
Abstract
Deep learning models have been developed for various predictions in glioma; yet, they were constrained by manual segmentation, task-specific design, or a lack of biological interpretation. Herein, we aimed to develop an end-to-end multi-task deep learning (MDL) pipeline that can simultaneously predict molecular alterations and histological grade (auxiliary tasks), as well as prognosis (primary task) in gliomas. Further, we aimed to provide the biological mechanisms underlying the model’s predictions. We collected multiscale data including baseline MRI images from 2776 glioma patients across two private (FAHZU and HPPH, n = 1931) and three public datasets (TCGA, n = 213; UCSF, n = 410; and EGD, n = 222). We trained and internally validated the MDL model using our private datasets, and externally validated it using the three public datasets. We used the model-predicted deep prognosis score (DPS) to stratify patients into low-DPS and high-DPS subtypes. Additionally, a radio-multiomics analysis was conducted to elucidate the biological basis of the DPS. In the external validation cohorts, the MDL model achieved average areas under the curve of 0.892–0.903, 0.710–0.894, and 0.850–0.879 for predicting IDH mutation status, 1p/19q co-deletion status, and tumor grade, respectively. Moreover, the MDL model yielded a C-index of 0.723 in the TCGA and 0.671 in the UCSF for the prediction of overall survival. The DPS exhibits significant correlations with activated oncogenic pathways, immune infiltration patterns, specific protein expression, DNA methylation, tumor mutation burden, and tumor-stroma ratio. Accordingly, our work presents an accurate and biologically meaningful tool for predicting molecular subtypes, tumor grade, and survival outcomes in gliomas, which provides personalized clinical decision-making in a global and non-invasive manner.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
Glioma, a highly prevalent brain tumor, ranks as the third leading cause of cancer-related deaths worldwide1. The fifth edition of the WHO Classification of Tumors of the Central Nervous System (CNS) published in 2021 introduced significant changes to the classification of CNS tumors2. This edition incorporates both histopathological and genotypic characteristics to facilitate an “integrated” diagnosis2. The therapeutic efficacy and prognosis of gliomas heavily rely on their genotypic and histopathological profiles, with particular emphasis on factors such as the IDH mutation status, 1p/19q co-deletion status, and histological grade3. However, accurate detection of this information typically necessitates invasive procedures such as biopsy or surgery4. Therefore, non-invasive evaluation of patient prognosis guided by genotypic and histopathological biomarkers holds considerable promise for personalized postoperative treatment strategies.
Radiological imaging provides a global characterization of tumors and is routinely employed for diagnosis, staging, response evaluation, and patient monitoring in cancer care. The advent of artificial intelligence (AI), encompassing machine learning and deep learning, has revolutionized the analysis of imaging data, uncovering disease-specific patterns often with accuracy rivaling or surpassing human experts5,6. The integration of AI into medical practice has opened new possibilities for improving the diagnosis and treatment of gliomas. Despite this progress, many prior studies have adopted narrow, task-specific models that lack a comprehensive grasp of inter-task relationships7. This narrow focus can undermine the efficacy of these approaches8. Multitask deep learning (MDL), in contrast, offers numerous benefits including shared feature representations, synergistic learning, improved generalization, reduced computational demands, and higher data efficiency when simultaneously addressing multiple related tasks9.
Deep learning models, primarily driven by data, often obscure the mechanistic rationale behind their predictions from practitioners. This disconnection from underlying biological processes results in models that are not interpretable, significantly hindering their clinical applicability10. Revealing the biological basis underlying predictive models not only strengthens clinical decision-making but also lays a foundation for more profound investigations11. For instance, radiogenomic analysis bridges the gap between mesoscopic radiomic or deep features and microscopic molecular features, such as somatic mutation and gene expression profiles12. This approach lends biological relevance to previously abstract computational imaging features13.
In this current study, we aimed to develop and validate an multi-task deep learning (MDL) framework. This framework aims to concurrently predict molecular alterations (IDH mutation and 1p/19q co-deletion status) and histological grade (three auxiliary tasks), along with prognosis (primary task), from radiological images. Additionally, we investigated the biological foundations of the deep features and the Deep Prognosis Score (DPS) generated by the MDL model, employing multi-omics analysis, encompassing pathomics, transcriptomics, and proteomics.
Results
Overview of study design and patient characteristics
The study design is outlined in Fig. 1. We trained an MDL model utilizing MRI images to classify molecular alterations, and histological grade, and predict patient prognosis. This integrative approach, combining histopathology and prognosis prediction, is predicated on their closely related nature, as evidenced by established mechanistic relationships. To elucidate the biological basis of the MDL model, especially focusing on the prognosis prediction task, we conducted multi-omic analyses on the The Cancer Genome Atlas (TCGA) dataset14.

FAHZU dataset: patients from The First Affiliated Hospital of Zhengzhou University; HNPH dataset: patients from Henan Provincial People’s Hospital; TCGA dataset: patients from The Cancer Genome Atlas; UCSF dataset: patients from the University of California San Francisco; EGD dataset: patients from The Erasmus Glioma Database. MDL multi-task deep learning, DPS deep prognosis score, ROC receiver operator characteristic, WSI whole slide image, PRO protein, CNV copy number variation, MET Methylation. FAHZU hospital and MRI scanner icons were sourced from Vecteezy.com. HNPH hospital, database, and computer icons were sourced from Freepik. Sequencing instrument icon was sourced from BioRender.com.
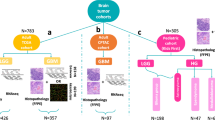
Figure 2 depicts the patient inclusion pathway and outlines the cohorts designated for each task. In total, 2776 patients were included for predicting IDH mutation status, 1p/19q co-deletion status, and histological grade, while 2242 independent patients were selected for the prognosis prediction task. Patient and tumor characteristics for both the training and validation cohorts are presented in the Table 1.

FAHZU dataset: patients from The First Affiliated Hospital of Zhengzhou University; HNPH dataset: patients from Henan Provincial People’s Hospital; TCGA dataset: patients from The Cancer Genome Atlas; UCSF dataset: patients from the University of California San Francisco; EGD dataset: patients from The Erasmus Glioma Database. Abbreviations: WSI whole slide image, PRO protein, CNV copy number variation, MET Methylation.
Quantitative evaluation of auto-segmentation accuracy
The MRI images from 2916 patients (Training Cohort [TC], n = 1352; Internal Validation Cohort [IVC], n = 579; External Validation Cohort 1 [EVC1], n = 242; External Validation Cohort 2 [EVC2], n = 500; and External Validation Cohort 1 [EVC3], n = 243) were automatically segmented using the pre-trained mmFormer15. As shown in Supplementary Fig. 1, the mmFormer demonstrated high segmentation performance with median Dice coefficients of 0.828 (95% confidence interval [CI]: 0.820–0.836) and 0.828 (95% CI: 0.815–0.837) in the TC and IVC, 0.920 (95% CI: 0.912–0.926) in the EVC1, 0.883 (95% CI: 0.873–0.891) in the EVC2, and 0.833 (95% CI: 0.810–0.847) in the ECV3. The findings underscore the robustness and accuracy of mmFormer in automatically segmenting glioma lesions across diverse patient cohorts. To illustrate the advantage of mmFormer, we conducted comparative studies by benchmarking it against other previously published segmentation methods, including U-HeMIS16, U-HVED17, and 3D-UNET18,19. Overall, the mmformer demonstrates better segmentation performance (Supplementary Table 4), indicating the relatively high accuracy of the segmentation algorithm compared to human annotation.
MDL model predicts molecular alterations and histological grade
Overall, the proposed deep learning model achieved high accuracy in predicting IDH mutation status, 1p/19q co-deletion status, and histological grade in the TC (Table 2 and Fig. 3a–c). In the IVC, the model demonstrated notable accuracy, with area under receiver operating characteristic curve (AUC) values of 0.872 for IDH mutation status prediction, 0.845 for 1p/19q co-deletion status prediction, and 0.842 for histological grade prediction. Moreover, the model exhibited similarly high levels of discriminative performance in the EVCs 1, 2, and 3. Specifically, it achieves AUCs ranging from 0.892 to 0.903 for IDH status, 0.710–0.894 for 1p/19q status, and 0.850–0.879 for histological grade prediction, as depicted in Table 2 and Fig. 3a–c. In addition, the corresponding confusion matrices (Fig. 3d–f) demonstrated good concordance between the model’s predictions and the actual diagnostic categories for IDH mutation, 1p/19q co-deletion, and histological grade.

a–c Receiver operating characteristic (ROC) curves for the prediction of IDH mutation status, 1p/19q co-deletion status, and histological grade, respectively. d–f Corresponding confusion matrices show the pairwise comparison; diagonal: number cases of correctly classified; off-diagonal: number of cases incorrectly classified.
MDL model predicts prognosis outcomes
In the primary task of prognosis prediction, the MDL model demonstrated significant efficacy. It achieved a C-index of 0.712 (95% CI: 0.689–0.734) in the TC, 0.704 (95% CI: 0.671–0.737) in the IVC, 0.723 (95% CI: 0.670–0.777) in the EVC1, and 0.671 (95% CI: 0.629–0.712) in the EVC2. The model successfully categorized patients into low- and high- DPS subtypes using the optimal cutoff point of -0.49 for the DPS in the TC. Supplementary Figure 2 displays the differences in molecular pathology and deep prognosis features between patients with low-DPS and high-DPS subtypes. This stratification of DPS revealed significant differences in overall survival (OS) across the four cohorts (Fig. 4a). Additionally, notable differences in progression-free survival (PFS) between these subtypes were observed, with all P-values being <0.001 in the TC, IVC, and EVC1 (Supplementary Fig. 3). The calibration curves (Fig. 4b) demonstrate the model’s well-calibrated performance across all cohorts. Additionally, the receiver operating characteristic (ROC) curves reflect the model’s good sensitivity and specificity in predicting OS (Fig. 4c).

a Kaplan–Meier curves show the overall survival (OS) of patients in the low-DPS and high-DPS subtypes. The OS of patients in the high-DPS group was significantly shorter compared to the low-DPS group in the training and validation cohorts. b Calibration curves depict the agreement between the actual and predicted survival probabilities. c Time-dependent receiver operating characteristic curves at the given points of 1 year, 3 year, and 5 year. DPS, deep prognosis score.
Supplementary Figure 4 shows the MRI images and corresponding feature maps along with the predicted molecular alterations, histological grade, and DPS for four representative cases. Visually the feature maps appear that in patients with a good prognosis, the model mainly focuses on the tumor core, while in patients with a poor prognosis, it mainly focuses on the edema area of the tumor.
Furthermore, the prognostic value of the DPS was assessed in each patient subgroup defined by similar clinicopathologic risk factors, such as IDH mutation status, 1p/19q co-deletion status, and histological grade. DPS successfully provided additional stratification for OS in all these subgroups (Supplementary Fig. 5).
Comparison of multi-task model with single-task models
We developed four independent single-task models for predicting IDH status, 1p/19q status, grade, and OS, respectively. Then, we compared the performance of the multi-task model with that of the single-task models. The results showed that the accuracy of the multi-task model was marginally better than that of the single-task model in predicting 1p/19q status (Supplementary Table 5). The C-index for predicting OS using single-task model was 0.705 (95% CI: 0.654-0.752), 0.693 (95% CI: 0.615-0.762), 0.686 (95% CI: 0.554-0.794), and 0.678 (95% CI: 0.586-0.758) in the TC, IVC, EVC1, and EVC2, respectively. These values were lower than those obtained with the multi-task model. However, the multi-task model did not demonstrate obvious advantages in predicting IDH status and grade compared to the single-task model (Supplementary Table 5).
Pathomics analysis
The tumor-stroma ratio (TSR), which is intrinsically related to tumor invasion and metastasis, has gained increasing attention as an independent predictor of tumor prognosis20. In our analysis, we found a significantly lower TSR in the high-DPS subtype compared to the low-DPS subtype (p < 0.01) (Supplementary Fig. 6). However, similar distinctions were not evident in the tumor-necrosis and tumor-inflammation ratios (Supplementary Fig. 6).
Functional annotations and pathway enrichment analyses
In the TCGA cohort, a differential expression analysis identified 309 differentially expressed genes, highlighting significant variations in gene expression between the low- and high-DPS subtypes (Supplementary Data 1).
To delve into the biological significance of these prognostic subtypes, Gene Set Enrichment Analysis (GSEA) was performed (Fig. 5b and Supplementary Data 2). This analysis revealed the activation of various pathways such as epithelial-mesenchymal transition (EMT), interferon alpha response, interferon-gamma response, angiogenesis, E2F targets, IL6-JAK-STAT3 signaling, inflammatory response, and hypoxia. In contrast, the KRAS signaling pathway appeared suppressed (Fig. 5c). To establish a relationship between the 16 prognostic deep features and biological functions, the enrichment score of each biological function was calculated for every patient in the TCGA cohort using single-sample gene set enrichment analysis (ssGSEA)(Supplementary Data 3). A significant correlation was observed between the deep features and biological functions such as EMT, coagulation, and angiogenesis (Fig. 5d and Supplementary Data 4).

a Cohort information in the EVC1, which provides information about the matched multi-omics data in the EVC1 radio-multi-omics cohort. Each column represents a case and a colored column indicates a case with corresponding multi-omics data. b The top enriched pathways are ordered by the normalized enrichment score in the high-DPS subtype (in red) and low-DPS subtype (in blue). c Gene set enrichment analysis shows the significantly enriched pathways with different colors. d Bubble plots show the correlation between prognostic deep features and tumor biological processes. DPS deep prognosis score, EVC1 external validation cohort 1, MRI Magnetic resonance imaging, WSI whole slide imaging, CNV copy number variation.
Immune infiltrating patterns
Using the “ESTIMATE” package, we inferred the stromal score, immune score, estimate score, and tumor purity of glioma specimens (Supplementary Data 5). Notably, the stromal cell score, immune score, and estimate score were significantly higher in the high-DPS subtype than in the low-DPS subtype. This suggests that patients in the high-DPS subtype displayed lower tumor purity. Correspondingly, high-DPS was associated with increased stromal and immune scores, but decreased glioma purity, indicating a higher level of stromal and immune cells in the tumor microenvironment (TME) of high-DPS tumors (Fig. 6a).

a The immune score and estimate score calculated by ESTIMATE were significantly correlated with DPS. b The infiltrating levels of 28 subpopulations of immune cells in high- and low-DPS subtypes based on ssGSEA. c Cell abundance scores were calculated with ssGSEA and compared between the two subtypes. Statistical significance was denoted as ***p < 0.001; **p < 0.01; *p < 0.05. d The heatmap displays the β values for the top 44 CpG sites with the highest DNA methylation variability. e Box plot shows the distribution of TCGA well-defined subtypes based on DNA methylation in the DPS subgroups. f The average methylation levels differed between DPS subgroups. g Violin diagram shows the difference in TMB between DPS subgroups. DPS deep prognosis score, TMB tumor mutation burden, ssGSEA single-sample gene set enrichment analysis.
Subsequently, we employed ssGSEA to explore disparities in immune cell enrichment and immune-related pathway activity between the two prognostic subtypes within the TCGA cohort (Supplementary Data 6). Specifically, we compared the enrichment scores of immune cells and the activities of immune-related pathways. This comparison revealed significant differences in 25 out of 28 immune cell types between the two subtypes. The high-DPS subtype exhibited notably higher levels of immune cell infiltration, particularly in activated CD4 + T cells, activated dendritic cells, CD56 bright natural killer cells, central memory CD4 + T cells, central memory CD8 + T cells, effector memory CD8 + T cells, gamma delta T cells, immature B cells, myeloid-derived suppressor cells, memory B cells, natural killer cells, natural killer T cells, plasmacytoid dendritic cells, T follicular helper cells, type 1 T helper cells, and type 2 T helper cells (Fig. 6b, c).
Distinct epigenetic features
In our investigation of epigenetic alterations, differential analysis was performed using the R package limma 3.44.3. This analysis identified 6,045 differentially methylated CpG sites between the two prognostic subtypes (Supplementary Data 7). Among these, 44 CpG sites showed a logFC > 0.47 and p < 0.0001 (Fig. 6d). The TCGA cohort has classified diffuse gliomas into six distinct methylation clusters (LGm1 to LGm6)21. In our study, the high-DPS subtype was predominantly enriched in the clusters LGm4 to LGm6, which are characterized by genome-wide hypo-methylation. Conversely, clusters LGm1 to LGm3 were more prevalent in the low-DPS subtype (Fig. 6e). A significant difference in DNA methylation levels was observed between the low-DPS and high-DPS subtypes (Fig. 6f and Supplementary Data 8).
Genomic and transcriptomic alterations
To elucidate the somatic mutation landscape within the two risk subtypes, we analyzed the top 20 frequent mutations for both subtypes, revealing distinct mutational landscapes (Supplementary Fig. 7A, B). Mutual exclusion and cooperation analysis among mutated genes indicated a higher frequency of mutation co-occurrence in the low-DPS subtype (Supplementary Fig. 7C, D). Additionally, significant mutual exclusion of mutations such as IDH-PTEN, IDH-EGFR, TP53-CIC, and CIC-ATRX was observed in the low-DPS subtype (Supplementary Fig. 7D).
DNA copy number aberration frequency plots for the distinct prognostic subtypes revealed that high-DPS tumors exhibit more significant changes, including more frequent gains in chromosome 7 and more frequent losses in chromosome 10 (Supplementary Fig. 7E, F).
Furthermore, the tumor mutational burden (TMB) has been recognized as a predictor of patient response to immune checkpoint blockade in various types of cancer22. We computed the TMB for each glioma specimen and observed a positive association between the DPS and mutation burden (Fig. 6g).
Differentially expressed proteins
Our analysis of Reverse Phase Protein Assay(RPPA) data from the TCGA dataset indicated distinct protein expression patterns between the prognostic subtypes (Supplementary Data 9). Significantly higher levels of epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) were observed in the high-DPS subtype. Conversely, proteins such as extracellular signal-regulated kinase 2 (ERK2) were found to be more highly expressed in the low-DPS subtype (Supplementary Fig. 7G).
OncoPredict for targeted drug sensitivity analysis
To explore potential therapeutic strategies, we used the OncoPredict package, which predicts the sensitivity scores of targeted drugs based on their IC50 values (Supplementary Data 10). In the high-DPS subtype, Selumetinib (a MEK1/2 inhibitor) and ULK1_4989 (a ULK1 inhibitor) were predicted to be effective. For the low-DPS subtype, promising candidates for targeted therapies included Lapatinib (a tyrosine kinase inhibitor), NVP-ADW742/Linsitinib (an IGF-1R inhibitor), and Vorinostat (an HDAC inhibitor) (Supplementary Fig. 8).
Discussion
In this study, we successfully developed and validated an MDL pipeline. This pipeline uniquely enables concurrent prediction of molecular profiling, histological grade (auxiliary tasks), and prognosis (primary task) using diagnostic MRI images, demonstrating stable and accurate performance. The model was extensively validated in international multi-center cohorts, encompassing 2776 patients. Remarkably, this model is capable of identifying two distinct prognostic subtypes prior to therapy. This identification holds the potential to guide personalized decision-making and post-surgery surveillance. Further, through in-depth multi-omic data analyses, we demonstrated that the imaging model-derived DPS was highly correlated significantly with several key biological aspects. These include activated oncogenic pathways, immune infiltrating patterns, specific protein expression, methylation level, TMB, and TSR. Moreover, our study identified potential targeted drugs tailored for patients classified as low-DPS and high-DPS.
Our work builds upon extensive evidence supporting the crucial role of IDH mutation, 1p/19q co-deletion status, and histological grade in prognosis evaluation and therapy planning23,24,25. Typically, acquiring these biomarkers necessitates invasive procedures such as biopsy or surgery, which may be prone to sampling errors. In contrast, radiological imaging offers a noninvasive, global assessment of the entire tumor, enabling repeated acquisitions during treatment. Thus, imaging-based deep learning models have been extensively trained with significant promise. However, existing methods have been largely based on single-task approach without considering the interrelationships among various tasks9. For instance, Zhang et al.26 introduced a multi-level feature exploration and fusion network to identify IDH-related features for IDH prediction. Lee et al.27 developed a three-dimensional conventional neural network-based survival prediction model using whole-brain MRI for glioma patients. Our recent publication28 took a step forward by developing an imaging-based deep learning model for glioma molecular subtyping and integrating these subtypes with clinical variables to evaluate patient prognosis. While this sequential design enhanced the relationship between molecular subtyping and prognosis, it did not identify additional prognostically relevant information beyond recognized biomarkers.
The use of MDL approaches in the field of medical image analysis has gained increasing traction29,30,31. An MDL framework, as demonstrated in our study, enhances the efficacy and generalization ability of the model. By adopting a multi-task learning strategy, rather than a sequential approach, our model effectively addresses the interrelations among different tasks. For instance, in the context of glioma, IDH-wildtype and 1p/19q co-deletion are known to be mutually exclusive32 and IDH-wildtype tumors are typically associated with a grade IV classification2. Although some studies have employed MDL networks to predict genetic and histological features of glioma18,33, they have not incorporated accurate prognostic stratification. To our knowledge, our study is the first to design an MDL model that concurrently predicts molecular alterations, histological grade, and prognosis in glioma patients. Our deep learning model, trained to predict survival outcomes, captures prognosis-related information beyond the scope of molecular alterations and histological grade.
Despite the remarkable advancements and potential of deep learning tools in medical imaging, the challenge of interpretability remains. This limitation hinders the transition of these tools from research settings to clinical practice, especially in high-stakes applications such as treatment planning. Bridging this gap requires establishing an internal connection between quantitative imaging features and their biological basis, facilitated by interdisciplinary technologies. Recently, integrating biological meaning into digital image features has gained increasing interest11. For instance, a study linked radiomic features extracted from preoperative MRI images of glioma with key gene modules and biological pathways, such as immune regulation, and cellular functions using RNA-seq34. This approach enhanced both the reproducibility and interpretability of the findings by focusing on gene interactions rather than individual genes. Another study explored the use of radiomics for stratifying glioblastoma patients into distinct prognostic groups, investigating the molecular profiles of radiomics-defined phenotypes35. Specific genomic signatures were identified for each radiomic phenotype. Furthermore, Li et al.34 performed bioinformatic analysis to understand the implications of MRI-based radiomic features, uncovering strong correlations with immune system processes, especially tumor macrophage infiltration. This correlation was further corroborated through single-cell RNA-seq and immunohistochemical staining. In a recent study36, we developed a deep survival score from diffusion tensor imaging to predict OS in glioma patients and initiated an investigation into the biological pathways underlying this signature using RNA-seq analysis. The link between radiomic features and TME provides a plausible explanation for the accurate prognostic predictions in glioma patients using radiomic models. However, the complex interplay underlying radiomic features cannot be fully decoded by radiogenomic analysis alone.
Unlike previous studies, we sought to uncover the biological basis of our deep learning model using a multi-omic approach. This involved integrating information from various pan-omic disciplines such as pathomics, transcriptomics, and proteomics to achieve a more comprehensive understanding of the model predictions. At the cellular level, our radiopathomic analysis demonstrated a negative correlation between the DPS and TSR. This correlation is consistent with previous studies that have established the prognostic value of TSR in many solid tumors20,37,38. Typically, tumors with a low-TSR are more prone to invasion and aggressiveness, which may be partly due to the process of EMT. At the molecular level, the high-DPS subtype exhibited a higher mutation load compared to the low-DPS subtype, suggesting genomic instability. Radiogenomic analysis revealed that molecular pathways characterizing tumor invasion and metastasis, such as EMT, angiogenesis, and hypoxia, were enriched in the high-DPS subtype. EMT and angiogenesis are key morphogenetic processes in cancer progression39, and EMT can be driven by hypoxia40, a condition associated with various biological processes such as cell proliferation, EMT, angiogenesis, therapy resistance, and metastasis41. Proteomic analysis indicated elevated expression of proteins such as EGFR and HER2 in the high-DPS subtype, while proteins such as ERK2 exhibited higher expression in the low-DPS subtype. These findings suggest that ErbB-targeted therapies might be effective for high-DPS tumors, whereas targeting ERK2 could be a strategy for low-DPS tumors. In terms of immune response, our analysis suggested that the high-DPS subtype might correlate with an immune ‘Hot’ phenotype, whereas the low-DPS subtype could be characterized as an immune ‘Cold’ phenotype. Interestingly, the high-DPS subtype, despite being associated with active immune responses, had a worse prognosis than the low-DPS subtype, aligning with studies where high-risk patients may experience immune evasion42.
One limitation of our study is its retrospective design, which carries the potential for unavoidable selection biases. To mitigate this concern, we conducted extensive validations using international datasets, and it was encouraging to observe that the deep learning model demonstrated robust performance across various validation cohorts. Another limitation is the exclusion of functional MRI techniques such as diffusion- and perfusion-weighted imaging from our analysis. These techniques can provide valuable insights into tumor cellularity and vascularity, complementing conventional imaging modalities36,43,44. However, their application is not widespread in clinical practice, being more commonly employed for research purposes. Additionally, the multi-omic analyses in our study were conducted exclusively using the TCGA dataset. While the TCGA provides a rich resource, it may not fully encapsulate the diversity found in the general population. It is also important to note that our multi-omic analysis primarily relied on post hoc correlations. While these correlations are informative, they do not establish a mechanistic model. Lastly, the current understanding of the relationship between computerized imaging features and biological processes is largely correlational. There remains a substantial knowledge gap regarding causality in this relationship. Bridging this gap is critical and calls for further collaborative research efforts and investigations.
In summary, our study introduced a biologically interpretable MDL pipeline, uniquely designed to concurrently predict molecular biomarkers, histology grade, and survival outcomes in glioma patients. The DPS, derived from imaging data, emerges as a potent tool for prognostic stratification. Through an integrated multi-omics analysis, we were able to uncover the underlying biological basis of the prognostic phenotypes identified by our deep learning model. Looking forward, prospective studies and clinical trials are needed to validate the clinical applicability of the MDL pipeline.
Methods
Patients and cohorts
The First Affiliated Hospital of Zhengzhou University (FAHZU) dataset included 1,726 adult glioma patients treated at FAHZU from January 2011 to December 2021. The Henan Provincial People’s Hospital (HPPH) dataset consisted of 223 glioma patients from HPPH between January 2016 and December 2021. After excluding 18 patients, the remaining 1931 patients from the two private datasets were integrated and randomly assigned to TC and IVC in a 7:3 ratio.
EVC1 comprised 213 glioma patients, with matched MRI images and multi-omics data sourced from TCGA Low Grade Glioma Collection (TCGA-LGG) and the Glioblastoma Multiforme Collection (TCGA-GBM)14. EVC2 was comprised of 410 glioma patients from the University of California San Francisco Preoperative Diffuse Glioma MRI (UCSF-PDGM) dataset45. EVC3 consisted of 222 glioma patients from The Erasmus Glioma Database (EGD)46.
For the purposes of this study, the FAHZU, HPPH, TCGA, UCSF, and EGD datasets were utilized for the prediction of molecular markers. Additionally, the FAHZU, HPPH, TCGA, and UCSF datasets were used for prognosis prediction. The TCGA dataset was curated to validate the biological significance of the MDL model.
MR imaging protocols
All patients in the study underwent MRI scans prior to initiating anti-cancer treatment. Due to the retrospective nature of this study, the MRI examinations adhered to local policies, resulting in the lack of a standardized imaging protocol. Detailed scanning parameters for each dataset are available in the Supplementary Notes1.
Genetic and histological features
For our in-house datasets, the genetic and histological information data were derived from the analysis of tumor tissues post-biopsy or surgery, specifically for the FAHZU and HNPH datasets. The methodologies for gene detection in these cases have been described in a previous publication47. The genetic and histological data for the public datasets are also available in the Supplementary Notes 2.
Automatic tumor segmentation
mmFormer15, a state-of-the-art Transformer-based method for 3D brain tumor segmentation, was utilized in this study. The mmFormer is distinguished by its hybrid modality-specific encoders, an inter-modal Transformer, and a decoder that performs progressive upsampling, integrating with modality-invariant features for robust segmentation. In our private datasets, imaging data were processed using the mmFormer for automated segmentation. For the public datasets, the segmentation masks were pre-existing and were verified for accuracy. For the private datasets (FAHZU and HNPH), the initial automated segmentation with the mmFormer was followed by manual revisions or corrections. These corrections were based on registered contrast-enhanced T1-weighted (CE-T1w) images and T2-fluid attenuated inversion recovery (T2-FLAIR) images. The manual adjustments were carried out layer-by-layer by two fellowship-trained neuroradiologists, with 7 and 10 years of experience in neuro-radiology. Their work was further validated by a senior neuroradiologist with 20 years of experience. Discrepancies in the segmentation were resolved through consensus. Notably, while the neuroradiologists were aware that all patients were diagnosed with glioma, they were blinded to any other clinical or genetic information.
Image normalization
After aligning the images to the centers of the volumes of interest (VOIs), we normalized the images and set the model input size to 88 × 112 × 88 voxels. For VOIs smaller than this predefined size, we implemented an expansion approach by evenly padding zeroes in various directions to meet the specified dimensions. Conversely, if the original VOIs exceeded this size, we attained the desired VOI dimensions through image cropping, ensuring consistency across all data samples. Additionally, to mitigate potential issues such as gradient explosion or dispersion during the model training phase, and to facilitate quicker model convergence, we employed standardization using a z-score normalization which is represented as:
Where \(X\) denotes the original, \(u\) represents the mean of the original image, \({\rm{\sigma }}\) is the standard deviation of the raw image, and \({X}^{{\prime} }\) represents the resultant normalized image.
MDL model construction
During model training, online augmentation techniques were employed for image augmentation in the TC. This approach enhances the generalizability of the model by reducing dependency on specific image attributes. Online augmentation, unlike offline augmentation, does not require local storage of augmented data, substantially reducing data storage space. It also offers flexibility by generating an unlimited amount of augmented data. The augmentation techniques included image rotation, scale transformation, and elastic deformation, increasing the diversity of training data and aiding the model in generalizing to unseen samples.
We utilized the 3D version of ResNet-10 as the network backbone for the MDL model. This model was designed to predict molecular markers, histological grade, and prognosis. To accommodate simultaneous predictions of the IDH mutation status, 1p/19q co-deletion status, and histological grade (II/III or IV), we modified the last fully connected layer of the original 3D ResNet-10 with three separate fully connected layers. A total of 512 deep features were extracted from this layer and shared across the three classification tasks. To enable the concurrent prediction of OS in glioma patients, we incorporated four additional branches into the model. The 512 shared deep features were condensed to 16 features through two fully connected layers utilizing the Relu activation function to prevent overfitting. Finally, the 16 features were combined with the prediction scores from the three auxiliary tasks to produce 19 imaging features, with the sigmoid function employed for predicting OS.
For model training, the Kaiming normal method48 was employed to initialize model parameters, with a learning rate of 0.001 and a batch size set of 32. We used cross-entropy as the loss function for the three subtasks:
where “task” refers to IDH mutation status, 1p/19q co-deletion status, and histological grade. \(\hat{Y}\) is the output probability, and \(Y\) is the ground truth.
Initially, the model was trained on the parameters of the three prediction branches, with the total loss being the sum of the losses from each branch:
Upon convergence of these branch models, the prognostic prediction branch model was trained. During this phase, the parameters of the first three branch models were frozen to prevent learning, updating only the prognostic branch parameters. The Cox model estimated the risk function, and the Cox loss function was defined as:
where \({T}_{i}\), \({E}_{i}\), and \({x}_{i}\) are the event time, event indicator, and baseline data for the \({i}^{th}\) observation, respectively. The product is defined over the set of patients with an observable event \({E}_{i}\) = 1. The risk set \({\mathfrak{R}}(t)=\{i:{T}_{i}\ge t\}\) is the set of patients still at risk of failure at time t.
Similar to the DeepSurv network49, the loss function was set to be the negative log partial likelihood:
where NE=1 is the number of patients with an observable event and \(\lambda\) is the ℓ2 regularization parameter. We then used gradient descent optimization to find the weights of the network which minimize Eq. 5.
Our method was implemented in Python 3.6. The deep learning model was implemented using Pytorch 0.4.0 and was accelerated on an NVIDIA GeForce RTX 3080 Ti GPU (12GB on-board memory).
Evaluation of the model accuracy for classification tasks
The discriminative ability of the MDL model was evaluated in terms of its capacity to predict IDH mutation status, 1p/19q co-deletion status, and histological grade. The evaluation was conducted across three distinct cohorts (EVC1, EVC2, and EVC3). A comprehensive set of evaluation metrics was employed to thoroughly assess the discriminative power of the model. These metrics included AUC, sensitivity, specificity, accuracy, negative predictive value (NPV), and positive predictive value (PPV). Furthermore, to quantify the pairwise classification accuracy among different classes confusion matrices were utilized.
Evaluation of the model accuracy for the prognosis prediction task
The proficiency of the MDL model in prognostic prediction, specifically in forecasting OS events, was assessed through measures of discrimination and calibration. We employed the Kaplan–Meier survival curve to evaluate the differences in survival outcomes between the two identified subtypes. To gauge the performance of the model in survival prediction, the Harrell’ concordance index (C-index) was utilized. Additionally, the AUC was used as a metric to assess the model’s accuracy in predicting 1 year, 3 year, and 5 year OS probabilities. Calibration curves were also plotted to compare the predicted survival probabilities with the actual observed probabilities for the event of interest. For the purpose of stratifying patients into prognostic phenotypes, the optimal cutpoint was determined for DPS in the TC using the maximally selected rank statistics from the ‘maxstat’ R package. This same threshold was then applied consistently across the validation cohorts. Consequently, patients were classified into two distinct prognostic groups: low-DPS and high-DPS. Patients in the low-DPS group had DPS values equal to or below the cutoff, while those in the high-DPS group had DPS values above the cutoff. This analysis encompassed a total of 2,242 patients across the TC, IVC, EVC1, and EVC2.
Uncovering the biological basis of the model
Recognizing the essential role of understanding the biological underpinnings behind deep learning model predictions for clinical application, we embarked on a comprehensive bioinformatic analysis. This analysis aimed to elucidate the cellular and molecular distinctions between the two prognostic subtypes identified by the model. To this end, we conducted in-depth analyses of multi-omic data, encompassing a broad spectrum of biological aspects. These included whole slide images, whole-exome sequencing, copy number alterations, tumor mutation burden, RNA sequencing (RNA-seq), methylation levels, and protein expression profiles.
Whole slide image collection and nuclear feature extraction
We obtained H&E pathologically stained sections of the TCGA samples from the GDC portal. Next, we performed nuclear segmentation and classification using the Hover-Net model, which was pretrained on the PanNuke dataset and could identify five types of nuclei: tumor, inflammatory, stromal, normal (nonneoplastic epithelial), and dead (necrotic) nuclei50. In total, we divided the input WSI images to 50,978,382 patches from 130 patients. It is important to note that tumors do not exist in isolation, and evaluating their microenvironment, which includes stromal, inflammatory, and necrotic components, can provide valuable insights into survival outcomes. Then, we extracted the areas of the tumor, stromal, inflammatory, and necrotic regions and calculated the ratios of tumor-stroma, tumor-inflammation, and tumor-necrosis (Supplementary Fig. 9). Tumor-stroma ratio was defined as the proportion of tumor tissue relative to surrounding stromal tissue. Tumor-inflammation ratio refers to the proportion of tumor tissue relative to inflammation tissue. Tumor-necrosis ratio was defined as the proportion of tumor tissue relative to necrosis tissue.
RNA sequencing data acquisition and processing
The patients in the TCGA dataset (n = 132) were divided into low- and high-DPS groups. The RNA-Seq data for the 132 glioma patients were obtained from the TCGA database, which accessed at https://portal.gdc.cancer.gov/. To identify significantly differentially expressed genes (DEGs) between the high- and low-DPS groups in the TCGA dataset, we employed the Wilcox test from the “DESeq2” package. The cut-off criteria for DEG selection were set at |log2FC | ≥ 2 and false discovery rate (FDR) < 0.05.
GSEA and ssGSEA
To investigate biological differences and gain insights into associated pathways, we utilized the “clusterProfiler” package51 for gene set enrichment analysis (GSEA). The gene set was obtained from the Hallmark gene set collection (h.all.v2023.1.Hs.symbols.gmt) within the Molecular Signatures Database52, accessible at https://www.gsea-msigdb.org/ gsea/msigdb/index.jsp. Furthermore, we performed functional annotation of deep features through Gene Set Variation Analysis (GSVA) and Pearson correlation analysis. The “GSVA” R software package53 was employed for gene set variation analysis, while the Hallmark gene set from the Molecular Signatures Database was used to explore alterations in signaling pathways. Significant correlations (P < 0.05) between deep features and functions or pathways were used for annotating the deep features.
Comprehensive analysis of immune characteristics
The “GSVA” package was employed for ssGSEA to compute infiltrating immune cell scores and evaluate immune-related pathways. The ESTIMATE algorithm was utilized to evaluate stromal status, immune status, and tumor purity for each sample. Furthermore, the Wilcoxon rank sum test was performed to compare immune infiltration levels between the high- and low-DPS subtypes. Pearson correlation analysis was used to identify correlations among the stromal cell score, immune score, estimate score, and DPS.
Analysis of genomic variation between low- and high-DPS subtypes
To investigate the somatic mutations associated with DPS, we utilized the R package “maftools” to generate waterfall plots. These plots illustrate the mutation patterns of the TCGA patients in the high- and low-DPS subtypes. The “somaticInteractions” function was used to identify mutually exclusive and co-occurring mutations among the most frequent ones. The “tmb” function was utilized to calculate the mutation load of each sample.
Copy-number analysis
Publicly available copy-number alteration seg files were downloaded by “TCGA-biolinks”. Recurrent arm-level and focal-level SCNAs of each subtype were identified using GISTIC 2.0 on GenePattern (https://cloud.genepattern.org/gp). GISTIC2.0 was executed with the following parameters: confidence.level = “0.90”, run.broad.analysis = “0”, focal.length.cutoff = “0.50”, arm.peel = “0”, gene.collapse.method = “extreme”. Additionally, we performed copy number variation (CNV) analysis on the top 20 genes showing the greatest differences between the low- and high- DPS subtypes.
RPPA analysis
RPPA data for the core TCGA samples were obtained from TCPA. Standard experimental procedures for RPPA were followed, using validated antibodies as described in a previous study54. In the case of GBMs, 171 primary antibodies were used, including 45 phospho-antibodies. For LGGs, 189 primary antibodies were used, including 51 phospho-antibodies. There were 141 protein antibodies that overlapped between the two arrays. Differential expression analysis between the two DPS subtypes was performed using the R package limma v3.52.2, with a FDR of 0.05 and a fold change (FC) cutoff of 0.4. The resulting heatmap illustrates the differences in expression patterns between the two DPS subtypes.
Identification of potential targeted drugs
The Genomics of Drug Sensitivity in Cancer (GDSC) is a publicly available dataset that provides information on drug sensitivity in cancer cells and molecular markers of drug response55, GDSC2 gene expression profile and corresponding drug response information were downloaded to generate a ridge regression model that could be applied to glioma transcriptomic data. Then the sensitivity scores were yielded to predict the half-maximal inhibitory concentration (IC50) of all drugs in glioma patients.
Statistical analysis
The development of the MDL model was carried out using Python version 3.6.0. Bioinformatics analyses of multi-omics data were conducted using R 4.2.3. For evaluating the classification performance of the MDL mode, we utilized the “reportROC” package in R. The survival prediction performance of the model was assessed using the “rms” package in R. Unless otherwise specified, Fisher’s exact test was used to calculate P-values in comparisons between two categorical variables. For comparisons between two continuous variables, we utilized the Wilcoxon rank sum test and the t-test. To address the issue of multiple testing and control the FDR, Q-values were calculated, except where noted otherwise. A P-value of < 0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The public datasets are available at the following links:
TCGA-LGG14: https://www.cancerimagingarchive.net/collection/tcga-lgg/
TCGA-GBM14: https://www.cancerimagingarchive.net/collection/tcga-gbm/
UCSF45: https://www.cancerimagingarchive.net/collection/ucsf-pdgm/
EGD46: https://xnat.bmia.nl/REST/projects/egd
The in-house dataset consists of confidential medical data not open to the public. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Code availability
Source code for the deep learning model is available at: https://github.com/zhang-AI-MI/GliomaDeepLearning.
References
Miller, K. D. et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin. 71, 381–406 (2021).
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-Oncology 23, 1231–1251 (2021).
Weller, M. et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 18, 170–186 (2021).
Jackson, R. J. et al. Limitations of stereotactic biopsy in the initial management of gliomas. Neuro-Oncology 3, 193–200 (2001).
Nagendran, M. et al. Artificial intelligence versus clinicians: systematic review of design, reporting standards, and claims of deep learning studies. BMJ 368, m689 (2020).
Esteva, A. et al. A guide to deep learning in healthcare. Nat. Med. 25, 24–29 (2019).
Luo, J., Pan, M., Mo, K., Mao, Y. & Zou, D. Emerging role of artificial intelligence in diagnosis, classification and clinical management of glioma. Semin. Cancer Biol. 91, 110–123 (2023).
Hosny, A., Parmar, C., Quackenbush, J., Schwartz, L. H. & Aerts, H. J. W. L. Artificial intelligence in radiology. Nat. Rev. Cancer 18, 500–510 (2018).
Zhao, Y., Wang, X., Che, T., Bao, G. & Li, S. Multi-task deep learning for medical image computing and analysis: a review. Comput. Biol. Med. 153, 106496 (2023).
Jiang, Y. et al. Biology-guided deep learning predicts prognosis and cancer immunotherapy response. Nat. Commun. 14, 5135 (2023).
Tomaszewski, M. R. & Gillies, R. J. The biological meaning of radiomic features. Radiology 298, 505–516 (2021).
Liu, Z. et al. Radiogenomics: a key component of precision cancer medicine. Br. J. Cancer 129, 741–753 (2023).
Sun, Q. et al. Biologic pathways underlying prognostic radiomics phenotypes from paired MRI and RNA sequencing in glioblastoma. Radiology 301, 654–663 (2021).
Bakas, S. et al. Advancing the cancer genome Atlas glioma MRI collections with expert segmentation labels and radiomic features. Sci. Data 4, 170117 (2017).
Zhang, Y. et al. mmFormer: Multimodal medical transformer for incomplete multimodal learning of brain tumor segmentation. In Medical Image Computing and Computer Assisted Intervention—MICCAI 2022 (eds. Wang, L., Dou, Q., Fletcher, P. T., Speidel, S. & Li, S.) 107–117 (Springer Nature Switzerland, Cham, 2022).
Havaei, M., Guizard, N., Chapados, N. & Bengio, Y. HeMIS: Hetero-modal image segmentation. In Medical Image Computing and Computer-Assisted Intervention – MICCAI 2016 (eds. Ourselin, S., Joskowicz, L., Sabuncu, M. R., Unal, G. & Wells, W.) 469–477 (Springer International Publishing, Cham, 2016).
Dorent, R. et al. Hetero-modal variational encoder-decoder for joint modality completion and segmentation. In Medical Image Computing and Computer Assisted Intervention—MICCAI 2019. (eds. Shen, D. et al.) 11765 (Springer International Publishing, Cham, 2019).
Decuyper, M., Bonte, S., Deblaere, K. & Van Holen, R. Automated MRI based pipeline for segmentation and prediction of grade, IDH mutation and 1p19q co-deletion in glioma. Comput. Med. Imaging Graph. 88, 101831 (2021).
Choi, Y. S. et al. Fully automated hybrid approach to predict the IDH mutation status of gliomas via deep learning and radiomics. Neuro Oncol. 23, 304–313 (2021).
Wang, K. et al. Tumor-stroma ratio is an independent predictor for survival in esophageal squamous cell. Carcinoma. J. Thorac. Oncol. 7, 1457–1461 (2012).
Ceccarelli, M. et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164, 550–563 (2016).
Goodman, A. M. et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 16, 2598–2608 (2017).
Yan, H. et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 (2009).
Reuss, D. E. et al. IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: a grading problem for WHO. Acta Neuropathol. (berl.) 129, 867–873 (2015).
Weller, M. et al. European association for neuro-oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 18, e315–e329 (2017).
Zhang, J. et al. Multi-level feature exploration and fusion network for prediction of IDH status in gliomas from MRI. IEEE J. Biomed. Health Inform. 29, PP (2023).
Lee, J. O. et al. Added prognostic value of 3D deep learning-derived features from preoperative MRI for adult-type diffuse gliomas. Neuro Oncol. 26, 571–580 (2023).
Yan, J. et al. Quantitative MRI-based radiomics for noninvasively predicting molecular subtypes and survival in glioma patients. NPJ Precision Oncol. 5, 72 (2021).
Fu, S. et al. Multi-task deep learning network to predict future macrovascular invasion in hepatocellular carcinoma. EClinicalMedicine 42, 101201 (2021).
Shao, W. et al. Multi-task multi-modal learning for joint diagnosis and prognosis of human cancers. Med. Image Anal. 65, 101795 (2020).
Liu, L., Dou, Q., Chen, H., Qin, J. & Heng, P.-A. Multi-task deep model with margin ranking loss for lung nodule analysis. IEEE Trans. Med. Imaging 39, 718–728 (2020).
Labussière, M. et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology 74, 1886–1890 (2010).
van der Voort, S. R. et al. Combined molecular subtyping, grading, and segmentation of glioma using multi-task deep learning. Neuro Oncol. 25, 279–289 (2023).
Li, G. et al. An MRI radiomics approach to predict survival and tumour-infiltrating macrophages in gliomas. Brain 145, 1151–1161 (2022).
Choi, S. W. et al. Multi-habitat radiomics unravels distinct phenotypic subtypes of glioblastoma with clinical and genomic significance. Cancers 12, 1707 (2020).
Yan, J. et al. Deep learning features from diffusion tensor imaging improve glioma stratification and identify risk groups with distinct molecular pathway activities. EBioMedicine 72, 103583 (2021).
Huijbers, A. et al. The proportion of tumor-stroma as a strong prognosticator for stage II and III colon cancer patients: validation in the VICTOR trial. Ann. Oncol. 24, 179–185 (2013).
Yoo, S.-Y. et al. Whole-slide image analysis reveals quantitative landscape of tumor–immune microenvironment in colorectal cancers. Clin. Cancer Res. 26, 870–881 (2020).
Shojaei, S. et al. Mesenchymal stem cell-derived exosomes enriched with miR-218 reduce the epithelial–mesenchymal transition and angiogenesis in triple-negative breast cancer cells. Eur. J. Med. Res. 28, 516 (2023).
Hapke, R. Y. & Haake, S. M. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer Lett. 487, 10–20 (2020).
Jawad, S. F. et al. The strict regulation of HIF-1α by non-coding RNAs: new insight towards proliferation, metastasis, and therapeutic resistance strategies. Cancer Metastasis Rev. 43, 5–27 (2023).
Gangoso, E. et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 184, 2454–2470.e26 (2021).
Guo, Y. et al. Improving noninvasive classification of molecular subtypes of adult gliomas with diffusion‐weighted MR imaging: an externally validated machine learning. Algorithm. J. Magn. Reson. Imaging 58, 1234–1242 (2023).
Pei, D. et al. Radiomic features from dynamic susceptibility contrast perfusion-weighted imaging improve the three-class prediction of molecular subtypes in patients with adult diffuse gliomas. Eur. Radiol. 33, 3455–3466 (2023).
Calabrese, E. et al. The university of California San Francisco preoperative diffuse glioma MRI dataset. Radiol. Artif. Intell. 4, e220058 (2022).
van der Voort, S. R. et al. The erasmus glioma database (EGD): structural MRI scans, WHO 2016 subtypes, and segmentations of 774 patients with glioma. Data in Brief 37, 107191 (2021).
Zhang, Z.-Y. et al. Prognostic value of preoperative hematological markers combined with molecular pathology in patients with diffuse gliomas. Aging (milano) 11, 6252–6272 (2019).
He, K., Zhang, X., Ren, S. & Sun, J. Delving deep into rectifiers: surpassing human-level performance on ImageNet classification. 2015 IEEE Int. Conf. Comput. Vision (ICCV) 1026–1034 (Santiago, Chile, 2015).
Katzman, J. L. et al. DeepSurv: personalized treatment recommender system using a Cox proportional hazards deep neural network. BMC Med. Res. Methodol. 18, 24 (2018).
Graham, S. et al. Hover-Net: simultaneous segmentation and classification of nuclei in multi-tissue histology images. Med. Image Anal. 58, 101563 (2019).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Castanza, A. S. et al. Extending support for mouse data in the molecular signatures database (MSigDB). Nat. Methods 20, 1619–1620 (2023).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinf. 14, 7 (2013).
Brennan, C. W. et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013).
Maeser, D., Gruener, R. F. & Huang, R. S. oncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 22, bbab260 (2021).
Acknowledgements
We acknowledge financial support from the National Key Research and Development Program of China (2023YFF1204600); the National Natural Science Foundation of China (82102140, 82227802, 82302306, 82302336); the Clinical Frontier Technology Program of the First Affiliated Hospital of Jinan University (No. JNU1AF-CFTP-2022-a01201); the Science and Technology Projects in Guangzhou (202201020022, 2023A03J1036, 2023A03J1038); the Science and Technology Youth Talent Nurturing Program of Jinan University (21623209); the Postdoctoral Science Foundation of China (2022M721349); Beijing Natural Science Foundation (L232132).
Author information
Authors and Affiliations
Contributions
X.W.: Writing—review and editing, Writing—original draft, Visualization, Validation, Software, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. S.Z.: Writing—review and editing, Writing—original draft, Visualization, Validation, Software, Resources, Methodology, Investigation, Funding acquisition, Formal analysis. Z.Z.: Writing—review and editing, Visualization, Validation, Resources, Investigation, Data curation. Z.H.: Writing—review and editing, Validation, Investigation. Z.X.: Writing—review and editing, Validation, Software, Methodology, Formal analysis. W.W.: Visualization, Validation, Resources, Data curation. Z.J.: Validation, Methodology, Investigation. J.Y.: Writing—review and editing, Validation, Investigation. Y.G.: Resources, Data curation. L.Z.: Writing—review and editing, Validation, Funding acquisition, Investigation. W.H.: Validation, Visualization, Resources, Funding acquisition. F.W.: Visualization, Resources, Funding acquisition. X.L.: Visualization, Supervision, Resources. D.Y.: Resources, Data curation. J.C. Supervision, Resources. J.Y.: Writing—review and editing, Supervision, Validation, Visualization, Resources, Investigation, Data curation. S.Z.: Writing—review and editing, Supervision, Validation, Visualization, Project administration, Resources, Funding acquisition, Investigation. B.Z.: Writing—review and editing, Writing—original draft, Visualization, Validation, Supervision, Resources, Project administration, Methodology, Investigation, Funding acquisition, Conceptualization.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
This study adhered to the ethical tenets of the Declaration of Helsinki and was approved by the institutional review boards of participating hospitals. Informed consent was waived due to the use of deidentified clinical data and the low risk of this investigation. Notably, the use of public data does not require approval from an ethics committee, as no identifiable patient information was involved.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, X., Zhang, S., Zhang, Z. et al. Biologically interpretable multi-task deep learning pipeline predicts molecular alterations, grade, and prognosis in glioma patients. npj Precis. Onc. 8, 181 (2024). https://doi.org/10.1038/s41698-024-00670-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-024-00670-2
- Springer Nature Limited