
Abstract
DP303c is a HER2-targeted ADC with a cleavable linker-MMAE payload. Previous in vitro studies demonstrated that DP303c showed similar or better antitumor activity than T-DM1 in xenograft models. This was a multicenter, dose escalation and dose expansion phase 1 study in China. Eligible patients were 18-75 years old with HER2-positive advanced solid tumors who were unable to benefit from standard therapy. DP303c was administered intravenously every 3 weeks, with accelerated titration at lower dose of 0.5 mg/kg and 3 + 3 design with dose levels of 1.0, 2.0, 3.0 or 4.0 mg/kg at dose escalation part, followed by the selected dose level at dose expansion part. The primary endpoints were safety and tolerability, as well as identification of recommended phase 2 dose. As of Feb 28, 2023, 94 patients were enrolled and received DP303c (dose escalation: n = 22; dose expansion: n = 72), of whom 68 patients had breast cancer. One dose limiting toxicity (Grade 3 eye pain) was observed at 4.0 mg/kg dose, and the maximum tolerated dose was not reached. The most common treatment-related adverse events at grade 3 or higher were blurred vison (16.0%), dry eye (6.4%), and peripheral neuropathy (5.3%). No treatment-related death occurred. Overall, among 91 efficacy evaluable patients, 39 patients (42.9%) achieved an objective response. Disease control was observed in 62 patients (68.1%). In 66 efficacy evaluable patients with breast cancer, 34 patients achieved an objective response (51.5%). Disease control was achieved in 51 patients (77.3%). Median PFS was 6.4 months. On a molar basis, DP303c Cmax at 3.0 mg/kg doses was 132-folder higher than that for free MMAE. DP303c demonstrated promising anti-tumor activity with acceptable safety in patients with pre-treated advanced HER2 positive solid tumors, especially in breast cancer. Based on safety and efficacy results, 3.0 mg/kg Q3W was determined as recommended phase 2 dose for DP303c. (Trial registration: ClinicalTrials.gov Identifier: NCT04146610).
Similar content being viewed by others


Introduction
Human epidermal growth factor receptor 2 (HER2) is overexpressed in 20%-25% of breast cancer, 20%–30% of ovarian cancer and 15–20% of gastric cancer, and associated with aggressive behavior, high risk of relapse and poor prognosis1,2,3,4,5,6,7. Several studies revealed that HER2 targeted therapy had been demonstrated to improve the survival prognosis of HER2 positive tumors8,9,10,11,12. Till now, there were several HER2 targeted therapies available in China, including trastuzumab, pertuzumab, anti-HER2 tyrosine kinase inhibitor (lapatinib, pyrotinib, niratinib), antibody-drug conjugates (ADC, trastuzumab emtansine, trastuzumab deruxtecan and disitamab vedotin). Despite the currently available agents, a substantial proportion of patients with advanced solid tumors developed progressive disease after the treatment of HER2 targeted therapy13,14, so there are unmet clinical needs for treatment of HER2 positive solid tumors.
ADCs has emerged as a rapidly growing anticancer therapy, which was designed as a highly effective drug-delivery system that enable the potent cytotoxins to cancer cells while sparing non-malignant cells, thereby limiting the risk of off-target adverse effects15. Five key elements that affects the activities of ADC were target antigen, antibody, linker, cytotoxic payload and conjugation methods. Till now, the development of ADC drugs could be subdivided into three generations, and greater progress have been made in identification of new antigen, full humanized antibody, new payload with optimal toxicity and bystander effects, and design of new linker to balance between stability and payload release15.
As a second generation HER2-targeted ADC, DP303c is composed of a human immunoglobulin G1 (IgG1) anti-HER2 antibody (DP001), an enzyme-based cleavable peptide-linker, and two tubulin polymerization inhibitors (MMAE)16. DP001, which was manufactured using a stable Chinese Hamster Ovary (CHO) cell line, had the same amino acid sequence with trastuzumab. Each DP303c antibody has an average of 2 MMAE molecules attached to DP001 by a site-specific conjugation technology (an engineered microbial transglutaminase, that meant site-specific conjugation through transamidation to residue Q295 in the constant region of heavy chain), which expect to improve binding stability, reduce the heterogeneity of ADC molecule, and provide a larger therapeutic window17. In contrast with T-DM1, DP303c displayed similar or greater inhibitory activity against cells with overexpression of HER2 and cells with a reduced levels of HER2 expression (e.g. SK-BR-3, HCC1954, NCI-N87, BT-474, SK-0V-3, JIMT-I and MDA-MB-468), with IC50 value of 0.065 to >1000 nM and 0.088 to >1000 nM, respectively16. Moreover, DP303c also showed potent anticancer activity in variable cell line-derived xenograft models16. Finally, release of MMAE from DP303c ranged from 0.003% to 0.599% on day 14 in rats16, monkeys and humans, which was lower than that of other VC-MMAE ADCs18,19,20, suggesting that DP303c had a high plasma stability. Here we report a first-in-human, phase 1, multicenter, open-label, dose-escalation and dose-expansion study to evaluate the safety and tolerability, pharmacokinetics, immunogenicity, and efficacy of DP303c in patients with HER2 positive advanced solid tumors (NCT04146610).
Results
Patient demographics and baseline characteristics
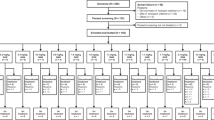
Between January 8, 2020 and February 28, 2023, a total of 147 patients underwent screening for eligibility. Among them, 94 were successfully enrolled and received at least one dose of DP303c treatment (dose escalation: n = 22; dose expansion: n = 72, Fig. 1 and Supplementary Fig. 1). The most common cancer type was breast cancer (n = 68, 72.3%), followed by colorectal cancer (n = 10, 10.6%) and gastric cancer (n = 9, 9.6%). The baseline characteristics of all patients and patients with breast cancer were summarized in Table 1 and Supplementary Table 1. All 94 patients received ≥1 prior line of systemic therapy. 65 patients with breast cancer (95.6%) received ≥2 prior lines of systemic therapy. 14 patients with breast cancer brain metastasis received previous treatment with trastuzumab (Supplementary Table 2). 57 patients (60.6%) received ≥4 cycles of DP303c at dose of 0.5 to 4.0 mg/kg. As of February 28, 2023, 85 patients discontinued the treatment, and nine patients were still on treatment. Most of the patients (n = 67, 78.8%) discontinued the treatment due to disease progression. All 94 patients were included in safety analysis set (SS) and full analysis set (FAS), and 91 patients were included in efficacy analysis set (EAS) (Supplementary Figure 2).

EAS efficacy analysis, FAS full analysis set, IS Immunogenicity set, PKCS pharmacokinetics concentration set, PKPS pharmacokinetics parameter set, SS safety analysis set.
Safety and tolerability
In dose escalation part, one dose limiting toxicity (DLT) (grade 3 eye pain) was observed in one of 22 patients at 4.0 mg/kg dose level, and maximum toxicity dose (MTD) was not reached. Based on combined data of antitumor activity, safety and pharmacokinetics profiles, 3.0 mg/kg every 3 weeks (Q3W) was chosen for further investigation in dose expansion phase with another 72 patients enrolled.
All 94 patients were included in SS and had experienced at least one treatment emergent adverse event (TEAE) and treatment related adverse event (TRAE). The most common TRAEs were corneal disease (n = 82, 87.2%), blurred vision (n = 58, 61.7%), dry eye (n = 54, 57.4%), peripheral neuropathy (n = 44, 46.8%), hypertriglyceridemia (n = 42, 44.7%), alopecia (n = 34, 36.2%), increased aspartate aminotransferase (AST) (n = 30, 31.9%), increased alanine aminotransferase (ALT) (n = 28, 29.8%), hypercholesterolemia (n = 26, 27.7%), anemia (n = 25, 26.6%), proteinuria (n = 25, 26.6%), hyponatremia (n = 24, 25.5%) and hyperglycemia (n = 23, 24.5%; Table 2). 34 patients (36.2%) experienced TRAEs of grade ≥ 3, and the most frequents grade ≥ 3 TRAEs were blurred vision (n = 15, 16.0%), dry eye (n = 6, 6.4%), and peripheral neuropathy (n = 5, 5.3%). Treatment related serious adverse events (SAEs) occurred in 6 patients (n = 6, 6.4%), including blurred vision (n = 3, 3.2%), peripheral neuropathy (n = 3, 3.2%) and decreased visual acuity (n = 1, 1.1%; Supplementary Table 4).
62 patients (66.0%) required dose interruption, mainly caused by corneal disease (n = 49, 52.1%), blurred vision (n = 31, 33.0%), dry eye (n = 19, 20.2%) and peripheral neuropathy (n = 18, 19.1%; Supplementary Table 4). Dose reductions, which occurred in 15 patients (16.0%), were due to corneal disease (n = 9, 9.6%), blurred vision (n = 7, 7.4%) and dry eye (n = 5, 5.3%). 10 patients (10.6%) discontinued the treatment because of AEs, of which 8 (8.5%) were related to DP303c. The most common TRAEs leading to treatment discontinuation included peripheral neuropathy in 5 patients (1 grade 2, 4 grade 3), myalgia (grade 3), muscle weakness (grade 3), pulmonary fibrosis (grade 1) and weight loss (grade 2) in 1 patient each (Supplementary Table 5).
Efficacy
As of Febuary 28, 2023, the median duration of follow-up was 12.0 (range 1.7–35.2) months. Of 91 patients evaluable for efficacy, 39 patients achieved an objective response (42.9%, 95% confidence intervals [CI] 32.5–53.7), including 32 confirmed response and 7 unconfirmed responses. One response (1/3, 33.3%) occurred at 1.0 mg/kg Q3W, 2 responses (2/6, 33.3%) at 2.0 mg/kg Q3W, 32 responses (32/75, 42.7%) at 3.0 mg/kg Q3W, four responses (4/6, 66.7%) at 4.0 mg/kg Q3W (Table 3). Disease control was observed in 62 patients (68.1%, 95%CI 57.5–77.5). Of 39 patients achieving complete response and partial response (PR), the median duration of response (DoR) was 11.0 [95% CI 4.0-not reached (NR)] months and the time to response (TTR) was 1.7 (range 1.2–8.4) months. Overall, 64 patients (68.1%) had progression-free survival (PFS) events, and the median PFS in all patients was 4.4 (95% CI 3.4–6.4) months.
Patients with breast cancer exhibited more promising clinical activity than other tumor types and were prioritized for further study. Of 66 efficacy evaluable patients with breast cancer, 34 patients (51.5%) achieved objective response, disease control was seen in 51 patients (77.3%, 95%CI 65.3–86.7). The median PFS in all patients with breast cancer was 6.4 (95% CI 4.1–8.5) months. As shown in waterfall plot (Fig. 2a), 52 patients (78.8%) had some degree of tumor shrinkage, of which 36 patients had ≥30% decrease in the sum of diameters of target lesions.

a Best percentage change from baseline in target lesion size; b Tumor responses per RECIST version 1.1 over time.
Prespecified subgroup analyses in patients with breast cancer showed consistent responses across several prognostic subgroups (Supplementary Figure 2), except that objective response rate (ORR) and disease control rate (DCR) in patients who were pretreated with anti-HER2 ADC (ORR: 40.9% [9/22, 95%CI 20.7–63.6], DCR: 63.6% [14/22, 95%CI 40.7–82.8]) were slightly lower than that in patients who were pretreated with trastuzumab (ORR: 53.1% [34/64, 95%CI 40.2–65.7], DCR: 79.7% [51/64, 95%CI 67.8–88.7]), pertuzumab (ORR: 53.6% [15/28, 95%CI 33.9–72.5], DCR: 78.6% [22/28, 95%CI 59.0–91.7]) and anti-HER2 tyrosine kinase inhibitor (ORR: 49.2% (29/59, 95%CI 35.9–62.5), DCR: 76.3% [45/59, 95%CI 63.4–86.4]) (Supplementary Table 7). Notably, Patients with brain metastasis (64.3%, 95%CI 35.1–87.2) have the similar ORR with those with lung metastasis (58.1%, 95%CI 39.1–75.5), bone metastasis (54.8%, 95%CI 36.0–72.7), liver metastasis (48.1%, 95%CI 28.7–68.1), and other metastases (52.9%, 95%CI 38.5–67.1) (Supplementary Figure 2).
In the analysis of the colorectal cancer and gastric cancer subgroup, ORR were 10.0% (1/10, 95%CI 0.3–44.5), 25.0% (2/8, 95%CI 3.2–65.1), DCR were 30.0% (3/10, 95%CI 6.7–65.2), 50.0% (4/8, 95%CI 15.7–84.3), respectively. Detailed efficacy results in the colorectal cancer, gastric cancer and other tumors subgroup were shown in Supplementary Figure 3.
In addition, PRs were also observed in patient with salivary gland cancer (n = 1) and patient with small intestine cancer (n = 1).
Pharmacokinetic and immunogenicity analysis
The concentration versus time curve for serum DP303c, total antibody and plasma free MMAE after the first administration of DP303c 1.0 to 4.0 mg/kg were shown in Fig. 3. The pharmacokinetics parameters of DP303c, total antibody and free MMAE were summarized in Supplementary Table 8. Overall, following the first administration of DP303c at 1.0 to 4.0 mg/kg, DP303c and total antibody demonstrated similarity in pharmacokinetics profiles, characterized by similar exposure levels, long half-life time and low clearance. At the dose level of 3.0 mg/kg, t1/2 of DP303c and total antibody were 2.98 and 2.81 days after the first dose of DP303c. On a molar basis, DP303c Cmax at 3.0 mg/kg dose was 132-folder higher than that for free MMAE, its payload.

a DP303c mean concentration-time curve; b Total antibody mean concentration-time curve; c Free MMAE mean concentration-time curve. A, 30 minutes prior to infusion; Time 0, immediately after the infusion. Error bars represent standard deviation.
Immunogenicity data was available in 89 patients after DP303c treatment at 1.0 to 4.0 mg/kg, and anti-DP303c antibody (ADA) positivity was only observed in 3.0 mg/kg group, the incidence of ADAs after treatment was 11.0% (8/73). DP303c exposure (Cmax, AUC0-t and AUC0-∞) stratified by ADA status was compared, and there was no remarkable difference in DP303c exposure between patients with ADA positive samples and those with ADA negative samples.
Discussion
This was a first-in-human study of DP303c to evaluate the safety and tolerability, pharmacokinetics, immunogenicity, and efficacy of DP303c in patients with HER2 positive advanced solid tumors. In this study, 94 patients with HER2 positive advanced solid tumors received DP303c monotherapy at the dose range of 0.5 to 4.0 mg/kg, and DP303c monotherapy had clinically meaningful and durable antitumor activity in advanced solid tumor, with an ORR of 42.9% (95%CI 32.5–53.7) and median DoR of 11.0 (95% CI 4.0–NR) months.
In this study, the most common TRAE was ocular toxicity, which was also frequently occurred after other ADCs treatment21. Ocular TRAEs were reported in 89/94 patients (94.7%), of which were grade 1 in 25 patients (26.6%), grade 2 in 44 patients (46.9%), grade 3 in 20 patients (21.3%). No grade 4 or 5 ocular TRAEs occurred. Corneal disease and blurred vision were the most common eye problem in this study, and of 58 patients who had blurred vision, 57 patients experienced concurrent corneal disease. The most common grade 3 ocular TRAEs were blurred vision, dry eye, corneal disease, cataract, decreased visual acuity and eye pain (Supplementary Table 6). Ocular toxicities were reversible. All grade 3 ocular TRAEs observed at higher doses (≥3.0 mg/kg) after the second administration (median occurrence time, 5.29 weeks). Of 20 patients who experienced grade 3 ocular AEs, 18 patients (90%) had totally recovered with dose interruption and artificial tears, one patient (5%) had improved after medical intervention, median recovery time was 5.57 weeks. During the whole study, only one patient developed ulcerative keratitis without perforation and resolved following the treatment with ofloxacin eye drops. No ocular AEs leading to treatment discontinuation occurred. The exact mechanism of ocular AEs was not fully understood, but was proposed to be an off-target delivery of unconjugated cytotoxin22. As an organ, the eye is particularly susceptible to off-target toxicities given its unique microenvironment. Preclinical trials showed that the expression of HER2 had been detected in human corneal, limbal and conjunctival epithelium23, thereby the ocular surface was likely vulnerable after anti-HER2 ADCs treatment due to its proliferative epithelial cells and numerous cell receptors that allowed antibody binding and release of cytotoxic payload24. Due to the prevalence of ocular adverse events, study protocol was amended in September 2020 and artificial tears were recommended before the administration of DP303c to prevent ocular toxicities.
Peripheral neuropathy was the secondary most common TRAE. Peripheral neuropathy was a frequent adverse event after MMAE-ADCs treatment, which might be attributed to non-specific uptake of the ADC in peripheral nerves and release of MMAE, disrupting microtubes and causing neurodegeneration25. Peripheral neuropathy TRAEs were reported in 44/94 patients (46.8%) in the present study, most were grade 1–2 (39/44, 88.6%). 5 patients experienced grade 3 peripheral neuropathy TRAEs. No grade 4 or 5 peripheral neuropathy TRAEs occurred. The proportion of patients who had grade 3 peripheral neuropathy TRAEs was lower for those in ≥3.0 mg/kg group (4/84, 4.8%) than those in <3.0 mg/kg group (1/10, 10%); further investigation is needed to establish the relative risk by dose.
Left ventricular ejection fraction (LVEF) decrease and interstitial lung disease (ILD) have been reported as serious adverse events after treatment with trastuzumab and trastuzumab emtansine. In this study, these TEAEs were reported in six patients, and all were grade 1. Two patients in ≥3.0 mg/kg group experienced CTCAE grade 1 LVEF decrease, presenting as ventricular compliance decreased and left ventricular hypokinesia, respectively; four patients (one in <3.0 mg/kg group and three in ≥3.0 mg/kg group) had CTCAE grade 1 ILD.
Pharmacokinetic analysis revealed that, at a dose of 1.0 to 4.0 mg/kg, the mean t1/2 of DP303c ranged from 1.80 to 3.69 days, mean t1/2 of total antibody was 2.04 to 3.08 days, and tend to be longer with increasing dose, which was characteristic of target-mediated drug disposition. Analysis of pharmacokinetic properties showed significant lower Cmax and AUC0-∞ of free MMAE compared to those of DP303c and total antibody, which indicated that DP303c was stable in the circulation.
The most favorable response with DP303c was observed in patients with breast cancer, with an ORR of 51.5% (95%CI 38.9–64.0) and median DoR of 11.0 (95% CI 4.0–NR) months. The ORR in patients with pretreated with anti-HER2 ADC was slightly lower, partially because there is cross-resistance between DP303c and previously available anti-HER2 ADC containing payloads of microtubule inhibitors. And the objective response in 9 of 14 evaluable patients with brain metastases was notable, DP303c seemed to show promising antitumor activity in patients with breast cancer brain metastases and worth further exploration. DP303c also demonstrated antitumor activity in patients with salivary gland cancer and small intestine cancer. Subsequent clinical trials are ongoing to further explore the potential benefits of DP303c monotherapy in HER2 positive breast cancer, gastric cancer, and ovarian cancer (NCT05334810, NCT05901935, NCT04826107, NCT04828616).
This study had limitations. Firstly, it was non-randomized, single-arm design with heterogeneous patient population. Therefore, our results should be interpreted with caution. Secondly, HER2 status was assessed in local laboratory, and had not to be confirmed centrally.
In summary, DP303c has shown promising anti-tumor activity with acceptable safety in patients with pre-treated HER2 positive advanced solid tumors, especially in HER2 positive breast cancer. Combined with safety, efficacy, and PK profiles of DP303c, 3.0 mg/kg Q3W was expanded in this study, and the results supported 3.0 mg/kg Q3W was determined as recommended phase 2 dose (RP2D).
Methods
Study design and participants
This was a multicancer, first-in-human, two-part (dose escalation and dose expansion), phase 1 study of DP303c done at 11 hospitals in China (Supplementary Figure 1).
Eligible patients were 18 to 75 years of age with pathologically/histologically confirmed advanced solid tumors who were unable to receive standard of care, or failed standard of care; were HER2 positive, defined as below: (i) for breast cancer and colorectal cancer, tumors have 3+ positive staining by immunohistochemistry (IHC), or IHC 2+ and gene amplification detected by in situ hybridization (ISH), or ISH positive whatever IHC results were; (ii) for gastric cancer and esophageal cancer, IHC 3+ , or IHC 2+ and gene amplification detected by ISH; had a life expectancy of 3 months or more with ECOG performance score of 0 or 1; had a measurable lesion at baseline as per RECIST version 1.1; had enough bone marrow and organ function. Key exclusion criteria included not recovering from toxic response in the previous anti-tumor treatment (>Grade 1 as per NCI-CTCAE 5.0), with the exception of alopecia and pigmentation; had a left ventricular ejection fraction of 40% or less during previous trastuzumab treatment, or trastuzumab discontinuation due to treatment related adverse events; stable brain metastasis.
The study protocol and informed consent were approved by the ethic committee of each site (1907204-8 for Fudan University Shanghai Cancer Center; 2021YL019 for The First Hospital of China Medical University; E2019416 for Tianjin Medical University Cancer Institute and Hospital; 2020-337-001 for Henan Cancer Hospital; HDFY-LL-2020-152 for Affiliated Hospital of Hebei University; 2021-112 for Hunan Cancer Hospital; 2021-020 for Zhongshan Hospital Fudan University; 2021-023 for The First Affiliated Hospital of Bengbu Medical College; 2021-033-02 for Nanjing Drum Tower Hospital; 2021-0003 for The First Affiliated Hospital of Henan University of Science & Technology; 20210139 for Liaoning Cancer Hospital and Institute); this study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Guideline for Good Clinical Practice. Written informed consents had been obtained from all the patients before the enrollment (NCT04146610).
Procedures
The dose escalation part used 3 + 3 design by starting at 0.5 mg/kg Q3W, with sequent dose levels of 1.0, 2.0, 3.0 or 4.0 mg/kg Q3W. 0.5 mg/kg was selected as the starting dose based on 1/10 human equivalent dose of the maximum toxicity dose (30 mg/kg) in the most sensitive species (rat). DLT were defined as per NCI-CTCAE version 5.0: i) Nonhematological toxicity: infusion related reaction remain grade ≥ 3 after intervention; any grade ≥ 3 nonhematological toxicity with the exception of transient grade 3 fatigue, headache, nausea which resolved to grade 1 or baseline levels, grade 3 diarrhea or electrolyte disturbance and resolved to grade 1 or baseline levels within 48 hours, grade 3 vomiting and resolved to grade 1 or baseline levels within 48 hours after intervention, grade 3 fever if manageable; ALT/AST elevations to 3 × ULN and bilirubin elevation to 2×ULN without known reasons; ii) Hematological toxicity: grade 4 neutropenia lasting ≥5 days; febrile neutropenia; grade 4 thrombocytopenia; grade 3 thrombocytopenia complicated by hemorrhage; grade 4 anemia. In dose expansion part, a maximum of 80 patients were enrolled to receive the selected dose level of DP303c for further evaluation of the safety and efficacy of DP303c in HER2 positive advanced solid tumors, as well as the establishment of the RP2D.
DP303c was administered intravenously on Day 1 of each cycle until disease progression, intolerable toxicity, start of new anticancer treatment, death, loss to follow-up, withdrawal of consent, investigator decision, whichever occurred first. Dose interruption or reduction (up to two reductions) was permitted to manage adverse events, and the dose level would not be below 0.3 mg/kg Q3W. If the toxicities had been resolved to grade 1 or lower or baseline levels, DP303c would be resumed. Dose re-escalation was not allowed upon resolution of toxicity.
Safety assessment included vital sign, physical examination, cardiac function, laboratory examination, pulmonary and ophthalmologic assessments. Adverse events were evaluated until 30 days after last administration and graded as per NCI-CTCAE version 5.0. Responses were assessed according to RECIST version 1.1 using CT or MRI at baseline, every 6 weeks during the treatment. After treatment discontinuation, the patients were followed up for progression free survival every 2 months.
Blood samples were collected for analysis of serum DP303c, total antibody and plasma free MMAE concentrations at pre-dose, within 5 minutes after the intravenous infusion, 1, 4, 24, 48, 168, 336 hours after administration. Serum DP303c and total antibody concentrations were determined by a validated enzyme-linked immunosorbent assay with lower and upper limits of 50 ng/mL and 1600 ng/mL, and plasma free MMAE concentrations were determined by a validated liquid chromatography–tandem mass spectrometry with lower and upper limits of 0.01 ng/mL and 5 ng/mL. The ADA was measured within 0.5 hours before infusion at cycle 1–6 and 30 days after last administration by using a validated electrogenerated chemiluminescence immunoassay with the sensitivity of 4.82 ng/mL and 5.73 ng/mL in the screening assay and confirmed assay, respectively.
Outcome
The primary endpoints were safety and tolerability, including the occurrence of DLT and determination of MTD, the incidence of AE/SAE, as well as the establishment of RP2D. Secondary endpoints included ORR, DCR, DoR, PFS, TTR, time to progression (TTP) as per RECIST version 1.1, pharmacokinetics profile and immunogenicity.
Statistical analysis
Safety was analyzed based on SS, who received at least one dose of DP303c; efficacy was analyzed based on FAS, who received at least one dose of DP303c and EAS, who received at least one dose of DP303c and had at least one tumor assessment recording after administration. Statistical analysis was performed using SAS 9.4 (SAS Institute, Cary, North Carolina, USA). ORR and DCR with 95% CI were calculated using Clopper Pearson exact method. DoR, PFS, TTR and TTP were estimated using Kaplan-Meier method. The pharmacokinetics parameters of DP303c, total antibody and free MMAE were determined by noncompartmental methods using WinNonlin version 8.4 (Certara, L.P., USA).
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on request.
References
Costa, R. L. B., Brian, J. & Czerniecki, B. J. Clinical development of immunotherapies for HER2+ breast cancer: a review of HER2-directed monoclonal antibodies and beyond. NPJ Breast Cancer 6, 10 (2020).
Ahn, S., Woo, J. W., Lee, K. & Park, S. Y. HER2 status in breast cancer: changes in guidelines and complicating factors for interpretation. J. Pathol. Transl. Med. 54, 34–44 (2020).
Fujimura, M. et al. HER2 is frequently over-expressed in ovarian clear cell adenocarcinoma: possible novel treatment modality using recombinant monoclonal antibody against HER2, trastuzumab. Jpn J. Cancer Res. 93, 1250–1257 (2002).
Koopman, T. et al. HER2 positivity in gastric and esophageal adenocarcinoma: clinicopathological analysis and comparison. J. Cancer Res Clin. Oncol. 141, 1343–1351 (2015).
Janjigian, Y. Y. et al. Prognosis of metastatic gastric and gastroesophageal junction cancer by HER2 status: a European and USA International collaborative analysis. Ann. Oncol. 23, 2656–2662 (2012).
Zhang, Z. et al. Overexpression of HER-2 protein is a high-risk factor for patients with surgically-resected stage T3 gastric adenocarcinoma. Clin. Lab 63, 115–125 (2017).
Li, H. et al. Relationship between HER2 overexpression and long-term outcomes of early gastric cancer: a prospective observational study with a 6-year follow-up. BMC Gastroenterol. 22, 238 (2022).
Giordano, S. H. et al. Systemic therapy for advanced human epidermal growth factor receptor 2-positive breast cancer: ASCO guideline update. J. Clin. Oncol. 40, 2612–2635 (2022).
Cortés, J. et al. Trastuzumab deruxtecan versus trastuzumab emtansine for breast cancer. N. Engl. J. Med. 386, 1143–1154 (2022).
Swain, S. M. et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): end-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 21, 519–530 (2020).
Xu, B. et al. Pyrotinib plus capecitabine versus lapatinib plus capecitabine for the treatment of HER2-positive metastatic breast cancer (PHOEBE): a multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 22, 351–360 (2021).
Sheng, X. et al. Open-label, multicenter, phase II study of RC48-ADC, a HER2-targeting antibody-drug conjugate, in patients with locally advanced or metastatic urothelial carcinoma. Clin. Cancer Res 27, 43–51 (2021).
Blangé, D., Stroes, C. I., Derks, S., Bijlsma, M. F. & van Laarhoven, H. W. M. Resistance mechanisms to HER2-targeted therapy in gastroesophageal adenocarcinoma: A systematic review. Cancer Treat. Rev. 108, 102418 (2022).
Sidaway, P. HER2-targeted agents overcome resistance. Nat. Rev. Clin. Oncol. 17, 133 (2020).
Fu, Z., Li, S., Han, S., Shi, C. & Zhang, Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct. Target Ther. 7, 93 (2022).
Hui, X. et al. An innovative site-specific anti-her2 antibody-drug conjugate with high homogeneity and improved therapeutic index. Onco Targets Ther. 15, 331–343 (2022).
Panowski, S., Bhakta, S., Raab, H., Polakis, P. & Junutula, J. R. Site specific antibody drug conjugates for cancer thetapy. MAbs 6, 34–45 (2014).
Li, C. et al. Clinical pharmacology of vc-MMAE antibody-drug conjugates in cancer patients: learning from eight first-in-human Phase 1 studies. MAbs 12, 1699768 (2020).
Deslandes, A. Comparative clinical pharmacokinetics of antibody-drug conjugates in first-in-human Phase 1 studies. MAbs 6, 859–870 (2014).
Mahmood, I. Clinical pharmacology of antibody-drug conjugates. Antibodies 10, 20 (2021).
Eaton, J. S., Miller, P. E., Mannis, M. J. & Murphy, C. J. Ocular adverse events associated with antibody-drug conjugates in human clinical trials. J. Ocul. Pharm. Ther. 31, 589–604 (2015).
Ali, A. et al. Emergence of ocular toxicities associated with novel anticancer therapeutics: What the oncologist needs to know. Cancer Treat. Rev. 105, 102376 (2022).
Liu, Z., Carvajal, M., Carraway, C. A., Carraway, K. & Pflugfelder, S. C. Expression of the receptor tyrosine kinases, epidermal growth factor receptor, ErbB2, and ErbB3, in human ocular surface epithelia. Cornea 20, 81–85 (2001).
Sharma, A. et al. Reversible HER2 antibody-drug conjugate-induced ocular toxicity. Can. J. Ophthalmol. 57, 118–126 (2022).
Best, R. L. et al. Microtubule and tubulin binding and regulation of microtubule dynamics by the antibody drug conjugate (ADC) payload, monomethyl auristatin E (MMAE): Mechanistic insights into MMAE ADC peripheral neuropathy. Toxicol. Appl Pharm. 421, 115534 (2021).
Acknowledgements
We thank all the patients who participated in this study. This study was supported by CSPC Zhongqi Pharmaceutical Technology (Shijiazhuang) Co., Ltd. We would like to thank Lei Wang for the editorial support, who is an employee of CSPC Zhongqi Pharmaceutical Technology (Shijiazhuang) Co., Ltd. Results from this study were shared as mini oral presentation at the ESMO 2023 annual congress.
Author information
Authors and Affiliations
Contributions
Xichun Hu and Silong Xiang were responsible for the conception of the study. Jian Zhang, Yiqun Du, Xialu Zhang, Suqiong Wu contributed to the design and acquired the data together with Yanchun Meng, Xiaojun Liu, Yuxin Mu, Yunpeng Liu, Yehui Shi, Jufeng Wang, Aimin Zang, Shanzhi Gu, Tianshu Liu, Huan Zhou and Hongqian Guo. Mengke Li provided statistical support. Silong Xiang, Xialu Zhang, Suqiong Wu, Huanghuang Qi involved in analysis and interpretation of the data. Yiqun Du drafted and revised the manuscript. All authors approved the final version as submitted.
Corresponding author
Ethics declarations
Competing interests
Silong Xiang, Xialu Zhang, Suqiong Wu, Huanhuan Qi and Mengke Li are full-time employees of CSPC Zhongqi Pharmaceutical Technology (Shijiazhuang) Co., Ltd. Other authors declare that they have no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, J., Du, Y., Meng, Y. et al. First-in-human study of DP303c, a HER2-targeted antibody-drug conjugate in patients with HER2 positive solid tumors. npj Precis. Onc. 8, 200 (2024). https://doi.org/10.1038/s41698-024-00687-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-024-00687-7
- Springer Nature Limited