
Abstract
The tumor microenvironment (TME) plays a pivotal role in tumor initiation, progression, and the formation of pre-metastatic niches. Pancreatic ductal adenocarcinoma (PDAC) is characterized by a dense fibrotic stroma containing a significant enriched population of cancer-associated fibroblasts (CAFs). The interplay between CAFs and tumor cells is crucial in driving tumor advancement and metastasis, underscoring the potential benefits of novel therapeutic strategies targeting stromal cells to improve patient survival. Pancreatic (pro)enzymes have shown efficacy in cancer treatment. In this study, we evaluated a formulation of Trypsinogen and Chymotrypsinogen A (PRP) on a model of PDAC-CAFs dynamics. Our findings demonstrated PRP multifaceted effects, including: (i) inhibition of the acquisition of a pro-tumoral phenotype by normal fibroblasts; (ii) enhanced expression of E-cadherin and decreased expression of epithelial-mesenchymal transition (EMT)-associated genes; (iii) blockade of the crosstalk between CAFs and cancer cells, leading to a decrease in the proliferation and migration rate of the pancreatic cancer cells; (iv) modulation of CAFs-induced alteration on endothelial cells; (v) induction of Meflin expression, associated with a less pro-tumoral phenotype; (vi) inhibition of the TGF-β pathway, reducing the activation of Smad2/3 proteins involved in fibroblast phenotypic alteration; (vii) selective induction of apoptosis in CAFs by PRP, as evidenced by increased expression of the BAX biomarker; (viii) impaired fibrotic tissue formation, as tested in an in vivo model of PDAC, by reducing the population of CAFs within the TME. Collectively, these results underscore the potential of PRP as a therapeutic candidate for disrupting the intricate interactions within the PDAC TME. Further research and clinical investigations are necessary to validate the translational potential of PRP as an adjunct therapy for PDAC.
Similar content being viewed by others

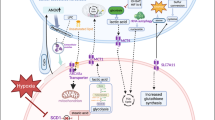
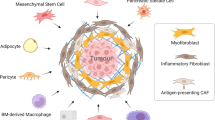
Introduction
Despite recent advancements in clinical management, pancreatic ductal adenocarcinoma (PDAC) remains one of the most aggressive and deadliest forms of cancer, projected to become the second leading cause of cancer-related deaths by 20301,2. PDAC is characterized by its late-stage diagnosis, limited treatment options, and poor prognosis3,4.
Tumors, including PDAC, are complex systems characterized by dynamic interactions between neoplastic cells and their surrounding environment known as the tumor microenvironment (TME), which comprises various cell types and extracellular components. Traditional perspectives often portrayed cancer cells as the sole drivers of malignancy, relegating the stromal compartment to a passive, supportive role5,6. However, recent studies have revealed the reciprocal communication and dynamic crosstalk between the tumor and stromal elements, indicating that both elements are equally involved in the tumorigenic process7,8. These findings have led to a paradigm shift in therapeutic approaches, moving beyond targeting cancer cells alone to address the intricate interactions within the tumor microenvironment. The importance of the TME in tumor initiation, maintenance, and progression is now widely acknowledged9,10,11, especially in tumors with high fibrotic content, such as PDAC12. Indeed, the complex interplay between PDAC cells and their surrounding stromal components is increasingly recognized as a hallmark feature of this disease12,13,14.
Within the TME, cancer-associated fibroblasts (CAFs) have emerged as pivotal orchestrators of tumor-stroma interactions, exerting a fundamental role in establishing the supportive niche crucial for sustaining PDAC growth and progression. These fibroblastic cells undergo phenotypic alterations in response to paracrine signals originating from cancer cells and neighboring stromal cells. Consequently, through the secretion of growth factors, cytokines, and extracellular matrix components, CAFs actively contribute to shaping a dynamic microenvironment that fosters tumor cell survival, proliferation, migration, and immune evasion15,16,17. Moreover, CAFs exhibit remarkable functional heterogeneity, contributing to the complexity and therapeutic resistance of the disease8,10. Rather than serving as a uniform stromal component, CAFs encompass diverse subpopulations with distinct phenotypes, molecular signatures and biological roles10. These include pro-tumorigenic functions such as promoting tumor cell proliferation, immune suppression and extracellular matrix remodeling, as well as tumor-restraining activities in certain contexts. This functional plasticity is influenced by dynamic interactions with cancer cells and other stromal and immune elements, highlighting the need for a nuanced understanding of CAF subsets when considering therapeutic strategies targeting the PDAC TME13,18.
Importantly, cancer cells participate in the activation of fibroblasts by secreting various growth factors, including transforming growth factor-β (TGF-β), platelet-derived growth factor (PDGF), and interleukin-6 (IL-6)10,18. Subsequently, the secretome of CAFs exerts profound effects on cancer cells, not only influencing the initiation of tumorigenesis but also crucially impacting intermediate stages such as angiogenesis, metabolic reprogramming towards a reverse Warburg phenotype, and the induction of epithelial–mesenchymal transition (EMT)18,19.
The current standard treatment for PDAC patients typically involves surgical resection and/or cytotoxic chemotherapy regimens, such as nab-paclitaxel plus gemcitabine (GEM) and FOLFIRINOX. However, pancreatic tumor surgical resection presents significant technical challenges, and the recurrence rate is alarmingly high. Despite systemic chemotherapy being the primary treatment option for most PDAC patients, drug resistance eventually develops in nearly all cases20,21. Recently, there has been growing attention on PDAC-TME-driven chemotherapy resistance, with numerous studies exploring how CAFs contribute to chemoresistance. In fact, CAFs create a physical barrier to GEM perfusion, affecting drug permeability, and secrete functional cytokines that induce immune suppression10,22. Moreover, CAFs contribute to GEM resistance through direct and paracrine signaling or by releasing deoxycytidine to competitively counteract GEM function within tumor cells23,24,25.
Reprogramming of CAFs and short-term inhibition of CAFs-predominant signaling pathways have been recognized as more effective strategies than complete ablation or chronic inhibition. Several studies have demonstrated that strategies focusing on CAF ablation are ineffective26,27. Therefore, there is a need for novel approaches to hinder CAFs’ tumor-supportive functions and their induction of chemoresistance. “CAF re-education therapy” has emerged as a promising approach to reduce PDAC fibrotic tissue, aiming to enhance drug uptake and inhibit the formation of the pre-metastatic niche10.
Enzyme therapy based on pancreatic (pro)enzymes regimens has been proven effective as an adjuvant anti-cancer therapy, leading to improvements in life expectancy and quality of life for cancer patients without associated side effects28. Previously, we demonstrated that treatment of human cancer cells with Trypsinogen (T) and Chymotrypsinogen A (C) in a 1:6 ratio (referred to as PRP) enhances cell adhesion, attenuates several EMT-associated markers, and increases expression of differentiation-associated markers, impacting their proliferative capacity and reducing malignant potential29. These effects align with the targets of novel chemo-differentiation therapeutic regimens for PDAC treatment30,31,32,33. Therefore, this study could confirm PRP both effects directly targeting tumour cells and modulating tumour-stromal interaction as well as stroma-stroma interaction.
Therefore, this study investigates the multifaceted roles of CAFs in PDAC, examining their diverse impacts on tumor-stroma interactions, disease progression, and therapeutic challenges. We assessed the efficacy of PRP formulation at various stages of malignancy. Through comprehensive in vitro and in vivo analyses, we examined the anti-tumor effects of the PRP formulation on different components of the TME, including cancer cells, cancer stem cells (CSCs), human umbilical cord-derived endothelial cells (HUVECs), normal-associated fibroblasts (NAFs), CAFs, and CAFs-educated fibroblasts (CEFs). These pancreatic (pro)enzymes could serve as a viable adjuvant treatment for cancer, representing a valuable strategy to impede the growth and development of the tumor microenvironment in solid pancreatic tumors. This approach opens up new opportunities for innovative and personalized therapeutic interventions.
Materials and methods
Pancreatic (pro)enzymes
The bovine pancreatic (pro)enzymes Chymotrypsinogen and Trypsinogen were supplied by AppliChem (UK). A stock solution containing 6 mg/ml of Chymotrypsinogen A and 1 mg/ml of Trypsinogen was prepared by dissolving the pancreatic enzymes in phosphate-buffered saline (PBS) (Sigma-Aldrich, St. Louis, MO), and stored at − 20 °C. For each experiment, the stock solution was diluted in medium to achieve the desired concentrations. The final concentrations of the pro-enzymes were selected based on literature data and previous clinical experiences34.
Cell culture
The human pancreatic cell line BxPC3 (ECACC 93120816) and MIA PaCa-2 (CRL-1420) were obtained from the American Type Culture Collection (ATCC) (Rockville, MD, USA). NAFs were isolated from human skin tissue obtained during abdominoplasty surgery and properly characterized (ethics committee reference: 0467-N-20); CAFs were obtained through various co-culture methods with tumor cells (protocols explained below); CEFs were generated by culturing NAFs in CAFs conditioned medium for 14 days. Primary human umbilical vein endothelial cells (HUVECs) were also obtained from the ATCC (PCS-100-010) and cultured in EGM-2 growth medium (Lonza, Basel, Switzerland) supplemented with 2% fetal bovine serum (FBS) and endothelial growth supplement (BD Biosciences, San Diego, CA).
All cell lines were routinely maintained in T75 culture flasks at 37 °C in a humidified atmosphere containing 95% air/5% CO2. The culture media used were: RPMI-1640 Medium for BxPC3, Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Grand Island, NY, USA) for MIA PaCa-2, NAFs and CAFs, a mixture of 30% DMEM and 70% medium obtained from CAFs for the growth of CEFs. All media were supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, Massachusetts, USA), 100 IU/ml penicillin (Invitrogen, Merelbeke, Belgium), and 100 µg/ml streptomycin (Invitrogen, Merelbeke, Belgium). HUVECs were cultured on collagen-coated Cytodex microcarrier beads (Sigma-Aldrich, St. Louis, MO). Cell dissociation for passaging was performed using the trypsin replacement TrypLE reagent (Life Technologies, Carlsbad, California, USA). Fibroblasts between passages 2–15 and HUVECs below passage 6 were used for all experiments.
Isolation of normal fibroblasts from human abdominal sample
All human samples used in this study were obtained after informed consent and authorization was provided from the Granada Provincial Ethics Committee (Ministry of Health and Families, Andalusia, Spain, reference: 0467-N-20). Abdominal skin samples of 9 cm2 in size were transported in DPBS from the operating room to the laboratory for the start of processing. In a laminar flow cabinet, samples were kept in washing solution (penicillin 600,000 Ul and streptomycin 600,000 Ul in DPBS) for 30 min. With the help of a scalpel and tweezers, the dermis was separated from the epidermis, mechanically processed and deposited in a 50mL falcon. Subsequently, 2 mg/ml of collagenase type I (Gibco) was added and incubated for 18–24 h at 37 °C while shaking. The solution was neutralized with double the volume of DMEM (10% FBS) and filtered through a 100 μm sterile filter, the resulting solution was centrifuged at 300 g for 10 min. Cells were seeded at a density of 100,000–140,000 cells/cm2 in DMEM. Medium changes were made every 2–3 days, until a confluency of > 80% was reached.
Determination of PRP IC50
To evaluate the inhibitory effect of PRP (chymotrypsinogen/trypsinogen) on pancreatic cancer cells, the cells were seeded at a density of 5 × 10⁴ cells/well in 100 µL of complete culture medium in 96-well plates. After 24 h to allow cell attachment, the cells were treated with increasing concentrations of PRP. The 1× concentration was defined as 0.123 mg/mL chymotrypsinogen and 0.0206 mg/mL trypsinogen. Serial dilutions were prepared to obtain final concentrations of 0.1×, 0.5×, 2.5×, 5×, 7.5×, 10× and 20×. Treatments were performed in triplicate for each condition. The cells were then incubated with PRP for 72 h. The same treatments were then reapplied at the corresponding concentrations for an additional 72 h. After this second treatment period (a total of six days), cell viability was assessed using the Alamar Blue assay. Briefly, 10% (v/v) Alamar Blue reagent (Invitrogen) was added to each well and the plates were incubated for 2–4 h at 37 °C. Fluorescence was then measured using a GloMax® Discover Microplate Reader (Promega) with an excitation/emission wavelength of 560/590 nm. Cell viability was normalised to the untreated control group.
Primary and secondary sphere formation assay
An enriched subpopulation of cancer stem-like cells from the BxPC3 pancreatic cancer (adenocarcinoma) cell line was cultured in our specific sphere-forming medium (WO2016020572A1)35 in ultralow attachment plates (Corning). Primary spheres were manually counted after 72 h, disaggregated with TrypLE, and re-seeded to form secondary spheres.
2D Co-culture assay
To perform indirect co-culture, six-well plates (353502, Corning, Durham, USA) with polystyrene transwell inserts (353090, Corning, Durham, USA) with a pore size of 0.4 microns were utilized. 80,000 NAFs were seeded into each well of the plate. After 24 h, CSCs derived from secondary spheres (originated from 300,000 BxPC3 cells) were added per transwell. 1.5 mL and 1 mL of DMEM medium were added to the well and transwell, respectively, with medium refreshed every three days.
For direct co-culture, 80,000 NAFs together with an enriched subpopulation of CSCs derived from secondary spheres (originated from 300,000 BxPC3 cells) were seeded per well of a 6-well plate with 2 mL of DMEM. Medium change was performed every 72 h until reaching 90% confluence. After 14 days, fibroblasts were isolated using TrypLE reagent as they required a shorter detachment time compared to the tumor cells and were characterized with specific CAF-expressing proteins.
For both types of co-cultures, a prior evaluation was carried out to establish which medium was the most optimal that both cell types shared. Among the options (RPMI, DMEM and sphere medium), it was determined to apply sphere medium with 1% FBS.
Cells were treated twice with PRP (T/C 0.07/0.42 mg/mL), 24 h after seeding and 72 h after the first treatment, under three conditions: (i) cancer cells were treated before co-culture with fibroblasts; (ii) PRP was added directly into the co-culture; and (iii) fibroblasts were treated after the co-culture, once they turned into CAFs.
3D direct co-culture assay
An enriched subpopulation of CSCs derived from secondary spheres (originated from 300,000 BxPC3 cells) was seeded per well of a 6-well plate until full confluence was achieved. Confluent monolayer cultures of fibroblasts and the BxPC3-derived from CSCs were detached from the wells using a micropipette tip and introduced into 15 mL tubes with 5 mL of DMEM, aiming to maintain the BxPC3 monolayer inside and the fibroblasts outside, simulating a protective barrier. Tubes were centrifuged at 300 g and 21 °C for 5 min to form pellets, which were then incubated at 37 °C with 5% CO2 for 24 h without agitation. The following day, tubes were gently tapped for pellet separation and medium change was performed while avoiding touching the pellet. Two PRP treatment models were implemented: maintaining the concentration used so far with established 72-hour intervals between treatments, or simulating a more frequent chemotherapy treatment with four doses instead of two, while halving the concentration. Each pair of pellets corresponds to a different experimental condition:
-
Condition A (control condition with no PRP treatment): pellets 1 and 2.
-
Condition B (PRP concentration of = T/C 0.07/0.42 mg/mL, 2 pulses, 72-hour.
intervals between treatments): pellets 3 and 4.
-
Condition C (PRP concentration of = T/C 0.035/0.21 mg/mL, 4 pulses, 48-hour.
intervals between treatments): pellets 5 and 6.
Cell proliferation was assessed using the Alamar Blue reagent. Fluorescence was measured over a 10-day period. At each time point, individual measurements were taken for each pellet, with independent replicate readings (Ex/Em = 560/590 nm) obtained using a plate reader (n = 2 per condition per time point). Raw fluorescence data are provided in Supplementary Table S1.
To quantify proliferation, fluorescence values were integrated over time for each individual pellet. As the measurement intervals were not equidistant, the area under the curve (AUC) was calculated using the trapezoidal method. This provided a cumulative measure of metabolic activity, used here as a proxy for cell proliferation, over the full 10-day experimental period. Individual AUCs, group means, unpaired Student’s t-test results, and associated p-values are reported in Supplementary Table S1.
For statistical analysis, individual AUC values were compared between experimental groups using an unpaired Student’s t-test. All analyses were performed using GraphPad Prism 9. Differences were considered statistically significant at p ≤ 0.05.
Conditioned medium (CM)
Culture medium was collected from different cell cultures under various conditions and labeled as follows:
-
i)
Normal Medium (NM): Depending on the cells, it could be DMEM or RPMI.
-
ii)
NAF CM: Medium collected from NAFs culture in T75 flasks.
-
iii)
NAF + PRP CM: NAFs were treated twice with PRP (T/C 0.07/0.42 mg/mL) on day 1 and day 4; on day 7, the media was changed, and media were collected from day 8 onwards.
-
iv)
CAF CM: Medium collected from CAFs culture in T75 flasks.
-
v)
CAF + PRP CM: CAFs were treated twice with PRP (T/C 0.07/0.42 mg/mL) on day 1 and day 4; on day 7, the media was changed, and media were collected from day 8 onwards.
Collected media were frozen and stored at −20 °C for use within the following 3 months. For longer storage periods, CM was stored at −80 °C.
CEFs establishment
100,000 NAFs were cultured in 7 mL of CAFs CM along with 3 mL of DMEM in a T75 flask, and the medium was changed every 72 h. After 14 days of culture, CEFs were established and characterized.
Angiogenic co-culture system
HUVECs were cultured in complete EGM-2 growth medium (Lonza, Basel, Switzerland) under standard cell culture conditions at 37 °C and 5% CO2. Cells were used between passages 4 and 6. Multiwell dishes (12-well plates) were coated with 300 µL of Matrigel (Collaborative; BD PharMingen, San Diego, CA) at 4 °C and incubated for 30 min at 37 °C. HUVECs (1.2 × 105 cells/well) were added to the Matrigel-coated wells in complete culture medium, and 24 h later, 80,000 NAFs and CAFs were seeded in chambers. The (pro)enzymes formulation was added after seeding the fibroblasts at a concentration of T/C: 0.07/0.42 mg/mL. After 14 days of co-culture, cells were photographed using a Leica DM 5500B fluorescent microscope (Solms, Germany).
LIVE/DEAD cell viability assay
Cell viability staining was performed by the LIVE/DEAD Viability/Cytotoxicity Kit (Molecular Probes), according to the manufacturer’s instructions. In brief, BxPC3 secondary spheres in different conditions were washed with PBS and labeled with the staining solution (1:4 calcein AM: EthD-1) (L3224, Invitrogen Kit). Images were taken by confocal microscopy (Nikon Eclipse Ti-E A1, USA) with a 10x magnification and analyzed using NIS-Elements software.
Proliferation assay
Culture medium was aspirated and replaced with a mixture of 100µL of AlamarBlue® (DAL1025, Invitrogen) stain per 1mL of medium. Cells were incubated at 37 °C for 1 h in darkness and 100µL of medium was poured into a well of a 96 plate and proceed with the fluorescence reading (560/590nm) (Synergy HT, BIO-TEK).
Colony formation assay
CSCs derived from secondary spheres, originating from 300,000 BxPC3 cells, were seeded in wells of a 6-well plate on day 0. Various culture conditions were tested: (i) RPMI medium (NM); (ii) RPMI medium + PRP (NM + PRP); (iii) NAF CM; (iv) NAF + PRP CM; (v) CAF CM; and (vi) CAF + PRP CM.
Cells were treated twice with PRP (T/C 0.07/0.42 mg/mL) on day 1 and day 4. Colony formation assays were conducted on day 7 and day 21 using Crystal Violet staining (C0775, Sigma-Aldrich, St. Louis, MO). Briefly, cells were washed twice in PBS and fixed with 4% paraformaldehyde (PFA) for 30 min. After three washes with PBS, cells were stained with 0.5% Crystal Violet in 5% methanol for 15 min at room temperature (RT) with gentle rocking. Individually stained colonies in each well were counted, and the colony formation fraction was calculated as follows: colony number/(number of cells seeded × plating efficiency), where plating efficiency was determined by the ratio of colony number to the number of cells seeded in the drug-free medium. Three independent experiments were conducted.
In vitro wound-healing assay
CSCs derived from secondary spheres, originating from 300,000 BxPC3 cells, were seeded in wells of a 12-well plate. Various culture conditions were tested: (i) RPMI medium (NM); (ii) RPMI medium + PRP (NM + PRP); (iii) NAF CM; (iv) NAF + PRP CM; (v) CAF CM; and (vi) CAF + PRP CM. For MIA PaCa-2 cell line, a total of 300,000 cells were seeded in wells of a 12-well plate. The culture conditions that we tested were: (i) DMEM medium and (ii) DMEM medium + PRP.
Once the cells reached 100% confluence, wounds were created by scraping the monolayer cells with a 200 µl pipette tip, and non-adherent cells were washed off with medium. At 0, 12, 24, 48, and 72 h after the creation of wounds, treated and control non-treated cells were observed, and migration images were captured with a 4x objective using phase-contrast microscopy. Cell migration was determined by the rate of cells moving towards the scratched area, and the scratched area was quantified using ImageJ™ software. All experiments were plated in triplicate wells and were carried out at least three times.
Treatment with TGF-βR1 inhibitor
CAFs obtained from co-culture were grown until reaching 80% confluence and then treated with Galunisertib (LY2157299, a TGF-βR1 inhibitor obtained from MedChemExpress, New Jersey, USA) at a concentration of 5 µM. The treatment duration was 24, 48, and 72 h post-progression towards a tumor-promoting phenotype before subsequent assays were conducted36.
Immunofluorescence analysis
For the Immunofluorescence analysis, the cells were fixed with 4% paraformaldehyde (PFA) in PBS for 30 min at room temperature (RT) and permeabilized with 0.5% Triton X-100 for another 30 min. Non-specific binding was blocked using a Blocking Reagent (11096176001, Roche, Basel, Switzerland) for 1 h at RT. Subsequently, cells were incubated overnight at 4 °C with primary antibodies: αSMA (ab7817, Abcam, Cambridge, UK), FAPα (ab28244, Abcam, Cambridge, UK), Smad2/3 (ab217553, Abcam, Cambridge, UK), Meflin (ab232986, Abcam, Cambridge, UK), VE-cadherin (ab33168, Abcam, Cambridge, UK), and CD31 (ab9498, Abcam, Cambridge, UK), Vimentin (sc-6260, Santa cruz biotechnology), Snail + Slug (ab180714, Abcam) and E-Cadherin (ab15148, Abcam).
On the following day, cells were washed three times with PBS and then incubated with secondary antibodies (Alexa, Abcam, Cambridge, UK) for 2 h at RT. After another three washes with PBS, cells were mounted with a fluoroshield mounting medium with DAPI (ab104139, Abcam, Cambridge, UK). Photographs were taken using a Leica DM 5500B (Solms, Germany) fluorescent microscope equipped with Meta Systems Isis and NIH ImageJ software (National Institutes of Health, MA, USA). The integrated fluorescence intensity for each cell line was determined by computing the average fluorescence intensity of the image, subtracting the background intensity, and normalizing the data. In Figs. 1 (αSMA and FAPα), 2 (αSMA and FAPα), 5 (VE-cadherin and CD31) and 9 (Smad2/3 and Meflin) double antibody staining was performed.
RNA isolation and quantitative real time RT-qPCR assay
Total RNA was extracted using the TRIZOL reagent following the manufacturer’s instructions (Life Technologies, Carlsbad, California, USA). cDNA was synthesized by reverse transcription of total RNA using the Reverse Transcription System kit (Promega, Madison, Wisconsin, USA) with oligo dT and random primers.
RT-qPCR was performed using the SYBR-Green PCR master mix (Promega, Madison, Wisconsin, USA) according to the manufacturer’s recommendations. Each reaction was performed in triplicate from cDNA dilutions. The comparative threshold cycle (Ct) method was used to calculate the amplification factor as specified by the manufacturer. PCR reactions were performed as follows: an initial denaturation at 95 °C for 2 min, followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s, and a final cycle of dissociation from 60 to 95 °C.
Human GADPH was used as an internal standard to normalize variations in RNA quality and the quantities of input cDNA. Different controls were used for each experiment, including assessment of reverse transcription performance, genomic DNA contamination, and PCR performance, using the ∆∆CT method of relative quantification. All samples were run in triplicate for each gene. A list of primers is summarized in Table 1.
Western blotting
Whole cell lysates were obtained using RIPA lysis buffer (sc-24948, Santa Cruz Biotechnology, Texas, USA). Cytosolic and nuclear proteins were extracted using a nuclear protein extraction kit (Beyotime Biotechnology Co., Jiangsu, China). Protein concentrations were determined using Bio-Rad detergent-compatible protein assays (Bio-Rad Laboratories, Hercules, California, USA). Equal amounts of protein (20 µg) were loaded onto 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) gels. After electrophoresis, proteins were transferred onto nitrocellulose membranes (162 − 0115, Bio-Rad Laboratories, Hercules, California, USA) for 30 min at 25 V and blocked in 5% non-fat milk for 1 h. Membranes were then incubated overnight with primary antibodies (1:1000 dilution) at 4 °C with shaking. After washing in Tris-buffered saline with Tween 20 (TBST), membranes were incubated with secondary antibodies (1:2000 dilution) for 2 h at RT, washed again in TBST, and detected using an enhanced chemiluminescence reagent kit (Beyotime Biotechnology Co., Jiangsu, China) on a LAS 3000 Imaging System (Fujifilm). Band intensities were determined using a gel image analysis system (Bio-Rad Laboratories, Hercules, California, USA) and normalized to β-actin. Protein band intensities were quantitated using Image-J software (NIH). Primary antibodies used were anti-Human/Mouse Smad2/3 (AF3797, R&D Systems Bio-Techne, Minneapolis, Minnesota, USA), anti-Human Phospho-Smad2/3 (S465/S467, R&D Systems Bio-Techne, Minneapolis, Minnesota, USA), and anti-β-actin (ab8227, Abcam, Cambridge, UK).
Analysis of apoptosis by Propidium iodide and Annexin V-FITC staining
Apoptosis was detected using the apoptosis detection kit for annexin V-FITC (88–8005−74, Thermo Fisher Scientific, Waltham, Massachusetts, USA). Both reagents annexin V-FITC and IP non-vital dye allowed the differentiation among: viable cells (no staining for both dyes), cells undergoing early apoptosis (positive for annexin V-FITC and negative for PI), cells undergoing late apoptosis (positive for both dyes), and necrotic cells (only showed IP staining). The results were representative of three independent experiments.
Animal care compliance
During the study, the care and use of animals were conducted in accordance with the principles outlined in the Guide for the Care and Use of Laboratory Animals, 8th Edition, 2010 (National Research Council).
In vivo anti-tumor xenograft studies
To minimize animal suffering, in accordance with the ARRIVE guidelines, the present study utilized tissues collected from mice that had been part of a previously published study37. NOD SCID Gamma (NSG) mice were supplied by the animal facility of Centro de Instrumentación Científica (CIC, University of Granada).
In brief, as reported in37, mice were housed and maintained at temperatures ranging from 20 °C to 24 °C, with a relative humidity of 50%, and a light-dark cycle of 14 to 10 h, with food and water provided ad libitum. Animals (n = 10 per group) were randomly assigned to control (1st group), pretreatment group (2nd group), and pretreatment + treatment group (3rd group). The treated groups received Trypsinogen and Chymotrypsinogen A at a dose of 83.3/500 mg/kg in combination in a single injection administered 3 days per week by intravenous injection into the tail vein at a dosing volume of 10 mL/kg. The volume of dosing solution administered to each animal was calculated and adjusted based on individual body weight measured immediately before dosing. Treatments were administered for 3 weeks before tumor induction (2nd group); and for 3 weeks before tumor induction and continued for 9.5 weeks after tumor induction (3rd group); the control group (1st group) was inoculated with physiological saline solution.
Tumors were generated by subcutaneous injections of BxPC3 CSCs, for which CSC spheres were disaggregated, and 10,000 counted viable cells per mouse were injected using 26-gauge needles. After 88 days all mice were sacrificed when showing clear signs of health decline in control mice (e.g., weight loss, ascites, or lethargy). Mice were anesthetized using isoflurane via inhalation. For euthanasia, they were sacrificed by an overdose of sodium pentobarbital, following ethical guidelines.
Paraffin-embedded blocks of all tumors were sectioned at 5 μm and subjected to immunofluorescence. After deparaffinization and rehydration, tumor sections were labeled with primary antibodies: αSMA (ab7817, Abcam, Cambridge, UK), FAPα (ab28244, Abcam, Cambridge, UK), Smad2/3 (ab217553, Abcam, Cambridge, UK), and Meflin (ab232986, Abcam, Cambridge, UK), at 4 °C overnight. Following washing with PBS, tumor sections were subsequently incubated with fluorescence-conjugated secondary antibody (Alexa, Abcam, Cambridge, UK) for 2 h at RT. The sections were then mounted with fluoroshield mounting medium with DAPI (ab104139, Abcam, Cambridge, UK). Images were captured using a confocal fluorescence microscope (Leica DM 5500B microscope).
Approval and accordance statement for human samples
All methods were carried out in accordance with relevant guidelines and regulations.
All experimental protocols were approved by the Vithas Hospital Granada (Plastic Surgery Service) Institutional Review Board approval (ethics committee number: 0467-N-20).
Approval and accordance statement for animal studies
All methods were carried out in accordance with relevant guidelines and regulations.
All experimental protocols were approved by the Institutional Committee for the Animal Care and Use of the University of Granada (code of the approved protocol: 03/07/2017/086).
Statistical analysis
All analyses were performed at least in triplicate in each of at least three independent experiments and exhibited mean ± standard deviations. Statistical significance was calculated using Student’s t-tests (*P < 0.05, **P < 0.01, ***P < 0.001), ($P < 0.05, $$P < 0.01, $$$P < 0.001) and (#P < 0.05, ##P < 0.01, ###P < 0.001). P < 0.05 was considered statistically significant. The Statistica 6.0 Program was used to analyze the results.
Results
Monolayer of BxPC3 and BxPC3-CSCs induce the transition from NAFs phenotype to CAFs phenotype
The first objective of this study was to establish CAFs. To achieve this, NAFs were co-cultured with monolayer BxPC3 cells or with BxPC3-derived cancer stem cells (BxPC3-CSCs) using a transwell system (Fig. 1A). Phase microscopy images revealed that both co-culture conditions induced an activation of NAFs towards a malignant-like phenotype. Specifically, a change in the size and shape of NAFs co-cultured with tumour cells was observed under phase-contrast microscopy. NAFs co-cultured with monolayer BxPC3 exhibited a larger and more star-shaped morphology (Fig. 1A, white arrows) compared to control NAFs, which appeared more elongated and thinner. Moreover, NAFs co-cultured with BxPC3-CSCs displayed an even larger size and a more stellate shape (Fig. 1A, white arrows).
An immunofluorescence assay targeting CAFs markers, alpha smooth muscle actin (αSMA) and fibroblast activation protein (FAPα), was conducted (Fig. 1B). αSMA staining was detected in both NAFs and CAFs, with no significance differences in expression between these cellular states. However, NAFs co-cultured with tumour cells showed an increased expression of the FAPα marker (235% in those co-cultured with monolayer BxPC3), which was more pronounced in fibroblasts co-cultured with CSCs (a 320%), both conditions compared to NAFs, as can be appreciated in the graphs showing fluorescence intensity of the labeled cells (Fig. 1E).
Subsequently, a RT-qPCR analysis was performed (Fig. 1F), revealing a significant increase in the expression of CAFs markers in NAFs cultured with monolayer BxPC3 (49-fold for FAPα; 33-fold for FSP; and 42-fold for SDF1), and it was more pronounced when fibroblasts were cultured with BxPC3-derived CSCs (111-fold for FAPα; 57-fold for FSP; and 74-fold for SDF1) both compared to NAFs expression. The significant increase in the expression of these genes after NAFs co-culture with BxPC3-derived CSCs, compared to NAFs, indicated that this approach leads to a more pronounced pro-tumoural CAF phenotype.
Pancreatic (pro)enzymes formulation inhibited CSCs induction of CAFs
The effect of PRP on the fibroblast activation potential of CSCs over NAFs was evaluated. To do so, three different sets of experiments were performed: (i) indirect co-culture of NAFs with BxPC3-CSCs that had been treated with PRP before the co-culture (represented in the figure as “before”); (ii) NAFs and BxPC3-CSCs co-culture with PRP added during the co-culture process (represented in the figure as “during”); and (iii) CAFs, obtained from the co-culture of NAFs and BxPC3-CSCs, that were treated with PRP after the co-culture (represented in the figure as “after”). Changes in the expression of CAF-related markers were studied in Fig. 2A and 2B.
The representative immunofluorescence images of NAFs co-cultured with BxPC3-CSCs indicated the acquisition of a CAF-like phenotype with increased expression of the protein FAPα when compared with NAFs (4800%). Interestingly, when the co-culture was performed with CSCs previously treated with PRP (before), the expression of FAPα dramatically decreased in a 85%, compared to untreated CAFs, indicating an inhibition of the phenotypic alteration potential of CSCs by PRP. On the other hand, the expression of FAPα also decreased in CAFs treated with PRP in a 56% (after), but it was less marked when the treatment was administered during the co-culture with a 33% of decrease (during), both compared to untreated CAFs. Meanwhile, no significant differences were found in αSMA expression in the different conditions (Fig. 2A and 2B).
Furthermore, RT-qPCR analysis performed on NAFs and CAFs samples indicated that PRP treatment clearly induced a down-regulation of genes related to CAFs (FAPα, FSP1, and SDF1) when the treatment was administered “before” or “after” co-culture (Fig. 2C). A significant decrease in expression of 72.46, 67.66, and 71.38% for FAPα, FSP1, and SDF1 respectively could be seen when the treatment was applied prior to co-culture, and 80.76, 23.60, and 30.81% for those CAFs that were treated after progression towards a tumor-promoting phenotype. The addition of PRP during co-culture also significantly down-regulated the expression of CAF markers: 49.70, 56.52, and 63.76%. Before, during and after conditions were compared to untreated NAFs/BxPC3 CSCs condition. No significant differences were observed in NAFs + PRP treatment for these CAFs-related genes expression compared to NAFs (Fig. 2C).
To try to replicate the interactions observed in the TME, we proceeded to directly co-culture BxPC3-CSCs with NAFs, both in a 2D model (without using a transwell system) and in a 3D model. Figure 3A-B demonstrate how the 2D direct co-culture strategy mirrored the results obtained with the transwell system (described earlier in Fig. 2), with a 78% of decrease in the expression of FAPα observed when NAFs were co-cultured with previously PRP-treated CSCs. Notably, treatment during co-culture led to a higher level of inhibition of the CSCs’ fibroblast activation potential (83% of decrease), indicating that the treatment could affect direct cell-cell interactions. On the other hand, no significant differences were found in αSMA expression in the different conditions.
Cell proliferation was assessed in a 3D direct co-culture model of fibroblasts and BxPC3 CSCs following administration of two different PRP treatment regimens (Figs. 3C–D): a higher dose of T/C = 0.07/0.42 mg/mL administered every 72 h (pellets 3–4), and a lower dose of T/C = 0.035/0.21 mg/mL administered every 48 h (pellets 5–6). The AUC for each pellet was calculated as described in the Methodology section. The individual AUC values were as follows: 20.43 and 21.20 for control condition A (pellets 1–2), 7.45 and 6.91 for condition B (high-dose PRP, pellets 3–4), and 11.67 and 13.67 for condition C (low-dose PRP, pellets 5–6). The average AUCs for each condition were 20.81 for condition A, 7.18 for condition B, and 12.67 for condition C (Supplementary Table S1). Treatment with the higher PRP dose led to a significant reduction in cell proliferation, with a 65.5% decrease compared to control. The lower-dose regimen also reduced proliferation by 39.2%. Statistical analysis using an unpaired Student’s t-test showed significant differences between condition B and control (p ≈ 0.0021), as well as between condition C and control (p ≈ 0.05). Importantly, direct comparison between the high-dose and low-dose regimens also revealed a significant difference (p < 0.05), confirming a clear dose-dependent inhibitory effect of PRP treatment on cell proliferation. These findings indicate that PRP treatment significantly decreases the proliferation of two key cell populations within the tumor microenvironment CAFs and CSCs.
PRP decreased the tumorigenic capacity of CAFs over tumoral cells
CAFs were obtained by indirect co-culture of NAFs and BxPC3-CSCs, using a transwell system, and used to investigate whether PRP treatment could influence the interaction between CAFs and tumor cells. Initially, disaggregated BxPC3-CSCs were co-cultured with CAFs and treated with PRP. The expression of EMT-related genes in BxPC3-CSCs was assessed after 14 days of co-culture and compared with a control culture of BxPC3-CSCs and a non-treated CAFs-BxPC3-CSCs co-culture.
Figure 4A shows that PRP treatment led to the up-regulation of E-cadherin. Notably, five other genes known to induce EMT were down-regulated following PRP treatment. Specifically, the transcription factor Snail, associated with metastatic cells undergoing EMT, showed a significant down-regulation (2.55-fold), as did SLUG expression (6.15-fold). Furthermore, other EMT markers, including N-cadherin (2.92-fold), Sox-2 (2-fold), and Vimentin (2.22-fold) exhibited decreased expression levels after PRP treatment. These findings align with our previous research38, underscoring PRP’s inhibitory effect on EMT in pancreatic tumor cells.
To validate the RT-qPCR findings, we conducted immunofluorescence assays (Supplementary Fig. 1), which demonstrated a 215%-more elevated Vimentin expression in the untreated CAFs–BxPC3-CSCs co-culture comparing BxPC3-CSCs. PRP treatment resulted in a lower, but also significant, increased of Vimentin levels (143%) comparing with BxPC3-CSCs. Conversely, E-cadherin was virtually absent in untreated co-cultures but was markedly upregulated following PRP administration in a 182% compared to BxPC3-CSCs. Moreover, PRP treatment significantly affected the protein levels of the EMT regulators SNAIL and SLUG, further substantiating its role in reversing the mesenchymal phenotype. Collectively, these results indicate that PRP effectively inhibits epithelial-to-mesenchymal transition in pancreatic tumor cells, corroborating the RT-qPCR data shown.
These results support the notion that certain factors produced by CAFs can influence neighboring cells within the TME. To prove this, a series of experiments were conducted using conditioned media obtained as described in the materials and methods section. Specifically, (i) RPMI was utilized as the control or normal medium (NM); (ii) NAF CM (medium from NAFs culture); (iii) NAF + PRP CM (medium from PRP-treated NAFs culture); (iv) CAFs CM (medium from CAFs culture); and (v) CAFs + PRP CM (medium from PRP-treated CAFs culture).
A wound healing assay was performed on monolayer BxPC3, grown after dispersion of BxPC3-CSCs, and the different culture mediums were added.
As shown in Figs. 4B, cells cultured in NM closed the gap by approximately 60% after 72 h. Migration was enhanced by NAF CM and CAF CM, reaching 85% and 100% closure, respectively, indicating that both fibroblast types secrete factors promoting tumor cell migration, compared to NM condition. PRP treatment markedly inhibited migration, reducing closure rates to 17%, 34%, and 50% in NM, NAF CM, and CAF CM, respectively, compared to untreated controls. These results suggest that PRP disrupts the pro-migratory signals from CAFs, significantly impairing cell motility.
To further assess PRP’s effects on migration and proliferation, the MIA PaCa-2 cell line harboring the KRAS G12C mutation was used. The IC50 of PRP was determined (Supplementary Fig. 2A), showing a significant viability reduction at 10x concentration. Using this dose, PRP delayed migration: untreated cells closed the gap by ~ 95% within 48 h, whereas PRP-treated cells required more than 72 h for complete closure (Supplementary Figs. 2B and 2C).
A crystal violet assay to study colony formation rate is shown in Fig. 4D. Disaggregated BxPC3 CSCs were grown in the different CMs and plate colony formation assay was performed after 7 and 21 days. During the first week, no significant difference was already observed in tumor cells seeded in NAF CM but there was in those seeded in CAF CM media, with a 1.28-fold increase in colony formation in the first CM and a 1.68-fold increase in the second CM compared to NM. When PRP was added to any of the three media, we observed a reduction in colony numbers of approximately 2.23-fold in NM medium, 1.95-fold in NAF CM medium, and 2.00-fold in CAF CM medium (compared to their respective untreated controls). At 21 days, colony formation increased by 1.09-fold in the NAF CM condition, no significant, and by 1.41-fold in the CAF CM condition, significant, compared to the NM condition. On the other hand, PRP treatment reduced colony formation at 21 days by 3-fold in the NM condition, 1.63-fold in the NAF CM condition, and 1.48-fold in the CAF CM condition, relative to their respective untreated controls. Therefore, the addition of NAF CM increased the number of tumor cell colonies, an increase that was even greater when the cells were cultured with CAF medium. The addition of PRP, on the other hand, negatively affected colony formation mainly in the NM medium and to a lesser extent, although also significantly, in the NAF CM and CAF CM media.
In addition, cell proliferation was assessed using the Alamar Blue protocol at 5 time points (days 1, 4, 7, 12, and 21), normalizing the values to day 1 (Fig. 4 F). For each condition, the AUC was calculated as an integrated measure of cumulative proliferation over time. The AUC was calculated using Simpson’s rule, allowing for more accurate estimation in irregular time series. The exact AUC values for each group have been included in Supplementary Table S2 to facilitate reproducibility.
Evaluating relative differences in this study, AUC was 62.8% lower in the + PRP NM condition, 16.9% higher in NAF CM, and 76.2% higher in CAF CM compared to NM. Proliferation decreased by 48.13% when measured in NAF + PRP CM compared to NAF CM. Similarly, there was a 56.58% decrease when measured in CAF PRP CM compared to CAF CM (Table S2).
Taken together, these results indicate that CAFs effectively regulate the EMT process in tumor cells and enhance their invasiveness and proliferation via paracrine signaling that PRP treatment is able to hamper.
The interaction between CAFs and non-tumoral cells within the TME can be modulated by PRP
To investigate the potential generation of heterogeneous cancer-promoting fibroblasts in the TME through the education of NAFs by CAFs, NAFs were incubated with CAFs CM, with and without PRP treatment.
Figure 5A illustrates the results of the RT-qPCR analysis, which evaluated the expression of genes associated with CAFs (FAPα and SDF-1), inflammatory response (IL-1, IL-6, and CXCL8), and immunomodulatory response (EGR2). The gene expression levels in CAFs, NAFs cultured in CAF CM were compared with those in NAFs alone, while NAFs cultured in CAF + PRP CM was compared with NAFs cultured in CAF CM. The results revealed that the expression of all six genes was up-regulated in NAFs cultured with CAFs CM. Specifically, CAF-related genes FAPα and SDF-1 showed a 7-fold and 6-fold up-regulation, respectively, compared to NAFs. Additionally, the expression levels of IL-1, IL-6, CXCL8, and EGR2 were also significantly increased (22-fold, 13-fold, 10-fold, and 5-fold, respectively) compared to NAFs. These findings suggest that factors secreted by CAFs into the medium were sufficient to activate normal fibroblasts, essentially “educating” the NAFs to develop into pro-inflammatory fibroblasts, hereafter referred to as CEFs. It is worth noting that CEF cells exhibit an inflammatory phenotype, expressing CAF-specific markers, but do not fully develop a complete pro-tumorigenic phenotype.
Interestingly, when NAFs were cultured in CM from CAFs treated with PRP, gene expression significantly decreased, compared to CEFs. Specifically, there was a 71% and 28% down-regulation of FAPα and SDF1, and a 74% decrease in IL-1, 72% in IL-6, 67% in CXCL8, and 47% in EGR2.
These results suggest that PRP treatment effectively disrupts the paracrine signaling of CAFs aimed at inducing a pro-inflammatory phenotype in surrounding tissue fibroblasts, thereby inhibiting the chain of phenotypic alteration among fibroblasts within the TME and surrounding tissue that leads to fibrosis.
During tumor initiation and progression, the TME relies on a dynamic and expanding vascular network, with endothelial cells playing a key role in neo-angiogenesis. In this study, we explored the ability of CAFs to promote HUVEC proliferation through paracrine signaling and evaluated whether PRP treatment could modulate this effect.
Immunofluorescence analysis (Figs. 5B–C) revealed that, in monoculture, HUVECs expressed basal levels of the endothelial markers VE-cadherin and CD31. Following PRP treatment, these cells exhibited a modest 9% decrease in VE-cadherin and a significant 36% reduction in CD31 expression compared to untreated HUVECs. When HUVECs were co-cultured with NAFs or CAFs, the expression of both markers significantly increased relative to monocultures, with CAFs eliciting a stronger pro-angiogenic response. Specifically, VE-cadherin increased by 14% and 19%, and CD31 by 36% and 39% in HUVECs co-cultured with NAFs and CAFs, respectively.
Interestingly, PRP treatment markedly suppressed the expression of these markers in co-culture systems. In HUVECs co-cultured with NAFs, PRP reduced VE-cadherin by 62% and CD31 by 67%, while in HUVECs co-cultured with CAFs, reductions of 47% in VE-cadherin and 57% in CD31 were observed, compared to their respective untreated co-cultures. These results suggest that PRP may modulate tumor-associated angiogenesis by limiting endothelial activation driven by both normal and cancer-associated fibroblasts.
Pancreatic (pro)enzymes formulation promotes a Cancer-Restraining CAFs like phenotype
To assess the potential of PRP treatment in “re-educating” CAFs, we examined the expression of Meflin, a protein associated with cancer-restraining CAFs that has been shown to enhance the sensitivity of PDAC to chemotherapeutics39. In Fig. 6A, a substantial expression of Meflin is observed in NAFs, while CAFs exhibit significantly lower expression of this protein, decreasing after quantitative analysis from 97 to 17 (5.71x) the MOD (Fig. 6B). Interestingly, a significantly higher abundance of Meflin + CAFs is detected after PRP treatment, with greater Meflin expression in cells that received the treatment during or after the progression towards a tumor-promoting phenotype (4.88x and 4.76x compared to CAFs). Additionally, administering PRP treatment before malignancy is sufficient to increase 2.65x Meflin expression, compared to CAFs. These results suggest that PRP enhances the subpopulation of cancer-restraining CAFs.
Furthermore, Meflin gene expression was assessed by RT-qPCR (Fig. 6C). It is evident that PRP treatment significantly enhances the expression of Meflin, supporting the notion of a shift towards a less pro-tumoral phenotype (Meflin + CAFs) following PRP administration. Specifically, there are notable changes in Meflin gene expression, with increases 2.3x, 1.83x and 1.63x compared to non-treated CAFs when the treatment was applied before, during, or after the progression towards a tumor-promoting phenotype, respectively.
It is particularly interesting that the data suggests that PRP can not only act as an inhibitory or preventive treatment against fibroblast activation, but also seems to promote a phenotypic change specifically targeting CAF cells, as no significant differences were observed in any of these experiments on the NAF population when treated with PRP.
PRP interferes with the TGF-β pathway in CAFs through the reduction of SMAD2/3 expression and phosphorylation
TGF-β signaling plays a pivotal role in various biological processes that promote tumorigenesis and metastasis, including the phenotypic alteration of fibroblasts within the TME. The canonical TGF-β pathway involves the expression and subsequent phosphorylation of Smad2/3 proteins40. To investigate whether PRP inhibits fibroblast activation and progression towards a tumor-promoting phenotype by interacting with the TGF-β pathway, we assessed the impact of PRP treatment on CAFs (Fig. 7A-E) and compared it with the addition of the TGF-β commercial inhibitor TGF-βRI (LY2157299) (Fig. 7F-J)41. Figure 7A shows the gene expression of Smad2 and Smad3 by RT-qPCR in NAFs, CAFs, and PRP treated CAFs. Fibroblast activation led to a significant up-regulation of Smad2 and Smad3 (87-fold and 60-fold respectively) compared to NAFs. PRP treatment significantly reduced the gene expression of both Smad2 and Smad3 by up to 4.2-fold and 5.3-fold respectively in treated CAFs compared to untreated CAFs. Furthermore, immunofluorescent labeling of Smad2/3 proteins revealed that PRP treatment appeared to reduce the translocation of Smad2/3 into the nucleus. PRP treatment not only prevented Smad2/3 nuclear translocation but also decreased its cytoplasmic expression. A 74% decrease in labeling was observed (in nucleus and cytoplasm) in treated CAFs compared to non-treated CAFs (Fig. 7B-C). Finally, we analyzed the expression of p-Smad2/3 and total Smad2/3 proteins by Western blotting in NAFs, untreated CAFs and CAFs + PRP (Fig. 7D-E). A 5x increase in Smad2/3 protein expression was observed in CAFs compared to NAFs, as well as a 4.7x increase in p-Smad2/3 protein expression. Specifically, Smad2/3 expression decreased by 60% in CAFs after receiving treatment, as well as p-Smad2/3 by 44% compared to untreated CAFs (Fig. 7D-E).
Analyzes were repeated with the commercial inhibitor to compare its effect with that obtained with PRP. Similarly, treatment with TGF-βRI resulted in a reduction of Smad2 and Smad3 gene expression by [5.71-fold] and [4.5-fold] respectively in CAFs compared to untreated CAFs (Fig. 7F). TGF-βRI also reduced the Smad2/3 expression in CAFs and blocked its translocation into the nuclei compared to untreated CAFs. In this case, the fluorescent labeling observed in treated CAFs decreased by 69% compared to untreated CAFs (Fig. 7G). We also analyzed the expression of p-Smad2/3 and total Smad2/3 proteins by Western blotting in TGF-βRI treated CAFs, NAFs and non-treated CAFs (Fig. 7I). Treatment with the TGF-β inhibitor also demonstrated a reduction in the expression of Smad2/3 and p-Smad2/3, by 31% and 46% respectively in CAFs (Fig. 7I).
The analysis confirmed that PRP repressed the TGF-β pathway by inhibiting Smad2/3 phosphorylation, preventing their translocation into the nucleus, which is necessary for the formation of the functional Smad complex. These results demonstrate that PRP effectively represses the canonical TGF-β pathway in CAFs by reducing Smad2/3 gene and protein expression, inhibiting their phosphorylation, and preventing nuclear translocation. This inhibition is comparable, though not identical, to that achieved with the commercial TGF-βRI inhibitor, indicating that PRP attenuates TGF-β/Smad signaling and potentially limits fibroblast activation toward a tumor-promoting phenotype.
PRP does not induce apoptosis in NAFs but increases apoptotic susceptibility in CAFs
Previous findings demonstrated the significant reduction of CAF markers and their tumorigenic functions upon PRP treatment. Here, it is delved deeper into the specific effects of PRP on the CAFs subpopulation. Annexin and PI double staining via cytometry revealed that PRP-treated NAFs did not undergo apoptosis or necrosis significantly (Fig. 8A). In contrast, PRP induced a significant reduction in the number of CAFs by promoting late apoptosis (from 9% to 21%) and necrosis (from 3% to 47%) (Fig. 8A).
Furthermore, we examined the expression of apoptosis-related genes, specifically Bcl-2 as an anti-apoptotic gene and BAX as a pro-apoptotic gene, through RT-qPCR (Fig. 8B). Consistent with our previous findings, PRP-treated NAFs showed no significant change in the expression of either gene compared to untreated NAFs. However, PRP-treated CAFs exhibited a notable up-regulation of BAX compared to untreated CAFs [4.07-fold]. Conversely, the expression of the anti-apoptotic gene Bcl-2 was down-regulated in PRP-treated CAFs [8.77-fold] compared to untreated CAFs.
Finally, Fig. 8C presents a comparative analysis of the “apoptotic susceptibility” value, indicating that CAFs exhibit a more than 35x increase in this value following PRP treatment. These results underscore the selective apoptotic susceptibility of CAFs to PRP treatment, highlighting its potential to induce programmed cell death specifically in the CAFs population within the TME.
In vivo study of the effect of PRP on TME
An in vivo study was conducted to elucidate the impact of PRP on TME formation (Fig. 9A). Three groups were established: a control non-treated group (1st group; mice without treatment), a pre-treated group (2nd group; mice treated before induction), and a pre-treated and treated group (3rd group; mice treated before and after tumor induction). Xenograft tumors were induced by subcutaneous inoculation of BxPC3-CSCs into immunodeficient mice and subjected to the treatment regimen as described in the materials and methods section and Fig. 9A.
After extraction, tumors were sectioned and immunofluorescent analyzed using markers for NAFs (αSMA), fibrosis (FAPα and Smad2/3), and “re-education” (Meflin) (Fig. 9B). αSMA labeling revealed the typical distribution of fibroblasts surrounding tumor cells, with no significant differences observed between the tested groups (Fig. 9B and 9F).
However, the expression of the CAF marker FAPα was notably reduced in tumors pretreated with PRP (2nd group) compared to untreated tumors (1st group) (85% decreased). Interestingly, the presence of CAFs was significantly diminished in tumors receiving double treatment (3rd group) compared to untreated tumors (93% decreased) (Fig. 9C and 9F).
The distribution of Smad2/3 within the tumor tissue was also affected by PRP treatment, particularly in tumors from mice treated before and after tumor induction (group 3), where a marked decrease in Smad2/3 expression was observed (68% decreased in group 2 and 87% reduced in group 3 compared to the 1 st group), along with a notable reduction in the fibrotic barrier (Fig. 9D, white arrows and Fig. 9F).
Consistent with previous findings, the tumor-reversion marker Meflin exhibited high expression in tumor sections from treated mice, with a significant increase observed in the 3rd group (2 times increased in 2nd group and 8 times increased in 3rd group compared to the 1 st group) (Fig. 9E and 9F).
In conclusion, PRP may in vivo efficacy impair fibrotic tissue formation, reducing the population of CAFs within the TME.
Discussion
This study aims to examine the impact of a (pro)enzymes pancreatic formulation on the intricate molecular interplay between pancreatic cancer cells and stromal cells. Using the pancreatic cell line BxPC3 we observed that PRP treatment attenuates the capacity of both non-stem cells and CSCs to stimulate the conversion of NAFs into CAFs. This attenuation is evidenced by the diminished expression of CAF-associated cytokines such as SDF142, along with CAF-specific markers like FSP143 and FAPα44. Notably, while αSMA has been traditionally identified as a CAF-specific marker, in previous studies42,45, our findings demonstrate that NAFs also expressed αSMA. This divergence in CAFs recognized markers could be attributed to the unique expression profiles of TME-related cells, such as CAFs, which are influenced by their precursor cell type and the molecular factors/pathways governing their “malignant education”46. Fibroblast activation is intricately linked to the intrinsic molecular dynamics of each cancer subtype. Indeed, various distinct CAF subpopulations with specific expression profiles and functional behaviors have been delineated within the same type of cancer, including PDAC47, lung adenocarcinoma48, and hepatocellular carcinoma49. Moreover, the relative abundance of each CAF subpopulation may fluctuate during different stages of tumor progression50.
Furthermore, the direct treatment of CAFs with PRP prompted the lowering of CAFs-related markers (SDF1, FSP1, and FAPα), whereas NAFs-associated markers like αSMA expression were not significantly altered compared with untreated controls. This is a relevant fact since it has been recently documented important functional disparities between FAPα- and αSMA-expressing CAFs in PDAC50. Interestingly, a study showed that there are two non-overlapping CAF subpopulations, with FAPα + CAFs exhibiting pro-tumorigenic properties, whereas αSMA + CAFs present an anti-tumor behavior50. Additionally, the authors used the high ratio αSMA/FAPα-expressing CAFs as a good prognosis factor related to increased overall survival in PDAC patients50. Similarly, the abundance of αSMA + CAFs has been directly correlated with non-recurrent PDAC, while the increased presence of FAPα + CAFs has been related to peritoneal metastases51. In the same line, Chiavarina and colleagues established different CAF subgroups in hepatocellular carcinoma based on specific expression profiles and their particular parental cell type, including hepatic stellate cells, vascular smooth muscle cells, and hepatic portal fibroblasts49. This work remarked that hepatic portal fibroblast-derived CAFs exhibited an anti-tumor activity through the secretion of prolargin49. These results underscore the critical role of CAF phenotypic diversity and plasticity within the tumor microenvironment. Accordingly, interpretations of CAF biomarker data, such as the lack of change in αSMA expression between NAFs and CAFs or following PRP treatment, must be approached with caution, given the profound heterogeneity inherent to CAF populations. Notably, αSMA overexpression by CAFs is increasingly recognized not as a marker of normal tissue but as a feature associated with brain metastases arising from diverse primary cancers, including those of the lung, colon, breast, and liver52.
While our findings provide valuable insights into the effects of PRP on fibroblasts, it should be noted that the fibroblasts used in this study were derived from human skin rather than pancreatic tissue. Fibroblasts from different anatomical sites may display variations in their phenotypic markers and functional responses, which could influence their behavior under treatment conditions. Nonetheless, skin fibroblasts represent a widely accessible and informative model, particularly in our experimental setting where they were co-cultured with pancreatic cancer stem cells, a condition that promoted their activation and acquisition of tumor-associated features. This approach provides relevant indications of fibroblast responses within a tumor-like context, although future studies incorporating primary pancreatic fibroblasts will be important to further validate and extend these observations.
Moreover, we found that PRP treatment triggered the upregulation of Meflin in treated CAFs, which may be considered as a relevant event since it has been previously noted that this factor could be categorized as a tumor-restraining CAFs biomarker and that the abundance of Meflin-expressing CAFs was positively correlated to tumors with well-differentiated histology, favorable clinical outcomes, and earlier stages of disease progression in PDAC patients53. In this context, pharmacologically enhancing Meflin expression in CAFs has been proposed as a strategy to convert them into tumor-suppressive cells, aiming to sensitize PDAC to chemotherapy54. Additionally, we observed that NAFs exhibited an elongated, spindle-like morphology, contrasting with the star-like shape of CAFs obtained after co-culture with BxPC3 cells. This finding is consistent with previous studies reporting similar morphological changes during CAF transformation, as observed in NAFs co-cultured with gastric cancer cells45,46.
Nevertheless, although these CSC-induced CAFs appeared slightly resistant to PRP-reprogramming effects, the results were still significant, indicating promising therapeutic potential for the pancreatic (pro)enzymes formulation.
Therefore, these findings support the idea that the PRP formulation may prompt the phenotypic re-education of treated CAFs, transitioning from a tumor-supportive phenotype to a more tumor-restrictive behavior. Indeed, the phenotypic plasticity of CAFs in response to certain external cues has been well-documented. For example, the transition from a proinflammatory-like phenotype towards a myofibroblast-like phenotype induced by TGF-β has been demonstrated in PDAC-related CAFs8. Importantly, our results demonstrate that the potential to induce the CAFs phenotypic switch can also be extrapolated to the in vivo level, as shown by the decreased FAPα and Smad2/3 expression and increased Meflin staining in the mouse xenograft model. Here, we observed a reduction in the amount of fibrotic tissue surrounding the tumors in mice that were treated with PRP. This can be attributed to the significant decrease in TGFβ−1, which is closely linked to the transformation of normal fibroblasts into CAFs55. The TME can also lead to the development of new CSCs from non-stem cancerous cells by releasing factors such as IL6, HGF, or TGFβ−110. Treatment with PRP before and after the procedure may decrease TGFβ−1 levels, which could impede the engraftment of CSCs, formation of their niche, and even subpopulation activation. These results are consistent with our previous in vivo study, in which we found that tumour incidence was markedly reduced in both treated groups (pre-treatment and pre-treatment + treatment) compared to controls, indicating a clear suppressive effect of PRP on CSC tumour engraftment. Moreover, tumour volume measurements over a 34-day period demonstrated a significant inhibition of tumour growth in both treated groups, with no significant difference observed between them. Histological analyses further supported these findings, revealing increased necrotic areas and reduced stromal fibrosis in PRP-treated tumours. Masson’s Trichrome staining showed a notable decrease in extracellular matrix deposition, and immunofluorescence analysis demonstrated reduced expression of the stemness marker CD44 in PRP-treated tumour tissues37. Collectively, these data highlight the antitumour and anti-stemness properties of PRP in vivo.
Interestingly, our results also indicate that the PRP compound selectively induces CAF apoptosis without significantly affecting NAF viability. This finding may be associated with alterations in the BAX/Bcl-2 expression ratio in non-treated and treated CAFs compared to that exhibited by NAFs. In this context, other studies on colorectal cancer have demonstrated the alteration of the BAX/Bcl-2 expression ratio in stromal cells within the TME, leading to aberrant growth rates56. Such expression changes in CAFs, particularly the overexpression of the anti-apoptotic factor Bcl-2, may contribute to the generation of extensive fibrosis associated with pancreatic cancer by preventing CAF apoptosis and growth control. Importantly, PRP may counteract this circumstance through the enhancement of pro-apoptotic BAX along with the inhibition of Bcl-2 expression in treated CAFs, while no significant changes were observed in NAFs exposed to the PRP mixture.
Our preclinical findings support PRP as a potential therapeutic agent for PDAC, offering insights into its mechanisms of action and its impact on the TME affecting both tumor-stromal as well as stroma-stroma interactions. Furthermore, considering previous studies and those obtained in Figs. 3 and 4 of the current study, we confirmed that PRP has also a direct effect on tumor cells. The dose-dependent reduction in cell proliferation indicates that PRP may modulate key pathways involved in these interactions and stemness properties, although further mechanistic studies are needed. The significant p-values support the robustness of these changes, highlighting the therapeutic potential of PRP in disrupting components of the TME that facilitate tumor growth and progression. In line with these findings, our data indicate that PRP treatment effectively reduces cell proliferation in the co-culture model, and this effect is dependent on the concentration regimen applied. Specifically, the high-dose protocol achieved a significantly stronger inhibition than the low-dose protocol. This additional analysis confirms a dose-dependent relationship, suggesting that higher concentrations of PRP more efficiently disrupt the proliferative capacity of CSCs and fibroblasts within the TME. These findings contribute valuable insights into the application of PRP as a multi-targeted approach to impair tumor support structures, warranting further investigation into its efficacy and underlying mechanisms in vivo. In this regard, a study on gastric cancer revealed that NAFs educated by CAFs, CEFs, presented an alternative protumoral phenotype characterized by the absence of αSMA and a high expression of pro-inflammatory cytokines, including IL-1b, IL-6, IL-33, LIF, or CRLF1, along with an upregulation of CXCL857. Specifically, our results indicate that treatment of CAFs with PRP drastically reduced their ability to potentiate the expression of CXCL8 together with pro-inflammatory cytokines like IL1 or IL6 in NAFs57, suggesting a reduced capacity of treated CAFs to induce the generation of CEFs. Indeed, these findings are consistent with previous investigations describing the anti-inflammatory role of pancreatic (pro)enzymes28. In a similar context, it is noteworthy to indicate that treated CAFs have a significantly lower ability to induce the proliferation of endothelial cells like HUVECs57, which may be especially relevant given the well-established protumoral role of angiogenesis in tumor progression. Moreover, additional functional properties of CAFs have been indicated by other researchers, highlighting their interaction with other TME-derived cells, including tumor-associated macrophages (TAMs). For instance, the reprogramming of blood monocytes towards an immune-suppressive and protumorigenic phenotype by triple-negative breast cancer-derived, FAPα-expressing CAFs has recently been identified58,59. Similarly, the polarization of M0 macrophages into tumor-promoting M2 TAMs by CAF-derived CM has been demonstrated in studies of hepatocellular and cervical cancer60,61. Importantly, the authors also showed that M2 TAMs generated by CAFs exhibited a particular secretome different from that of M2 TAMs directly induced by cancerous cells, thus reflecting a similar broad heterogeneity compared to that of their own CAFs60. In fact, it has been revealed that TAMs polarization can be specifically influenced by a particular CAF phenotype or subgroup60.
Several therapeutic approaches have been described to mitigate tumor-associated fibrosis, as it may exert tumor-supportive roles beyond pro-tumorigenic chemical signaling. A dense fibrous extracellular matrix, directly correlated with the abundance of CAFs, can decrease intra-tumor blood perfusion, thereby hindering the delivery of anti-cancer drugs61. Additionally, CAFs themselves may promote a chemoresistant phenotype in PDAC cells through various mechanisms, including the secretion of exosomes containing specific miRNAs62.
Therapeutic strategies targeting TME fibrosis can be classified into three main groups: (i) targeting extracellular matrix-related molecules; (ii) directly targeting CAFs; and (iii) targeting upstream pro-fibrotic signals10,63. According to this classification, PRP treatment fit into the three groups, as it interferes in the TGF-β pathway, induces the apoptosis/reprogramming of CAFs and reduces the capacity of cancerous cells to generate CAFs, likely by altering the pro-fibrotic signals derived from pre-treated tumor cells. Indeed, this event holds special relevance, as other studies have suggested that solely targeting the TME of PDAC may not be sufficient to effectively combat this disorder1,64.
It is important to highlight the reprogramming, but not drastically cytotoxic, effects of PRP on the general fibroblast population. For instance, total blockade of sonic hedgehog signaling in a mouse model of PDAC led to the general depletion of all CAF subpopulations along with stromal fibrosis, resulting in more aggressive pancreatic tumors with heightened proliferation7. Similarly, research has demonstrated adverse outcomes, such as accelerated tumor progression, increased cancer aggressiveness, and reduced overall survival in mouse models of pancreatic cancer following the complete eradication of all CAF subpopulations15. Özdemir and colleagues proposed the drawbacks of eliminating all CAF subpopulations, including tumor-suppressive CAFs, which could elucidate the conflicting outcomes observed with anti-fibrotic cancer therapies15. Supporting this notion, other studies have argued that reprogramming or normalizing the TME, rather than eliminating it entirely, might be more beneficial, as observed in pancreatic tumors65.
A recent study has emphasized the reprogramming of CAFs, tumor-infiltrating immune cells, the extracellular matrix, and the normalization of tumor-associated vasculature as a promising therapeutic strategy for long-term cancer management66,67,68.
We aimed to gain insight into how PRP treatment might induce the reprogramming of CAFs, focusing on the inhibition of the TGF-β pathway. This approach may align with studies by Lan et al., showing promising outcomes of anti-TGF-β drugs as therapy for cancer-associated fibrosis69. Our findings here may be consistent with previous research from our laboratory, where PRP treatment altered the TGF-β pathway in BxPC-3 CSCs, reducing their aggressiveness37. Specifically, we observed significant reductions in fibrotic desmoplasia associated with pancreatic tumors in nude mice following PRP treatment. Furthermore, our results might indicate that PRP treatment could partially inhibit the canonical TGF-β/Smad axis in CAFs. This partial inhibition may be significant considering the functional switch observed in this pathway during tumor progression, transitioning from a tumor-suppressive to a tumor-supportive role70,71. Thus, modulating rather than completely inhibiting this pathway may be crucial, in a manner that preserves specific CAF subpopulations within the TME10. Our data show that PRP significantly reduces Smad2/3 expression, phosphorylation, and nuclear translocation, without fully abolishing TGF-β signaling, likely reflecting the biological relevance of PRP in the TME, where complete suppression of TGF-β signaling could be detrimental55. This partial inhibition might explain, at least in part, the reprogramming of CAFs towards a less pro-tumoral phenotype. Nevertheless, it is important to note that this study does not completely demonstrate the involvement of TGF-β/Smad molecular axis inhibition in the CAF’s phenotypic switch promoted by PRP treatment. Such limitation should be addressed in future studies in order to gain further insights into PRP molecular mechanisms of action, for instance by checking whether Smad2/3 overexpression could cancel PRP effects or whether the specific inhibition of CAF’s TGF-β/Smad pathway (using commercial inhibitors) could mimic PRP effects regarding CAF’s phenotypic switch.
Treated CAFs showed reduced abilities to induce tumor cell proliferation, migration, and the EMT, processes closely linked to the TGF-β pathway. Our data also suggest a possible molecular link between cancer cells and CAFs via the TGF-β pathway, proposing a potential therapeutic target for both cell types with a single formulation. Moreover, other pro-fibrotic pathways involved in CAF transformation/activation, such as platelet-derived growth factor (PDGF), hedgehog (HH), or connective tissue growth factor (CTGF), may partially contribute to the observed reprogramming effects of PRP treatment72,73,74. For decades, activated fibroblasts were thought to be cancer cells’ accomplices in promoting tumor growth. However, fibroblasts appear to act as involuntary regulators of tumor growth, either positively or negatively, depending on the origin of the fibroblast on one hand, and on the other hand, the context in which it is found, such as the stage of tumor progression. To make thing worse, CAFs exhibit remarkable heterogeneity and plasticity, allowing CAFs from the same tumor to arise through different pathways, have different phenotypes, and have functionally different effects on tumor cells and the microenvironment. Nonetheless, it is clear that controlling, reducing or re-educating the CAFs subpopulation within the tumor is critical to improving current treatments10. This opens up avenues for future studies.
Conclusions
In conclusion, our results suggest that pancreatic (pro)enzymes treatment holds promising therapeutic potential. PRP effectively inhibits a specific subset of CAFs, which are crucial components of the TME, disrupting cellular interactions and impacting tumor progression.
Moreover, our findings indicate that PRP treatment reduces the expression of CAF-expressing proteins while increasing the expression of Meflin, suggesting an anti-fibrotic effect. Furthermore, PRP treatment suppresses tumorigenicity, endothelial cell proliferation, and cancer cell proliferation, contributing to a less aggressive phenotype in PDAC cells. Importantly, PRP does not affect normal stromal fibroblasts at concentrations effective against cancer-associated cells, supporting its potential as an alternative or adjunct therapy.
Additionally, PRP treatment leads to a reprogramming of CAFs, shifting them towards a less pro-tumoral phenotype rather than eliminating them entirely. This reprogramming effect is accompanied by a reduction in the ability of CAFs to influence other stromal cell types towards a pro-tumorigenic phenotype.
Lastly, our study suggests that PRP may down-regulate the TGF-β/Smad signaling axis in CAFs, a pathway known to be over-expressed in CAFs and implicated in PDAC progression. While the exact molecular mechanism requires further investigation, PRP’s partial inhibition of this pathway appears to contribute to its anti-tumorigenic effects.
In conclusion, our preclinical data underscore the potential of PRP as a therapeutic strategy for PDAC, elucidating its mechanisms of action and its influence on the tumor microenvironment, including both tumor-stroma and stromal-stromal interactions. These results offer important insights into the use of PRP as a multifaceted approach to disrupting tumor-supporting structures, highlighting the need for further in vivo studies to evaluate its effectiveness and to better understand its underlying biological mechanisms. Further research is warranted to explore the clinical translation of these findings and to elucidate the full therapeutic potential of PRP in PDAC treatment.

Indirect Co-culture of Monolayer BxPC3 or BxPC3-CSCs with Fibroblasts generates Cancer-Associated Fibroblasts. (A) Representative images of phase-contrast micrograph of the bottom of the dish after removing the chamber containing BxPC3 grown in monolayer or BxPC3-derived CSCs. CAFs displayed a larger and more star-shaped morphology (white arrow), contrasting with the elongated and thinner shape of NAFs (white arrow). (Original magnification 40x). Scale bar represents 500 μm. (B-D) Representative images of immunofluorescence staining with αSMA (B, green) and FAPα (C, red) antibodies. Nuclei were counterstained with DAPI (D, blue). (Original magnification 10x). Scale bar represents 247 μm. (E) Quantification of fluorescence intensity normalized by cell nuclei. Error bars indicate ± SD. Comparison with NAFs: ***P < 0.001, ns = no significance. (F) RT-qPCR analysis of CAFs-related genes. Error bars indicate ± SD. Comparison with NAFs: **P < 0.01, ***P < 0.001. The assays were conducted in at least three independent experiments.

PRP interferes with the malignancy process of BxPC3-CSCs cells over fibroblasts. (A) Indirect co-culture (transwell-system) of NAFs and BxPC3-CSCs cells. Representative immunofluorescence images of αSMA (green) and FAPα (red) expression in normal and cancer-associated fibroblasts without and with PRP treatment before, during, and after the indirect co-culture. Nuclei were counterstained with DAPI (blue). (Original magnification: 20x). Scale bar represents 123.7 μm. (B) Quantification of fluorescence intensity normalized by cell nuclei. Error bars indicate ± SD. Comparison with NAFs: ***P < 0.001, ns = no significance. Comparison with NAFs/BxPC3 CSCs: $$P < 0.01, $$$P < 0.001, ns = no significance. (C) RT-qPCR analysis of FAPα, FSP1, and SDF1 genes expression in NAFs and CAFs without and with PRP treatment before, during, and after the indirect co-culture. Error bars indicate ± SD. Comparison with NAFs: ***P < 0.001, ns = no significance. Comparison with NAFs/BxPC3 CSCs: $P < 0.05, $$P < 0.01, $$$P < 0.001. The assays were conducted in at least three independent experiments.

CAFs established after direct co-culture undergo a significant impairment in their CAF-like phenotype upon PRP addition. (A) Direct co-culture (in a well) of NAFs and BxPC3-CSCs cells. Representative immunofluorescence images of αSMA (green) and FAPα (red) expression in cancer-associated fibroblasts without and with PRP treatment before, during, and after the direct co-culture. Nuclei were counterstained with DAPI (blue). (Original magnification: 10x). Scale bar represents 500 μm. (B) Quantification of fluorescence intensity normalized by cell nuclei. Error bars indicate ± SD. Comparison with NAFs/BxPC3 CSCs: ***P < 0.001, ns = no significance. (C) Direct co-culture (pellet formation in a tube): tubes 1–2: controls with no treatment; tubes 3–4: PRP treatment consisting in two doses (T/C 0.07/0.42 mg/mL) every 72-hour; tubes 5–6: PRP treatment consisting in four doses (T/C 0.035/0.21 mg/mL) every 48 h. (D) Graph displaying cell count in the 3D direct co-culture assays. Proliferation was measured before the addition of new treatment with fresh medium. Comparison with proliferation rate in pellets 1–2: *P < 0.05, **P < 0.01. Comparison with proliferation rate in pellets 5–6: $P < 0.05. The experiments were conducted in duplicate. Error bars indicate ± SD.

Tumorigenic Capacity of CAFs over Tumor Cells. (A) Expression of EMT related genes in disaggregated BxPC3-CSCs cells after co-culture with or without CAFs and with or without PRP treatment. Comparison with BxPC3 CSCs: *P < 0.05, **P < 0.01, ***P < 0.001, ns = no significance. The experiment was conducted in triplicate. Error bars represent ± SD. (B) Quantification of cell migration distances at 0, 12, 24, 48, and 72 h after scratching by wound healing assay of BxPC3-CSCs cells grown in normal Bx medium (NM), NAFs conditioned medium (NAF CM), and CAFs conditioned medium (CAF CM) without and with PRP treatment. Original magnification 4x. Images analyzed with ImageJTM software. Scale bar represents 100 μm. (C) Percentage of cell migration determined by the rate of cells moving towards the scratched area over time using ImageJTM software. Comparison with NM: **P < 0.01, ***P < 0.001. Comparison with NAF CM: $$$P < 0.001. Comparison with CAF CM: ## P < 0.01. Data represent mean ± SD of three independent experiments. Statistical significance was determined by one-way ANOVA followed by Tukey’s post hoc test to compare conditions at each time point. (D) BxPC3-CSCs colony formation assay by crystal violet staining at 7 and 21 days after seeding in NM, NAF CM, and CAF CM without or with PRP treatment. (E) Percentage of clonogenicity rate determined by the rate of stained cells at 7 and 21 days using ImageJTM software. Data represent mean ± SD of three independent experiments. Statistical significance was determined by one-way ANOVA followed by Tukey’s port hoc text. Comparison with NM: *P < 0.05, **P < 0.01, ns = no significance. Comparison with NAF CM: $P < 0.05, $$P < 0.01. Comparison with CAF CM: #P < 0.05, ##P < 0.01. (F) Alamar blue cell viability and proliferation assay in BxPC3-CSCs under the same conditions as in (B) and (D). The graph illustrates the fluorescence levels of the redox indicator throughout 21 days after seeding, as a complementary study of D. Comparison with NM: *P < 0.05, **P < 0.01, ***P < 0.001. Comparison with NAF CM: $$P < 0.01. Comparison with CAF CM: ##P < 0.01. Results are expressed as mean cell number (x10^3) ± SD of three independent experiments. Statistical significance was determined by one-way ANOVA followed by Tukey’s post hoc test to compare conditions at each time point. Error bars represent ± SD.

Tumorigenic capacity of CAFs over normal cells. (A) RT-qPCR determination of the expression of the genes: FAPα, SDF-1, IL-1, IL-6, CXCL8 and EGR2 in control NAFs, CAFS, NAFs grown in CAF CM (CEFs) and NAFs grown in CAF + PRP CM (CEFs + PRP). Comparison with NAFs: *P < 0.05, **P < 0.01, ns = no significance. Comparison with CEFs: $P < 0.05, $$P < 0.01, $$$P < 0.001, ns = no significance. (B) Representative images of VE-cadherin and CD31 immunofluorescence labeling in HUVECs after 14 days of different co-culture conditions with or without PRP addition. (Original magnification: 20x). Scale bar represents 123 μm. (C) Quantification of fluorescence intensity normalized by cell nuclei. Comparison with HUVECs: *P < 0.05, **P < 0.01, ns = no significance. Comparison with HUVECs + NAFs: $$P < 0.01, $$$P < 0.001. Comparison with HUVECs + CAFs: ##P < 0.01. The assays were repeated in at least three independent experiments. Error bars indicate ± SD.

PRP induces expression of Meflin. (A) Representative immunofluorescence images depicting Meflin expression (in red) corresponding to NAFs, CAFs and CAFs before, during, and after PRP treatment. Nuclei were counterstained with DAPI (in blue) (Original magnification: 40x). Scale bar represents 247 μm. (B) Quantification of fluorescence intensity normalized by cell nuclei. Comparison with NAFs: ***P < 0.001, ns = no significance. Comparison with CAFs: $$P < 0.01, $$$P < 0.001. (C) RT-qPCR analysis of Meflin gene expression in NAFs, CAFs and CAFs before, during, and after PRP treatment. Comparison with NAFs: **P < 0.01, ns = no significance. Comparison with CAFs: $$$P < 0.001. The experiments were conducted in triplicate. Error bars indicate ± SD.

PRP modulates fibroblast phenotypic alteration by interfering with the TGF-β signaling pathway. (A) RT-qPCR analysis of Smad2 and Smad3 gene expression in NAFs, CAFs, and CAFs treated with PRP. Comparison with NAFs: **P < 0.01, ***P < 0.001. Comparison with CAFs: $$$P < 0.001. (B) Representative immunofluorescence images of Smad2/3 expression (green) in NAFs, CAFs, and CAFs treated with PRP. Nuclei were counterstained with DAPI (blue). (Original magnification: 40x). Scale bar represents 123 μm. (C) Quantification of fluorescence intensity normalized by cell nuclei. Comparison with NAFs: ***P < 0.001. Comparison with CAFs: $$$P < 0.001. (D) Western blot analysis of Smad2/3 and phosphorylated Smad2/3 (P-Smad2/3) in NAFs, CAFs, and CAFs treated with PRP. β-actin was used as an internal control. (E) The relative expression of Smad2/3 and P-Smad2/3 normalized to β-actin was quantified using Image-J software NIH. Comparison with NAFs: ***P < 0.001. Comparison with CAFs: $P < 0.05, $$P < 0.01. (F-J) Experiments were repeated treating CAFs with the pharmacological inhibitor Galunisertib (TGFβR1 inhibitor). F) RT-qPCR analysis of Smad2 and Smad3 gene expression in NAFs, CAFs, and CAFs treated with TGFβR1 inhibitor. Comparison with NAFs: **P < 0.01, ***P < 0.001. Comparison with CAFs: $$$P < 0.001. (G) Representative immunofluorescence images of Smad2/3 expression (green) in NAFs, CAFs, and CAFs treated with TGFβR1 inhibitor. Nuclei were counterstained with DAPI (blue). (Original magnification: 40x). Scale bar represents 123 μm. (H) Quantification of fluorescence intensity normalized by cell nuclei. Comparison with NAFs: ***P < 0.001. Comparison with CAFs: $$$P < 0.001. (I) Western blot analysis of Smad2/3 and phosphorylated Smad2/3 (P-Smad2/3) in NAFs, CAFs, and CAFs treated with TGFβR1 inhibitor. β-actin was used as an internal control. (J) The relative expression of Smad2/3 and P-Smad2/3 normalized to β-actin was quantified using Image-J software NIH. Comparison with NAFs: ***P < 0.001. Comparison with CAFs: $P < 0.05, $$P < 0.01. The assays were repeated in at least three independent batches. Error bars indicate ± SD.

PRP increases late apoptosis and necrosis in CAFs. (A) Representative images from flow cytometry after annexin V-fluorescein staining of NAFs and CAFs with or without PRP treatment showing the percentage of cells in each state. (B) Comparison with NAFs: *P < 0.05, ***P < 0.001, ns = no significance. Comparison with CAFs: $$P < 0.01, $$$P < 0.001. The assays were repeated in at least three independent batches. Error bars indicate ± SD. (C) Table showing the obtained BAX/Bcl-2 protein expression ratio for each experimental condition.

Xenografts in vivo study. The in vivo study was designed to determine the antitumor activity of PRP against CAFs within the TME of pancreatic CSCs-induced tumors. BxPC3-CSCs were disaggregated before injection into the flanks of nude mice. The mice were divided into three groups: 1 st group: control mice; 2nd group: mice pre-treated before tumor induction; and 3rd group: mice treated before and after tumor induction. (A) Scheme of the in vivo study design to determine anti-tumor activity of PRP. (B-E) Representative images of tumor sections staining with the antibodies αSMA (green), FAPα (red), Smad2/3 (green) and Meflin (red) (Original magnification: 20x for αSMA, FAPα, Meflin and 40x for Smad2/3). Arrows highlight SMAD2/3 immunolabeling localized in fibroblasts. Scale bar represents 123 μm. (F) Quantification of fluorescence intensity, normalized to nuclear staining, was performed using ImageJ and restricted exclusively to the stromal compartment. Tumor cell islets were excluded from the analysis to ensure stromal specificity. Comparison with 1 st Group: *P < 0.05, ***P < 0.001, ns = no significance. Statistical significance was calculated for each marker separately among groups. Error bars indicate ± SD.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Abbreviations
- αSMA:
-
Alpha Smooth Muscle Actin
- CAFs:
-
Cancer-associated Fibroblasts
- CCL:
-
Chemokine (C-C motif) Ligand
- CEFs:
-
CAF-educated Fibroblasts
- CM:
-
Conditioned Medium
- CSCs:
-
Cancer Stem Cells
- Ct:
-
Threshold Cycle
- CTGF:
-
Connective Tissue Growth Factor
- CTLA4:
-
Cytotoxic T Lymphocyte associated Protein-4
- CTLs:
-
Cytotoxic T Lymphocytes
- CXCL:
-
Chemokine (C-X-C motif) Ligand
- DAPI:
-
4’,6-diamidino-2-phenylindole
- DCs:
-
Dendritic Cells
- DNA:
-
Deoxyribonucleic Acid
- ECM:
-
Extracellular Matrix
- EMT:
-
Epithelial-to-mesenchymal Transition
- FACS:
-
Fluorescence-activated Cell Sorting
- FAK:
-
Focal Adhesion Kinase
- FAPα:
-
Fibroblast Activation Protein-α
- FBS:
-
Fetal Bovine Serum
- FSP-1:
-
Fibroblast-specific Protein 1
- GEM:
-
Gemcitabine
- HH:
-
Hedgehog
- HUVECs:
-
Human Umbilical Vein-derived Endothelial Cells
- IF:
-
Immunofluorescence
- IL:
-
Interleukin
- JAK/STAT:
-
Janus kinase-Signal Transducer and Activator of Transcription
- MCs:
-
Mast Cells
- MDSCs:
-
Myeloid-derived Suppressor Cells
- MMPs:
-
Metalloproteinases
- MSCs:
-
Mesenchymal Stem Cells
- MTT:
-
3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide
- NAFs:
-
Normal-associated Fibroblasts
- NK:
-
Natural Killer
- NM:
-
Normal Medium
- PBS:
-
Phosphate-buffered Saline
- PD-1:
-
Death Protein 1
- PD-L1:
-
Programmed Death Receptor Ligand-1
- PDAC:
-
Pancreatic Ductal Adenocarcinoma
- PDGF:
-
Platelet-derived Growth Factor
- PFA:
-
Paraformaldehyde
- PRP:
-
Chymotrypsinogen and Trypsinogen 6:1 formulation
- QoL:
-
Quality of Life
- rDCs:
-
Regulatory DCs
- RT:
-
Room Temperature
- SDF-1:
-
Stromal Cell-Derived Factor 1
- TAMs:
-
Tumor-associated Macrophages
- TANs:
-
Tumor-associated Neutrophils
- TBST:
-
Tris-buffered Saline with Tween
- TGF-β:
-
Transforming Growth Factor beta
- TME:
-
Tumor Microenvironment
- TN-C:
-
Tenascin-C
- Treg:
-
Regulatory T cells
- VEGF:
-
Vascular Endothelial Growth Factor
References
Ho, W. J., Jaffee, E. M. & Zheng, L. The tumor microenvironment in pancreatic cancer — clinical challenges and opportunities. Nat. Reviews Clin. Oncol. 17 (9), 527–540. https://doi.org/10.1038/s41571-020-0363-5 (2020).
Siegel, R. L., Miller, K. D., Fuchs, H. E., Jemal, A. & Cancer statistics CA: A Cancer Journal for Clinicians. 2022; 72(1):7–33. (2022). https://doi.org/10.3322/caac.21708
Chhoda, A. et al. Late-stage pancreatic cancer detected during high-risk individual surveillance: A systematic review and meta-analysis. Gastroenterology 162 (3), 786–798. https://doi.org/10.1053/j.gastro.2021.11.021 (2022).
Sarantis, P., Koustas, E., Papadimitropoulou, A., Papavassiliou, A. G. & Karamouzis, M. V. Pancreatic ductal adenocarcinoma: treatment hurdles, tumor microenvironment and immunotherapy. World J. Gastrointest. Oncol. 12 (2), 173–181. https://doi.org/10.4251/wjgo.v12.i2.173 (2020).
Rodriguez-Aznar, E., Wiesmüller, L., Sainz, B. & Hermann, P. C. EMT and stemness—key players in pancreatic cancer stem cells. Cancers 11 (8), 1136. https://doi.org/10.3390/cancers11081136 (2019).
Elyada, E. et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 9 (8), 1102–1123. https://doi.org/10.1158/2159-8290.cd-19-0094 (2019).
Bulle, A. & Lim, K-H. Beyond just a tight fortress: contribution of stroma to epithelial-mesenchymal transition in pancreatic cancer. Signal. Transduct. Target. Therapy. 5 (1). https://doi.org/10.1038/s41392-020-00341-1 (2020).
Biffi, G. et al. Il1-induced JAK/STAT signaling is antagonized by TGFΒ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 9 (2), 282–301. https://doi.org/10.1158/2159-8290.cd-18-0710 (2019).
Biffi, G. & Tuveson, D. A. Diversity and biology of cancer-associated fibroblasts. Physiol. Rev. 101 (1), 147–176. https://doi.org/10.1152/physrev.00048.2019 (2021).
Toledo, B., Picon-Ruiz, M., Marchal, J. A. & Perán, M. Dual role of fibroblasts educated by tumor in cancer behavior and therapeutic perspectives. Int. J. Mol. Sci. 23 (24), 15576. https://doi.org/10.3390/ijms232415576 (2022).
Chakkera, M., Foote, J. B., Farran, B. & Nagaraju, G. P. Breaking the stromal barrier in pancreatic cancer: advances and challenges. Biochimica et biophysica acta (BBA) - Reviews on cancer. ;1879(1):189065. (2024). https://doi.org/10.1016/j.bbcan.2023.189065
Cannone, S. et al. Cancer associated fibroblast (CAF) regulation of PDAC parenchymal (CPC) and CSC phenotypes is modulated by ECM composition. Cancers 14 (15), 3737. https://doi.org/10.3390/cancers14153737 (2022).
Liu, X. et al. 3D heterospecies spheroids of pancreatic stroma and cancer cells demonstrate key phenotypes of pancreatic ductal adenocarcinoma. Translational Oncol. 14 (7), 101107. https://doi.org/10.1016/j.tranon.2021.101107 (2021).
Götze, J. et al. Tumor-stroma interaction in PDAC as a new approach for liquid biopsy and its potential clinical implications. Front. Cell. Dev. Biology. 10 https://doi.org/10.3389/fcell.2022.918795 (2022).
Yang, D. et al. Cancer-associated fibroblasts: from basic science to anticancer therapy. Exp. Mol. Med. 55, 1322–1332. https://doi.org/10.1038/s12276-023-01013-0 (2023).
Thomas, D. & Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer. 18 (1). https://doi.org/10.1186/s12943-018-0927-5 (2019).
Kalluri, R. & Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer. 6 (5), 392–401. https://doi.org/10.1038/nrc1877 (2006).
Östman, A. & Augsten, M. Cancer-associated fibroblasts and tumor growth – bystanders turning into key players. Curr. Opin. Genet. Dev. 19 (1), 67–73. https://doi.org/10.1016/j.gde.2009.01.003 (2009).
Sahai, E. et al. A framework for advancing our Understanding of cancer-associated fibroblasts. Nat. Rev. Cancer. 20 (3), 174–186. https://doi.org/10.1038/s41568-019-0238-1 (2020).
Toledo, B., González-Titos, A., Hernández-Camarero, P. & Perán, M. A brief review on chemoresistance; targeting cancer stem cells as an alternative approach. Int. J. Mol. Sci. 24 (5), 4487. https://doi.org/10.3390/ijms24054487 (2023).
Nevala-Plagemann, C., Hidalgo, M. & Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Reviews Clin. Oncol. 17 (2), 108–123. https://doi.org/10.1038/s41571-019-0281-6 (2019).
Hu, C. et al. CIRCFARP1 enables cancer-associated fibroblasts to promote gemcitabine resistance in pancreatic cancer via the LIF/stat3 axis. Mol. Cancer. 21 (1). https://doi.org/10.1186/s12943-022-01501-3 (2022).
Dalin, S. et al. Deoxycytidine release from pancreatic stellate cells promotes gemcitabine resistance. Cancer Res. 79 (22), 5723–5733. https://doi.org/10.1158/0008-5472.CAN-19-0960 (2019).
Kim, H. J. et al. Reprogramming of cancer-associated fibroblasts by apoptotic cancer cells inhibits lung metastasis via notch1-WISP-1 signaling. Cell Mol. Immunol. 19 (12), 1373–1391. https://doi.org/10.1038/s41423-022-00930-w (2022).
Ham, I. H., Lee, D. & Hur, H. Cancer-Associated Fibroblast-Induced resistance to chemotherapy and radiotherapy in Gastrointestinal cancers. Cancers (Basel). 13, 1172. https://doi.org/10.3390/cancers13051172 (2021).
Glabman, R. A., Choyke, P. L. & Sato, N. Cancer-associated fibroblasts: tumorigenicity and targeting for cancer therapy. Cancers 14 (16), 3906. https://doi.org/10.3390/cancers14163906 (2022).
de Sostoa, J. et al. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J. Immunother. Cancer. 7 (1). https://doi.org/10.1186/s40425-019-0505-4 (2019).
González-Titos, A. et al. Trypsinogen and chymotrypsinogen: potent anti-tumor agents. Expert Opin. Biol. Ther. 21 (12), 1609–1621. https://doi.org/10.1080/14712598.2021.1922666 (2021).
Perán, M. et al. A formulation of pancreatic pro-enzymes provides potent anti-tumor efficacy: A pilot study focused on pancreatic and ovarian cancer. Sci. Rep. 7 (1). https://doi.org/10.1038/s41598-017-14571-x (2017).
Burris, H. A. 3rd. et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 15(6), 2403–2413. https://doi.org/10.1200/JCO.1997.15.6.2403 (1997) (PMID: 9196156).
Toledo, B. et al. Treatment resistance in pancreatic and biliary tract cancer: molecular and clinical Pharmacology perspectives. Expert Rev. Clin. Pharmacol. https://doi.org/10.1080/17512433.2024.2319340 (2024).
Fine, R. L. et al. The gemcitabine, docetaxel, and capecitabine (GTX) regimen for metastatic pancreatic cancer: a retrospective analysis. Cancer Chemother. Pharmacol. 61 (1), 167–175 (2008).
Von Hoff, D. D. et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl. J. Med. 369 (18), 1691–1703 (2013).
Leipner, J. & Saller, R. Systemic enzyme therapy in oncology. Drugs 59 (4), 769–780. https://doi.org/10.2165/00003495-200059040-00004 (2000).
Jiménez, G. et al. Mesenchymal stem cell’s secretome promotes selective enrichment of cancer stem-like cells with specific cytogenetic profile. Cancer Lett. 429, 78–88. https://doi.org/10.1016/j.canlet.2018.04.042 (2018).
Peng, S. et al. Metabolomics reveals that CAF-derived lipids promote colorectal cancer peritoneal metastasis by enhancing membrane fluidity. Int. J. Biol. Sci. 18 (5), 1912–1932. https://doi.org/10.7150/ijbs.68484 (2022).
Hernández-Camarero, P. et al. Pancreatic (pro)enzymes treatment suppresses BXPC-3 pancreatic cancer stem cell subpopulation and impairs tumor engrafting. Sci. Rep. 9 (1). https://doi.org/10.1038/s41598-019-47837-7 (2019).
Perán, M., Marchal, J. A., García, M. A., Kenyon, J. & Tosh, D. Vitro treatment of carcinoma cell lines with pancreatic (pro)enzymes suppresses the EMT programme and promotes cell differentiation. Cell. Oncol. 36 (4), 289–301. https://doi.org/10.1007/s13402-013-0134-8 (2013).
Iida, T. et al. Pharmacologic conversion of cancer-associated fibroblasts from a protumor phenotype to an antitumor phenotype improves the sensitivity of pancreatic cancer to chemotherapeutics. Oncogene 41 (19), 2764–2777. https://doi.org/10.1038/s41388-022-02288-9 (2022).
Ligorio, M. et al. Stromal microenvironment shapes the intratumoral architecture of pancreatic cancer. Cell 178 (1). https://doi.org/10.1016/j.cell.2019.05.012 (2019).
Melisi, D. et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer. 119 (10), 1208–1214. https://doi.org/10.1038/s41416-018-0246-z (2018).
Ba, P. et al. Curcumin suppresses the proliferation and tumorigenicity of cal27 by modulating cancer-associated fibroblasts of TSCC. Oral Dis. 26 (7), 1375–1383. https://doi.org/10.1111/odi.13306 (2020).
Sugai, M. et al. Correlation of tumor microenvironment-related markers with clinical outcomes in patients with squamous cell carcinoma of the lung. Translational Lung Cancer Res. 11 (6), 975–990. https://doi.org/10.21037/tlcr-22-10 (2022).
Liu, Y. et al. Fibroblast activation protein targeted therapy using [177lu]fapi-46 compared with [225AC]FAPI-46 in a pancreatic cancer model. Eur. J. Nucl. Med. Mol. Imaging. 49(3), 871–880. https://doi.org/10.1007/s00259-021-05554-2 (2021).
Cao, X-K., Xie, B., Shao, Y. & Lin, J. Cytokine-driven positive feedback loop organizes fibroblast transformation and facilitates gastric cancer progression. Clin. Transl. Oncol. 24 (7), 1354–1364. https://doi.org/10.1007/s12094-022-02777-z (2022).
Hernández-Camarero, P., López-Ruiz, E., Marchal, J. A., Perán, M. & Cancer A mirrored room between tumor bulk and tumor microenvironment. J. Experimental Clin. Cancer Res. 40 (1). https://doi.org/10.1186/s13046-021-02022-5 (2021).
Hu, B. et al. Subpopulations of cancer-associated fibroblasts link the prognosis and metabolic features of pancreatic ductal adenocarcinoma. Annals Translational Med. 10 (5), 262–262. https://doi.org/10.21037/atm-22-407 (2022).
Kim, D. et al. Identification and characterization of cancer-associated fibroblast subpopulations in lung adenocarcinoma. Cancers 14 (14), 3486. https://doi.org/10.3390/cancers14143486 (2022).
Chiavarina, B. et al. Fibroblast-derived prolargin is a tumor suppressor in hepatocellular carcinoma. Oncogene 41 (10), 1410–1420. https://doi.org/10.1038/s41388-021-02171-z (2022).
McAndrews, K. M. et al. Identification of functional heterogeneity of carcinoma-associated fibroblasts with distinct IL6-mediated therapy resistance in pancreatic cancer. Cancer Discov. 12 (6), 1580–1597. https://doi.org/10.1158/2159-8290.cd-20-1484 (2022).
Karamitopoulou, E. et al. Spatially restricted tumor-associated and host-associated immune drivers correlate with the recurrence sites of pancreatic cancer. Gut 72 (8), 1523–1533. https://doi.org/10.1136/gutjnl-2022-329371 (2023).
Akanda, M. R. et al. Different expression and clinical implications of cancer-associated fibroblast (CAF) markers in brain metastases. J. Cancer. 14 (3), 464–479. https://doi.org/10.7150/jca.80115 (2023).
Mizutani, Y. et al. Meflin-positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res. 79 (20), 5367–5381. https://doi.org/10.1158/0008-5472.can-19-0454 (2019).
Miyai, Y. et al. Meflin-positive cancer-associated fibroblasts enhance tumor response to immune checkpoint Blockade. Life Sci. Alliance. 5 (6). https://doi.org/10.26508/lsa.202101230 (2022).
Yoon, H. et al. TGF-β1-mediated transition of resident fibroblasts to cancer-associated fibroblasts promotes cancer metastasis in Gastrointestinal stromal tumor. Oncogenesis 10 (2). https://doi.org/10.1038/s41389-021-00302-5 (2021).
Kunac, N., Filipović, N., Kostić, S. & Vukojević, K. The expression pattern of bcl-2 and Bax in the tumor and stromal cells in colorectal carcinoma. Medicina 58 (8), 1135. https://doi.org/10.3390/medicina58081135 (2022).
Itoh, G. et al. Cancer-associated fibroblasts educate normal fibroblasts to facilitate cancer cell spreading and t-cell suppression. Mol. Oncol. 16 (1), 166–187. https://doi.org/10.1002/1878-0261.13077 (2021).
Timperi, E. et al. Lipid-associated macrophages are induced by cancer-associated fibroblasts and mediate immune suppression in breast cancer. Cancer Res. 82 (18), 3291–3306. https://doi.org/10.1158/0008-5472.can-22-1427 (2022).
Toledo, B. et al. Deciphering the performance of macrophages in tumor microenvironment: a call for precision immunotherapy. J. Hematol. Oncol. 17 (1), 44. https://doi.org/10.1186/s13045-024-01559-0 (2024).
Chen, S. et al. Cancer-associated fibroblast-induced m2-polarized macrophages promote hepatocellular carcinoma progression via the plasminogen activator inhibitor-1 pathway. Int. J. Oncol. 59 (2). https://doi.org/10.3892/ijo.2021.5239 (2021).
Sheng, Y. et al. Cancer-associated fibroblasts exposed to high-dose ionizing radiation promote M2 polarization of macrophages, which induce radiosensitivity in cervical cancer. Cancers 15 (5), 1620. https://doi.org/10.3390/cancers15051620 (2023).
Qi, R. et al. Cancer-associated fibroblasts suppress ferroptosis and induce gemcitabine resistance in pancreatic cancer cells by secreting exosome-derived ACSL4-targeting Mirnas. Drug Resist. Updates. 68, 100960. https://doi.org/10.1016/j.drup.2023.100960 (2023).
Hauge, A. & Rofstad, E. K. Antifibrotic therapy to normalize the tumor microenvironment. J. Translational Med. 18 (1). https://doi.org/10.1186/s12967-020-02376-y (2020).
Dong, G. et al. DDX18 drives tumor immune escape through transcription-activated STAT1 expression in pancreatic cancer. Oncogene 42 (40), 3000–3014. https://doi.org/10.1038/s41388-023-02817-0 (2023).
Whatcott, C. J., Han, H. & Von Hoff, D. D. Orchestrating the tumor microenvironment to improve survival for patients with pancreatic cancer. Cancer J. 21 (4), 299–306. https://doi.org/10.1097/ppo.0000000000000140 (2015).
Jiang, M., Gu, D., Liu, F., Lin, C. & Tian, L. Transforming cancer cells for long-term living with cancer: an inspiring new approach. Chin. J. Cancer Res. 35 (2), 108–125. https://doi.org/10.21147/j.issn.1000-9604.2023.02.03 (2023).
Goehrig, D. et al. Stromal protein βig-H3 reprogrammes tumor microenvironment in pancreatic cancer. Gut 68 (4), 693–707. https://doi.org/10.1136/gutjnl-2018-317570 (2018).
Lencioni, G. et al. Unravelling the complexities of resistance mechanism in pancreatic cancer: insights from in vitro and ex-vivo model systems. Sem. Cancer Biol. 106-107, 217–233. 10.1016:j.semcancer.2024.09.002 (2024).
Lan, Y. et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci. Transl. Med. 10 (424). https://doi.org/10.1126/scitranslmed.aan5488 (2018).
Gungor, M. Z., Uysal, M. & Senturk, S. The bright and the dark side of TGF-β signaling in hepatocellular carcinoma: Mechanisms, dysregulation, and therapeutic implications. Cancers 14 (4), 940. https://doi.org/10.3390/cancers14040940 (2022).
van Pelt, J. et al. Human pancreatic cancer patients with epithelial-to-mesenchymal transition and an aggressive phenotype show a disturbed balance in protein phosphatase type 2A expression and functionality. J. Translational Med. 21 (1). https://doi.org/10.1186/s12967-023-04145-z (2023).
Li, J., Zhi, X., Sun, Y., Chen, M. & Yao, L. The PDGF family is associated with activated tumor stroma and poor prognosis in ovarian cancer. Disease Markers 2022 Sept 262022:1–19. https://doi.org/10.1155/2022/5940049
Eibenschutz, L. et al. Evaluation of Hedgehog pathway Inhibition on nevoid basal cell carcinoma syndrome fibroblasts and basal cell carcinoma-associated fibroblasts: are vismodegib and sonidegib useful to target cancer-prone fibroblasts? Cancers 13 (22), 5858. https://doi.org/10.3390/cancers13225858 (2021).
Sang, Q. et al. Identification of prognostic gene expression signatures based on the tumor microenvironment characterization of gastric cancer. Front. Immunol. 13 https://doi.org/10.3389/fimmu.2022.983632 (2022).
Acknowledgements
Propanc Biopharma Inc. has funded the University of Jaen to carry out this research. Graphical abstract and Figure 9 A were created with biorender.com.
Funding
This work has been funded by Propanc Biopharma Inc.; by the Consejería de Transformación Económica, Industria, Conocimiento y Universidades, Junta de Andalucía, Modeling Nature (MNat, project number QUAL21-11) and the Chair “Doctors Galera-Requena in cancer stem cell research” (CMC-CTS963). BT was supported by “Youth Guarantee Program” ref: SNGJ_PTA4_M_008 Junta de Andalucía. BT was partly supported by the Short-Term Scientific Mission (STSM) grant of the COST Action CA21116 Transpan.
Author information
Authors and Affiliations
Contributions
MP and BT: conception and design of the study; BT, AT and PHC: collection, acquisition, analysis and data interpretation, methodology; CG, EL, AT and BT: *in vivo* study; BT, PHC and MP writing, drafting and editing the article. MP and JAM: funding acquisition. MP, EG and JAM: supervision, article review and final approval of submitted manuscript. All authors: final approval of submitted manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.


Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

{kind=link}
{kind=link}
Cite this article
Toledo, B., González-Titos, A., Hernández-Camarero, P. et al. Impact of pancreatic proenzymes on pancreatic ductal adenocarcinoma associated fibroblasts. Sci Rep 15, 43750 (2025). https://doi.org/10.1038/s41598-025-24863-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-24863-2
- Springer Nature Limited