interact with PLA2G7, among which celecoxib was identified as the most sensitive. Through computer simulation techniques, we further validated the binding potential of celecoxib with PLA2G7. Celecoxib, as a specific COX-2 inhibitor, has been shown to have significant potential in various inflammation-related diseases54,55,56,57. However, there are currently no clinical trials of celecoxib in IPF. Mechanistically, PLA2G7 breaks down membrane phospholipids to form arachidonic acid, which is then catalyzed by COX enzymes (COX-1 and COX-2) to convert into prostaglandins that promote inflammation and fibrosis. COX inhibitors reduce inflammation and improve lung function by inhibiting COX enzymes. Non-selective COX-1 and COX-2 inhibitors (such as ibuprofen) may cause gastrointestinal adverse reactions, cardiovascular events, and other side effects58. Therefore, celecoxib may be safer in the treatment of IPF. Additionally, we found that celecoxib has the strongest affinity for PLA2G7, even higher than the specific inhibitor of PLA2G7, Darapladib. In addition, we note that both celecoxib and darapladib can bind to GLN-352, and thus their binding to PLA2G7 may be competitive with each other. This is very interesting, suggesting that the dual targeting of celecoxib on PLA2G7 and COX-2 may be more effective than using PLA2G7 specific inhibitors (Darapladib) alone.
In conclusion, we developed an IRS that can be used for predicting the immune status and prognosis of IPF patients. Additionally, we identified the diagnostic and prognostic value of PLA2G7 in IPF and the clinical use of celecoxib in targeting PLA2G7 for IPF treatment. Nevertheless, our study has some limitations, and research involving larger patient cohorts is required to further validate the accuracy of the IRS. Moreover, combining more detailed clinical features may enhance the accuracy of IRS. Additional experiments and mechanistic analyses are needed to explore the effects of targeting PLA2G7 with celecoxib.
Data availability
The datasets supporting the conclusions of this article are available in the GEO database , GEO registration numbers are GSE70866, GSE110147, GSE10667, GSE128033. The single-cell sequencing data of mice are publicly accessible at https://ngdc.cncb.ac.cn/gsa with the identifier CRA011039 (https://ngdc.cncb.ac.cn/gsa/browse/CRA011039).
Abbreviations
- IPF:
-
Idiopathic Pulmonary Fibrosis
- PCA:
-
Principal component analysis
- PCs:
-
Principal components
- GEO:
-
Gene expression omnibus
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- ROC:
-
Receiver operating characteristic
- AUC:
-
Area under the curve
- RSF:
-
Random survival forest
- Enet:
-
Elastic network
- plsRcox:
-
Partial least squares regression for Cox
- SuperPC:
-
Supervised principal components
- GBM:
-
Generalised boosted regression
- survival-SVM:
-
Survival support vector machine
- GSEA:
-
Gene set enrichment analysis
- DO:
-
Disease ontology
- GS:
-
Gene significance
- MM:
-
Module membership
- WGCNA:
-
Weighted gene co-expression network analysis
- ssGSEA:
-
Single sample gene set enrichment analysis
- IRS:
-
Immune-related risk score
References
Martinez, F. J. et al. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers. 3, 17074 (2017).
Hilberg, O. et al. Epidemiology of interstitial lung diseases and their progressive-fibrosing behaviour in six European countries. ERJ Open. Res.8, 00597–02021 (2022).
Herazo-Maya, J. D. et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci. Transl. Med.5, 1 (2013).
Quinn, C., Wisse, A. & Manns, S. T. Clinical course and management of idiopathic pulmonary fibrosis. Multidiscip Respir Med.14, 35 (2019).
Van Manen, M. J. G., Geelhoed, J. J., Miranda., Tak, N. C. & Wijsenbeek, M. S. Optimizing quality of life in patients with idiopathic pulmonary fibrosis. Ther. Adv. Respir Dis.11, 157–169 (2017).
Lehmann, M. & Kolb, M. Another piece in the pirfenidone puzzle. Eur. Respir J.61, 2300240 (2023).
Noth, I., Oelberg, D., Kaul, M., Conoscenti, C. S. & Raghu, G. Safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis in the USA. Eur. Respir J.52, 1702106 (2018).
Karampitsakos, T., Juan-Guardela, B. M. & Tzouvelekis, A. Herazo-Maya, J. D. Precision medicine advances in idiopathic pulmonary fibrosis. eBioMedicine. 95, 104766 (2023).
Cui, F. et al. Air pollutants, genetic susceptibility and risk of incident idiopathic pulmonary fibrosis. Eur. Respir J.61, 2200777 (2023).
Peljto, A. L. et al. Idiopathic pulmonary fibrosis is associated with common genetic variants and limited rare variants. Am. J. Respir Crit. Care Med.207, 1194–1202 (2023).
Guo, S. et al. Integrative analysis reveals the recurrent genetic etiologies in idiopathic pulmonary fibrosis. QJM: Int. J. Med. hcad206. https://doi.org/10.1093/qjmed/hcad206 (2023).
Kropski, J. A. & Blackwell, T. S. Progress in understanding and treating idiopathic pulmonary fibrosis. Annu. Rev. Med.70, 211–224 (2019).
Heukels, P., Moor, C. C., Von Der Thüsen, J. H., Wijsenbeek, M. S. & Kool, M. Inflammation and immunity in IPF pathogenesis and treatment. Respir. Med.147, 79–91 (2019).
Ishikawa, G., Liu, A. & Herzog, E. L. Evolving perspectives on Innate Immune mechanisms of IPF. Front. Mol. Biosci.8, 676569 (2021).
Towards trustable machine learning. Nat. Biomed. Eng.2, 709–710 (2018).
Zhang, N. et al. An artificial intelligence network-guided signature for predicting outcome and immunotherapy response in lung adenocarcinoma patients based on 26 machine learning algorithms. Cell Prolif.56, e13409 (2023).
Ning, J. et al. Use of machine learning-based integration to develop an immune-related signature for improving prognosis in patients with gastric cancer. Sci. Rep.13, 7019 (2023).
Wang, L. et al. Comprehensive machine-learning survival framework develops a consensus model in large-scale multicenter cohorts for pancreatic cancer. eLife. 11, e80150 (2022).
Ning, J. et al. Use of machine learning-based integration to develop a monocyte differentiation-related signature for improving prognosis in patients with sepsis. Mol. Med.29, 37 (2023).
Prasse, A. et al. BAL Cell Gene expression is indicative of outcome and airway basal cell involvement in idiopathic pulmonary fibrosis. Am. J. Respir Crit. Care Med.199, 622–630 (2019).
Cecchini, M. J., Hosein, K., Howlett, C. J., Joseph, M. & Mura, M. Comprehensive gene expression profiling identifies distinct and overlapping transcriptional profiles in non-specific interstitial pneumonia and idiopathic pulmonary fibrosis. Respir Res.19, 153 (2018).
Konishi, K. et al. Gene expression profiles of Acute exacerbations of Idiopathic Pulmonary Fibrosis. Am. J. Respir Crit. Care Med.180, 167–175 (2009).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res.43, e47–e47 (2015).
Barbie, D. A. et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 462, 108–112 (2009).
Bagaev, A. et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell.39, 845–865e7 (2021).
Wilkerson, M. D. & Hayes, D. N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 26, 1572–1573 (2010).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innov.2, 100141 (2021).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet.25, 25–29 (2000).
Kanehisa, M. K. E. G. G. Kyoto Encyclopedia of genes and genomes. Nucleic Acids Res.28, 27–30 (2000).
Lv, J. et al. Dynamic atlas of immune cells reveals multiple functional features of macrophages associated with progression of pulmonary fibrosis. Front. Immunol.14, 1230266 (2023).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01767-y (2023).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 16, 1289–1296 (2019).
Squair, J. W. et al. Confronting false discoveries in single-cell differential expression. Nat. Commun.12, 5692 (2021).
Richeldi, L., Collard, H. R. & Jones, M. G. Idiopathic pulmonary fibrosis. Lancet. 389, 1941–1952 (2017).
Larson-Casey, J. L., Deshane, J. S., Ryan, A. J., Thannickal, V. J. & Carter, A. B. Macrophage Akt1 kinase-mediated Mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity. 44, 582–596 (2016).
Wang, J. et al. Nestin promotes pulmonary fibrosis via facilitating recycling of TGF-β receptor I. Eur. Respir J.59, 2003721 (2022).
Szabo, A., Stolz, L. & Granzow, R. Surface plasmon resonance and its use in biomolecular interaction analysis (BIA). Curr. Opin. Struct. Biol.5, 699–705 (1995).
Charoentong, P. et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell. Rep.18, 248–262 (2017).
Fastrès, A. et al. Osteopontin and fibronectin in lung tissue, serum, and bronchoalveolar lavage fluid of dogs with idiopathic pulmonary fibrosis and control dogs. Veterinary Intern. Med. JVIM. 16870. https://doi.org/10.1111/jvim.16870 (2023).
Sikkema, L. et al. An integrated cell atlas of the lung in health and disease. Nat. Med.29, 1563–1577 (2023).
Kumar, A. et al. Inhibition of PDIA3 in club cells attenuates osteopontin production and lung fibrosis. Thorax. 77, 669–678 (2022).
Khan, F. A., Stewart, I., Saini, G., Robinson, K. A. & Jenkins, R. G. A systematic review of blood biomarkers with individual participant data meta-analysis of matrix metalloproteinase-7 in idiopathic pulmonary fibrosis. Eur. Respir J.59, 2101612 (2022).
Adegunsoye, A. et al. Circulating plasma biomarkers of survival in antifibrotic-treated patients with idiopathic pulmonary fibrosis. Chest. 158, 1526–1534 (2020).
Morse, C. et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur. Respir J.54, 1802441 (2019).
Nissen, S. E. et al. Cardiovascular Safety of Celecoxib, Naproxen, or Ibuprofen for Arthritis. N Engl. J. Med.375, 2519–2529 (2016).
Duchemann, B. et al. Prevalence and incidence of interstitial lung diseases in a multi-ethnic county of Greater Paris. Eur. Respir J.50, 1602419 (2017).
Baeuerle, P. A. & Henkel, T. Function and activation of NF-kappaB in the Immune System. Annu. Rev. Immunol.12, 141–179 (1994).
Villarino, A. V., Kanno, Y. & O’Shea, J. J. Mechanisms and consequences of Jak–STAT signaling in the immune system. Nat. Immunol.18, 374–384 (2017).
Whalen, W. et al. Association of circulating cell-free double-stranded DNA and metabolic derangements in idiopathic pulmonary fibrosis. Thorax. 77, 186–190 (2022).
Chen, R. & Dai, J. Lipid metabolism in idiopathic pulmonary fibrosis: from pathogenesis to therapy. J. Mol. Med.101, 905–915 (2023).
Oh, M. et al. The lipoprotein-associated phospholipase A2 inhibitor Darapladib sensitises cancer cells to ferroptosis by remodelling lipid metabolism. Nat. Commun.14, 5728 (2023).
Wang, J. et al. Integrated analysis of single-cell and bulk RNA sequencing reveals pro-fibrotic PLA2G7high macrophages in pulmonary fibrosis. Pharmacol. Res.182, 106286 (2022).
Wang, J. et al. Characterizing cellular heterogeneity in fibrotic hypersensitivity pneumonitis by single-cell transcriptional analysis. Cell. Death Discov. 8, 38 (2022).
Tang, S. et al. Celecoxib ameliorates liver cirrhosis via reducing inflammation and oxidative stress along spleen-liver axis in rats. Life Sci.272, 119203 (2021).
Ji, Y. et al. Celecoxib attenuates hindlimb unloading-induced muscle atrophy via suppressing inflammation, oxidative stress and ER stress by inhibiting STAT3. Inflammopharmacol32, 1633–1646 (2024).
Fidahic, M., Jelicic Kadic, A., Radic, M. & Puljak, L. Celecoxib for rheumatoid arthritis. Cochrane Database Syst. Rev. (2017).
Alsaegh, H., Eweis, H., Kamel, F. & Alrafiah, A. Celecoxib decrease seizures susceptibility in a rat model of inflammation by inhibiting HMGB1 translocation. Pharmaceuticals. 14, 380 (2021).
Reed, G. W. et al. Effect of aspirin coadministration on the safety of Celecoxib, Naproxen, or Ibuprofen. J. Am. Coll. Cardiol.71, 1741–1751 (2018).
Acknowledgements
The study is reported in accordance with ARRIVE guidelines. This Project has been supervised and approved by Institutional Animal Care and Use Committee of Zhongshan Institute for Drug Discovery, SIMM, CAS.IACUC No. Approved:202312001.
Funding
This work was supported by National Natural Science Foundation of China (Grants 32271320); Creative Research Group of Zhongshan City, Lingnan Pharmaceutical Research and Innovation team (CXTD2022011).
Author information
Authors and Affiliations
Contributions
T.L, J.N, X.F, G.S and Q.F conceived and wrote the paper. T.L, J.N, X.F, G.S and Q.F analyzed the materials and drafted the manuscript. H.W carried out part of the data collection and collation. G.S and Q.F revised the whole paper. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All methods were carried out in accordance with relevant guidelines and regulations.
Consent for publication
All authors have agreed to publish this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, T., Ning, J., Fan, X. et al. Identification of immune patterns in idiopathic pulmonary fibrosis patients driven by PLA2G7-positive macrophages using an integrated machine learning survival framework. Sci Rep 14, 22369 (2024). https://doi.org/10.1038/s41598-024-73625-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-73625-z
- Springer Nature Limited