
Abstract
Ocean acidification, caused by anthropogenic CO2 emissions, is predicted to have major consequences for reef-building corals, jeopardizing the scaffolding of the most biodiverse marine habitats. However, whether corals can adapt to ocean acidification and how remains unclear. We addressed these questions by re-examining transcriptome and genome data of Acropora millepora coral holobionts from volcanic CO2 seeps with end-of-century pH levels. We show that adaptation to ocean acidification is a wholistic process involving the three main compartments of the coral holobiont. We identified 441 coral host candidate adaptive genes involved in calcification, response to acidification, and symbiosis; population genetic differentiation in dinoflagellate photosymbionts; and consistent transcriptional microbiome activity despite microbial community shifts. Coral holobionts from natural analogues to future ocean conditions harbor beneficial genetic variants with far-reaching rapid adaptation potential. In the face of climate change, these populations require immediate conservation strategies as they could become key to coral reef survival.
Similar content being viewed by others


Introduction
In the last 250 years, atmospheric partial pressure of carbon dioxide (pCO2) increased by 50% due to anthropogenic fossil fuel combustion, representing the fastest pCO2 change on Earth in millions of years1. Over this period, oceans absorbed around a third of the total anthropogenic CO2 emissions2,3, resulting in a pH reduction and a decrease of carbonate saturation in surface waters commonly referred to as ocean acidification4.
Many calcifying marine organisms such as reef-building corals, whose skeletons provide the scaffolding for the most biodiverse marine habitat (i.e., coral reefs), depend on carbonate to build their calcium carbonate structures5. Although ocean warming’s devastating effects are already evident in coral reefs throughout the tropics, ocean acidification is predicted to have major consequences for marine calcifying organisms in the following decades by increasing both the dissolution of present calcium carbonate skeletons and the difficulty of depositing new skeleton6. Moreover, the combined effects of global ocean acidification and warming appear to increase global reductions in net carbonate production and accretion of most coral reefs7.
According to these predictions, many reefs will degrade within the following decades unless corals are able to keep pace with the foreseen global environmental changes. Corals’ responses are limited to two main processes: poleward range expansions or persistence through adaptation and/or acclimatization8. Examples of poleward range expansions of tropical coral species have been reported in southern Japan in response to rising sea temperatures9, while mechanisms of rapid acclimatization of corals are still being unraveled. Rapid acclimatization usually takes place at the holobiont level and may include photosymbiont shuffling, bacterial community shifts and epigenetic modifications transmitted to the progeny through transgenerational plasticity10. Although acclimatization mechanisms will surely play an important role on coral long-term survival, the amount of standing genetic variation and the presence of adaptive alleles in coral metapopulations will be decisive in future coral reef persistence11,12.
Underwater volcanic CO2 seeps act as natural analogues of a future acidified ocean and provide unique opportunities to study long-term acclimatization and adaptation to ocean acidification13,14. Coral populations occurring in these in situ natural laboratories could harbor adaptive alleles conferring them with high resilience and/or resistance to ocean acidification. These adaptive alleles may have been selected by natural selection like in coral colonies living in naturally high-temperature microclimates15. For instance, temperate azooxanthellate solitary Mediterranean corals from a CO2 seep system of Italy exhibit high genetic differentiation in genes involved in calcification16. However, no study to date has used CO2 seeps to study genomic signals of local adaptation to ocean acidification in tropical reef-building corals.
Here, we used a publicly available transcriptomic dataset (from Kenkel and coauthors17) together with recent high-quality reference genomes to (i) unveil genomic targets of natural selection in the coral host, (ii) outline algal symbiont population differentiation and (iii) assess microbiome functionality changes in the reef-building coral Acropora millepora from the CO2 seep system of Milne Bay Province in Papua New Guinea (Fig. 1). This CO2 seep system has been relatively well studied, with changes reported in diversity and composition of corals and reef-associated macroinvertebrate communities at Dobu and Upa-Upasina reefs18,19. Seep and control sites have been identified at both Dobu and Upa-Upasina reefs, with pH ranging from 7.72 to 7.81 in the seep sites and from 7.98 to 8.01 in the control sites, respectively (see Methods and Fabricius and coauthors18 for a thorough description of the study site). Kenkel and coauthors17 compared gene expression profiles of Acropora millepora and their algal symbionts from control and seep environments in Dobu and Upa-Upasina reefs, identifying core transcriptomic responses involved in long-term acclimatization to ocean acidification. Here, we called Single Nucleotide Polymorphisms (SNPs) from Kenkel and coauthors’17 transcriptomic data using a recently sequenced high-quality reference genome of A. millepora20 to uncover genomic signals of natural selection and local adaptation to high pCO2 conditions. SNPs associated with environmental variables and SNPs showing signals of natural selection provide insight into the particular genomic regions that are under selective pressure, and have commonly been used to inform about the adaptive potential of coral species21. We hypothesize that these signals exist in genes involved in calcification and skeleton formation processes, on top of changes in gene expression. Symbiodiniaceae community composition in A. millepora does not differ between coral colonies from CO2 seeps and control environments in the seep system of Milne Bay Province, with Cladocopium goreaui (previously known as Symbiodinium Clade C, type C1) being the dominant species found in all coral colonies17,21. However, we hypothesize that intraspecific genetic differences exist between CO2 seep and control C. goreaui populations due to environmental or coral host selection of specific C. goreaui genotypes. To test this hypothesis, we called SNPs using C. goreaui transcriptomic data from Kenkel and coauthors17 with a pool-seq approach, and performed population genetic analyses. Finally, Morrow and coauthors22 identified microbial community changes driven by a reduction of symbiotic Endozoicomonas in A. millepora from CO2 seeps at Upa-Upasina reef. We hypothesize that these microbial community changes also drive changes in microbial transcriptional activity and extracted microbiome metatranscriptome from Kenkel and coauthors’17 transcriptome data to unveil specific functions that might differ between A. millepora microbiomes from CO2 seep and control sites.

Inset map shows the location of Normandby and Dobu Islands within the Papua Island region (white square). Sampling sites are colored as follows: teal for Dobu Reef, brown for Upa-Upasina Reef; darker colors for control sites, lighter colors for seep sites. Kenkel and coauthors’17 schematic workflow is presented at the top of the figure, shaded in gray. The schematic workflow of this study is below with the analyses conducted for the three holobiont compartment detailed: photosymbionts on the left, coral host in the center and microbiome on the right.
Results
Candidate adaptive SNPs and population structure of the coral host
The data downloaded from SRA project PRJNA362652 consisted of a total of 316.8 million raw reads and an average of 5.4 million sequences per individual17. A total of 40.7 million clean reads mapped the Acropora millepora reference genome and passed post-mapping filters, averaging 690,440 reads per individual, and 79,273 SNPs were obtained after variant calling and variant filtering. From these 79,273 loci, 977 SNPs were associated with pH in the RDA (Fig. 2a), 656 SNPs were detected as under selection by BayPass in basic mode (Fig. 2b), 3,235 SNPs were associated with pH in the BayPass run in covariate mode (Fig. 2c), and 35 SNPs were detected as under diversifying selection by BayeScan (Fig. 2d). The overlap of these four SNP lists resulted in 625 loci that were identified by at least two of the four analyses performed (Fig. 2e), which were then considered as candidate adaptive SNPs.

For the Manhattan plots (a–c) dark gray and light gray circles represent SNPs in odd and even chromosomes, respectively, loci under selection or associated with pH are represented by circles with strokes colored by chromosome. a Redundancy Analysis (RDA). b BayPass run in basic mode (BayPass XtX). c BayPass run in covariate mode (BayPass Env). d BayeScan results showing SNPs under diversifying selection in red circles. e Venn diagram representing overlapping results among the four analyses. The 625 SNPs identified by at least two of the four analyses are represented in bold.
Population structure analyses of the candidate SNPs under selection showed genetic differentiation among sites according to their pH value. The DAPC showed an ordination of the samples on the first DAPC axis following a pH gradient (Fig. 3a). Similarly, STRUCTURE results showed that cluster assignment proportions were driven by pH (Fig. 3c).

a DAPC results using the dataset of 625 candidate adaptive SNPs. b DAPC results using the dataset of 11,169 independent neutral SNPs. Both DAPC plots are colored following Fig. 1. c STRUCTURE results with K = 2 for the candidate adaptive SNP dataset (see delta K plot in Supplementary Fig. 2b). d STRUCTURE results with K = 2 for the independent neutral SNP dataset (see delta K plot in Supplementary Fig. 2a).
A dataset of 11,169 independent neutral SNPs was obtained after removing candidate adaptive SNPs and physically linked loci. Analyses of population structure and connectivity of the independent neutral SNP dataset contrastingly showed homogeneity and panmixia among the four sampling sites (Fig. 3b, d). In agreement with these results, global FST value was low (0.002) and the migration network analysis revealed high levels of connectivity among the four sites (Supplementary Fig. 1).
Loci under selection in the coral host
The combination of A. millepora reference genome annotations with blast searches resulted in the annotation of 589 out of the 625 candidate adaptive SNPs, which were located in 441 genes (Supplementary Data 1). The 441 candidate adaptive genes included genes with key functions in the calcification process and coral response to acidification, genes involved in the establishment and maintenance of symbiosis, and genes in common with other coral adaptation studies (Table 1).
The enrichment analysis identified 528 enriched GO terms among the candidate adaptive genes (Supplementary Data 2). The top 20 enriched GO terms for each category included the Biological Processes “regulation of adherens junction organization” and “immunological memory formation process”, the Cellular Component “9 + 2 motile cilium”, and the Molecular Function “coreceptor activity involved in Wnt signaling pathway” (see Fig. 4 for complete list).

a Biological processes, b cellular components, and c molecular functions. Enrichment scores (-log10(p-value)) are plotted for each GO term, with circle size representing the ratio between the number of candidate adaptive genes with a particular GO annotation and the number of genes in the A. millepora proteome with that GO annotation. Dotted lines represent p-value = 0.05, dashed lines represent p-value = 0.01. Numerical source is provided in Supplementary Data 3.
Cladocopium goreaui population differentiation
A total of 25.8 million reads mapped the C. goreaui reference genome, with an average of 437,714 reads per population pool (i.e., coral colony). A total of 2,707 SNPs were retained after SNP calling and filtering.
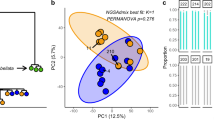
Population differentiation analyses of the C. goreaui pool-seq SNP dataset showed genetic differentiation between reefs and environments (Fig. 5). The DAPC showed that samples segregated by reef on the first DAPC axis (Fig. 5a, b). Samples from Dobu and Upa-Upasina split by environment on the second (Fig. 5a) and the third (Fig. 5b) DAPC axes, respectively. Pairwise FST analyses showed that C. goreaui population pools tend to present lower genetic differentiation within reef and within environment, and highlighted the presence of a highly differentiated seep-specific genetic cluster in Dobu Seep (Fig. 5c). In agreement with these results, the AMOVA showed that both space and environment explained a small but significant part of the genetic variation (p-values < 0.01) (Table 2).

DAPC results are shown on the first and second (a), and the first and third axes (b). c Heatmap plot of the pairwise FST matrix among C. goreaui population pools. Warm colors represent higher pairwise FST values, indicating high differentiation between population pools. Cold colors represent lower pairwise FST values, indicating low differentiation between population pools. Population pools are colored following Fig. 1 and coded in (c) as follows: Dobu Seep, DS; Dobu Control, DC; Upa-Upasina Seep, US; Upa-Upasina Control, UC.
Microbiome functional characterization
A total of 702,876 reads were taxonomically classified with Kraken2 using the NCBI taxonomic information, all of them belonging to Bacteria. The non-metric multidimensional scaling (NMDS) plot on the Bray-Curtis dissimilarities among colonies showed no clustering of samples, indicating similar abundances of metatranscriptomic reads from each taxonomic group regardless of reef and environment (Fig. 6a). In fact, the four sites presented similar percentages of metatranscriptomic reads from each phylum (Fig. 6b). Proteobacteria and Firmicutes were the dominant phyla representing ~50% and ~30% of the metatranscriptomic reads at each site, respectively, followed by Actinobacteria, Bacteroidetes, Cyanobacteria and Acidobacteria each accounting for ~3–6% of the metatranscriptomic reads.

a NMDS plot on the Bray-Curtis dissimilarities among colonies using the taxonomic classification of metatranscriptomic reads. b Percentages of microbial metatranscriptomic reads belonging to each Phylum at each sampling site. c NMDS plot on the Bray-Curtis dissimilarities among colonies using the functional classification of metatranscriptomic reads. Coral colonies are colored following Fig. 1. Numerical source is provided in Supplementary Data 4.
A total of 318,320 reads were functionally classified with Kraken2, all of them belonging to Bacteria. As for the taxonomic classification, the NMDS plot based on the functional classification showed no clustering of samples, indicating similar abundances of metatranscriptomic reads from each GO term regardless of reef and environment (Fig. 6c). Significant differences in read counts for each GO term were tested with Welch t-tests. After applying a Bonferroni correction, no GO term presented significantly different read counts between seep and control colonies.
Discussion
Our functional genomic analyses revealed that natural selection in a subset of coral host genes with key functions in calcification and skeleton formation appeared to be driving adaptation to ocean acidification in Acropora millepora in the CO2 seep system of Milne Bay Province. For instance, carbonic anhydrases play major roles in the physiology of coral calcification23,24 by catalyzing the interconversion of CO2 to bicarbonate ions and protons. Endothelin-converting enzyme, and CUB and peptidase-domain containing protein are part of the coral skeleton proteome25,26 and are involved in membrane processing, extracellular matrix/transmembrane and protein modification processes, as well as vesicle/secretion and metal binding26. Alongside these three enzymes, a relevant amount of genes encoding for coral skeletal proteins such as α-collagen, Protocadherin, Coadhesin, and Complement C3, also presented signals of adaptation to seep environments (Table 1). Additionally, signals of selection were detected in genes involved in the response to acidification (Table 1). These genes included Carbonic anhydrase 2, Fibropellin-1, Bone morphogenetic protein 7 and V-type proton ATPase subunit S1, which were differentially expressed in corals exposed to elevated pCO227,28,29.
Three of the main representative enriched GO terms, “regulation of adherens junction organization”, “9 + 2 motile cilium” and “coreceptor activity involved in Wnt signaling pathway” are intimately related to coral calcification. Intercellular junctions determine the ectodermal tissue permeability, regulating biomineralization by controlling the diffusion of molecules that can be transferred to the Extracellular Calcifying Medium (ECM)30,31. Enrichment of the GO term “9 + 2 motile cilium” suggests an important role in coral calcification of the cilia present on the ECM membrane of the coral calcifying cells32 Finally, the Wnt signaling pathway is a classical developmental signaling pathway that, in corals, controls development, growth, and calcification33,34,35. Our results demonstrate that despite the lack of population structure in the neutral SNP dataset, natural selection is occurring in core molecular pathways that control biomineralization, such as enzymes, skeletal proteins and ion channels. These results show that local adaptation is independent of migration under spatially varying selection when selective pressure is strong enough to counteract the effects of migration15,36,37,38,39. GO terms related to the immune system such as the BP “immunological memory formation process” and “response to other organism”, were also enriched in the list of candidate adaptive genes. Kenkel and coauthors17 suggested a potential impact of long-term acidification on the innate immune response of A. millepora at the CO2 seeps. Particularly, they found that the TNF receptor-associated factor (ig7766), which is involved in the innate immune response and the coral stress response40, was a top discriminatory gene for seep environment17. Interestingly, signals of selection and associations with seep environments at the TNF receptor-associated factor were found in the present study, confirming that changes in its expression are part of an adaptation mechanism to long-term elevated pCO2. Signals of environmental-driven adaptation were also found in eight additional differentially regulated loci from Kenkel and coauthors17, suggesting cis-regulation of gene expression in key components of the coral response to acidification, including calcification, transcription processes, and the immune system (Table 1).
Previous studies scanning for genomic signals of local adaptation and genotype-environment associations on reef-building corals were mostly focused on disentangling their adaptive responses to elevated temperature15,20,41,42. We found that some of the candidate adaptive genes here were shared with genes locally adapted in these studies, suggesting common targets of natural selection across environmental stressors. For instance, we found seven genes in common between our candidate adaptive genes and those conferring heat resistance in Acropora hyacinthus15 (Table 1). Additionally, our analyses revealed signals of adaptation in two Synaptotagmins, which also showed significant genetic differentiation between Platygyra daedalea colonies from the warm Persian/Arabian Gulf and the relatively cooler Gulf of Oman42. Synaptotagmins are membrane-trafficking calcium sensor proteins43 involved in maintaining calcium homeostasis during the coral stress response40. As Synaptotagmins’ expression varies with different types of cellular stress44,45, it is not surprising that they would also be the target of natural selection across different environmental stressors such as heat or elevated pCO2. Similarly, strong signals of differentiation were detected in the 26 S proteasome non-ATPase regulatory subunit in both our A. millepora from CO2 seep environments and in A. hyacinthus with different bleaching susceptibility41, albeit on different subunits (26 S proteasome non-ATPase regulatory subunit 7 and 11, respectively). Since the 26 S proteasome degrades damaged ubiquitin-conjugated proteins after any type of cellular stress46, different stressors may induce a similar selective pressure on the regulation of this protease machinery. Finally, many ribosomal proteins seem to be frequently targeted by natural selection under different long-term environmental stressors15,41. Signals of adaptation in ribosomal proteins may be due to the role of translational reprogramming during the stress response47,48 or to their extra-ribosomal functions as p53 regulators during cellular stress49.
Unlike whole transcriptome sequencing, tag-based RNAseq only produces short fragments of the transcriptome, complementary to the 3’-end of the transcripts50. Hence, our dataset could be missing other primordial gene regions bearing signals of adaptation that might have been detected with whole transcriptome or whole genome sequencing approaches. However, high linkage and low probability of recombination events within a single gene ensure that signals of selection found in tag-based RNAseq transcripts are representative of selection in that whole gene. In fact, because of the high chance of linkage within neighboring genomic regions, it is common practice in whole genome selection scans to produce thinned SNP datasets filtering physically linked SNPs within a few kbp, resulting in a similar exclusion of putative adaptive SNPs51,52.
Besides revealing signals of selection in the coral host genome, our results also showed how other compartments of the coral holobiont (Symbiodiniaceae photosymbionts and the bacterial microbiome) respond to long-term elevated pCO2. In fact, the pool-seq approach implemented in this study yielded more informative results than other commonly used methods (i.e., ITS2 DGGE profiling and RFLP17,21) about the effects of ocean acidification on algal photosymbionts. Here, we detected the presence of a highly differentiated seep-specific genetic cluster of C. goreaui in Dobu Seep site, and a combined effect of both space and environment in structuring C. goreaui populations, suggesting that symbiont shuffling may well be a long-term strategy for adaptation to acidification in A. millepora53.
Consistent with these results, we found signals of selection in coral host genes involved in the establishment and maintenance of symbiosis. For instance, 15 genes under selection known to be differentially expressed between symbiotic and apo-symbiotic coral larvae were detected in the CO2 seep colonies54,55 (Table 1). Additionally, 3 mutations in the gene coding for the Tachylectin-2 protein, a pattern recognition receptor of the innate immune response involved in photosymbiont acquisition56, were under selection; one of them causing an amino acid change (Ser264Gly). Finally, two additional genes, known to be differentially expressed between symbiotic and apo-symbiotic Aiptasia anemones, were under selection in the CO2 seep corals57 (Table 1). Accordingly, and as we mentioned above, GO terms related to the immune system were enriched among the candidate adaptive genes, which plays major roles in the establishment of the cnidarian-dinoflagellate photosymbiosis58,59. These results suggest adaptation in the regulation of photosymbiont communities in response to long-term acidification, which is considered a coral adaptation mechanism to environmental change10,51,60.
The characterization of the A. millepora microbiome revealed highly similar metatranscriptomes in coral colonies regardless of reef and environment, both taxonomically and functionally. These results indicate that despite significant microbial community changes between seep and control colonies22, the A. millepora microbial transcriptional activity is both taxonomically and functionally constant between the two environments. This could mean for instance, that the key functional activity of various taxa, such as the reduced Endozoicomonas at seep sites22, is maintained either by an extra transcriptional effort from the remaining Endozoicomonas or by other taxa taking over their functional activity. Significant coral microbiomes metatranscriptomic differences are usually found between healthy and heavily diseased coral colonies61,62. The fact that this pattern is not found between seep and control site A. millepora microbiomes most likely indicates that we are in the presence of adapted and acclimatized healthy holobionts at both sites. This interpretation is further supported by the minimal gene expression changes found between seep and control sites A. millepora coral hosts17 and the absence of significant differences in skeletal densities between A. millepora colonies from Upa-Upasina control and seep sites63, a ‘supertrait’ and an easy-to-measure indicator of coral health64,65.
Overall, our analyses clearly indicate that there are three distinct and synergistic adaptation mechanisms in three different compartments of the Acropora millepora holobiont in the CO2 seep system of Milne Bay Province. We found signals of selection in multiple coral host genes involved in calcification and symbiosis, structuring of Cladocopium goraeui genetic clusters and microbiome metatranscriptomic responses to long-term acidification. Remarkably, this study shows that A. millepora colonies living in CO2 seeps harbor genomic adaptations to the pCO2 levels foreseen by the end of the current century. These adapted natural populations may become key for the persistence of coral reefs, acting as a source of pre-adapted alleles that may facilitate rapid adaptation to climate change on a global scale, particularly in broadcast-spawning species such as A. millepora. Regardless, mitigation of anthropogenic CO2 emissions together with active conservation and restoration actions are essential for coral reef survival66, as adaptive responses alone will most likely be insufficient under the current rates of environmental change67 and the multi-factorial nature of climate change.
Methods
Study area, data processing and variant detection in the coral host genome
Milne Bay Province in Papua New Guinea hosts a well-studied shallow-water seep system of ~99% CO2 gas17,18,19,21,22 (see Fabricius and coauthors18 for a thorough description of the study site). Kenkel and coauthors17 (2018) sequenced tag-based RNAseq libraries for a total of 59 Acropora millepora colonies originating from two reefs in this system, Upa-Upasina and Dobu reefs. At each site, they sampled coral fragments from both a CO2 seep site and an adjacent control site <2.5 km apart: 14 colonies from Dobu CO2 seep site (pH = 7.72, 998 μatm pCO2), 15 from Dobu control environment (pH = 8.01, 368 μatm pCO2), 15 from Upa-Upasina CO2 seep site (pH = 7.81, 624 μatm pCO2), and 15 from Upa-Upasina control environment (pH = 7.98, 346 μatm pCO2)17 (Fig. 1). Control and seep sites had similar seawater temperature, salinity and geomorphology18.
Raw reads were downloaded from the NCBI SRA BioProject PRJNA36265268 and Single Nucleotide Polymorphisms (SNPs) from tag-based RNAseq data were called following GATK best practices and Rogier and coauthors69. First, raw reads were adapter and quality trimmed using specific perl scripts for tag-based RNAseq with default parameters (available at https://github.com/z0on/tag-based_RNAseq), followed by a quality check using fastqc 0.11.970. Clean reads were then mapped against the A. millepora high-quality reference genome20,71 using bwa-mem 0.7.1772, and sorted with samtools 1.1673. MarkDuplicates 2.18.25 in Picard tools (http://broadinstitute.github.io/picard/) was used to mitigate biases introduced during library preparation and minimize gene expression variations. Finally, SplitNCigarReads in GATK 4.2.4.174 was applied as the last post-processing step as recommended by GATK best practices.
Prior to calling variants with FreeBayes 1.175, sample names were added to each original individual BAM file and merged into one single stream with bamaddrg (https://github.com/ekg/bamaddrg). Only variants belonging to reads with mapping quality >20 were retained, using the FreeBayes flag -m 20. VCFtools 0.1.1676 was used to filter and retain biallelic SNPs (--remove-indels --min-alleles 2 --max-alleles 2) with a minimum quality score of 20 (--minQ 20) and present in at least 70% of the individuals (--max-missing 0.7). A minimum allele frequency filter of 0.05 (--maf 0.05) was set to avoid calling singleton SNPs. Our final A. millepora coral host dataset consisted of 79,273 SNPs.
Identification and annotation of loci under selection and genotype-environmental association analyses in the coral host
Four approaches were implemented to identify potential adaptive SNPs associated with pH: BayeScan 2.177, BayPass 2.378 in basic mode, BayPass in covariate mode, and a Redundancy Analysis (RDA) using the rda function in the vegan R package79. Due to the negligible population structure found among sites (see Results and Fig. 3) and in an effort to increase the number of individuals per condition as well as the model accuracy, individuals from both control sites and from both seep sites were pooled for the BayeScan and BayPass in basic mode analyses.
BayeScan was run with default parameters, using the -snp option, and a false discovery rate of 0.05 (FDR = 0.05). To control for potential population structure in the BayPass in basic mode analysis, a Linkage Disequilibrium (LD) pruned dataset was first obtained using plink 1.980 and plink 2.080. A first run was performed with the LD pruned dataset, after which the resulting omega matrix was used for the controlled run with the whole SNP dataset. To establish the threshold of the XtX statistic to consider a SNP as under selection for the BayPass in basic mode, a neutral SNP distribution was simulated with the simulate.baypass R function included in the BayPass package. Then, BayPass was run on the simulated data and, finally, SNPs under selection with an FDR = 0.05 were obtained by selecting the 95% quantile of the simulated XtX distribution.
To identify genotypes associated with pH changes, BayPass in covariate mode and an RDA were performed, using the pH value of each site as the covariate environmental data for both analyses. For the RDA, missing genotypes were first imputed with the most common genotype at each site, and the rda function was run on the imputed dataset. SNP loadings were extracted for the RDA axis 1, and the outlier threshold was established as 2.5 times the standard deviation, equaling a p = 0.01.
To avoid false positives, a SNP was considered adaptive if it appeared at least in two of the four analyses performed. This resulted in a total of 625 candidate adaptive SNPs, which were annotated using the reference genome annotations and SnpEff81. As tag-based RNAseq produces fragments of the transcriptome near the 3’-end of the transcripts50, many reads were expected to map 3`-UTR regions, which are generally poorly annotated in reference genomes. Indeed, 16.7% of the SNPs were annotated as intergenic SNPs by SnpEff using A. millepora genome annotations from Fuller and coauthors20. To improve annotations of these transcripts, BLASTN and BLASTX searches82 against the nr NCBI database were performed using default parameters. Although the exact mechanisms by which these genes operate might still be unknown, differentially expressed genes in transcriptomic studies are functionally involved in the response to the environment they were exposed to. Thus the function of the annotated candidate adaptive SNPs was investigated and when possible, determined using numerous previously published transcriptomic and functional coral studies.
A Gene Set Enrichment Analysis (GSEA) was performed to test whether there were over-represented Gene Ontology (GO) terms in the candidate adaptive SNP annotations compared to the whole genome. The proteome fasta file of the A. millepora reference genome was annotated using the PANNZER2 web server83. Then, a GSEA was performed with the GOEnrich_pannzer2 function in the PlantNGSTools R package84, using the GO annotations obtained with PANNZER2 and the list of candidate adaptive genes. Following the original description of the GSEA method85, only those gene sets with a false discovery rate <0.25 were selected (“useFDR = T, cut = 0.25”) in order to control for false positives. The REVIGO web server86 was used to cluster, remove redundant, and find representative enriched GO terms for each GO category (Biological Process, BP; Cellular Component, CC; Molecular Function, MF). Although some GO terms should be interpreted with caution in non-model organisms, the species-neutrality design of the GO database specifically allows the transfer of functional annotations from model genes to their non-models orthologues87.
Coral host population genetic analyses
To verify if corals from the two sampling sites were genetically distinct or homogeneous populations, population genetic analyses were performed. A neutral SNP dataset of 78,648 SNPs was obtained by excluding the 625 candidate adaptive SNPs using the VCFtools function --exclude-positions. Subsequently, to avoid the inclusion of physically linked loci, the neutral SNP dataset was thinned to ensure physical distance of at least 10 kbp (following e.g., Bennett and coauthors88; Mattingsdal and coauthors52; Zou and coauthors89) using the function --thin in VCFtools, resulting in a dataset of 11,169 independent neutral SNPs.
Population structure in both the independent neutral and the candidate adaptive SNP datasets was assessed using the Bayesian clustering approach in STRUCTURE 2.390 and the discriminant analyses of principal components (DAPC) implemented in the adegenet R package91. STRUCTURE was run for 200,000 MCMC iterations with a burn-in of 50,000 iterations, using the admixture model and setting the putative number of clusters (K) from 1 to 10 with 10 replicates for each run. STRUCTURE HARVESTER92 was used to find the most likely number of genetic clusters using the Evanno Delta K method93. Subsequently, CLUMPAK94 was used to average individual’s membership across replicates and graphically display STRUCTURE results. The DAPC analysis was performed using the sampling sites as a prior and choosing the optimal number of retained principal component axes using the xvalDapc function in the adegenet R package. Two and eight PC axes and two and three DA axes were retained for the neutral and the adaptive SNP datasets, respectively.
Additionally, for the independent neutral SNP dataset, a migration network was inferred using Nei’s GST method of the divMigrate function in the diveRsity R package95, and the global FST value was calculated using the basic.stats function in the hierfstat R package96.
Cladocopium goreaui endosymbiont population genetic analyses using a pool-seq approach
To test for photosymbiont genetic differentiation between sites, a pool-seq approach was implemented. In this approach, every coral colony corresponded to one population pool of algal endosymbionts. Both Noonan and coauthors21 and Kenkel and coauthors17 demonstrated that the Symbiodiniaceae type did not differ between control and CO2 seep environments, with Cladocopium goreaui (previously known as Symbiodinium Clade C, type C1) being the dominant species in both environments. Hence, clean reads from each colony were mapped against the Cladocopium goreaui reference genome obtained from http://symbs.reefgenomics.org97 using bwa-mem2 v. 2.2.198. Positions with a mapping quality <20 were filtered out with samtools view and files were sorted by reference position using samtools sort.
SNP calling of C. goreaui endosymbionts was performed using FreeBayes with the --pooled-coninuous flag to account for pooled populations of unknown number of individuals. VCFtools was used to filter and retain SNPs (--remove-indels) with a minimum quality score of 20 (--minQ 20), a minimum allele frequency of 0.05 (--maf 0.05) and present in at least 70% of the individuals (--max-missing 0.7). Our final C. goreaui photosymbiont dataset consisted of 2,707 SNPs. Population structure was assessed with a DAPC analysis as indicated above for the coral host, retaining 20 PC and three DA axes as optimized by xvalDapc. The vcf file was then transformed into a pooldata object using the vcf2pooldata function in the poolfstat 2.1.1 R package99. A pairwise FST table was calculated using the compute.pairwiseFST function in the poolfstat R package and the results were visualized with the pheatmap function in the pheatmap 1.0.12 R package100. Additionally, two analyses of molecular variance (AMOVA) were conducted using the amova function in the poppr 2.9.3 R package101 to test the partition of genetic variance in both a spatial and an environmental model, with p-values based on 1000 permutations. In the spatial model, the genetic variance was predicted by sampling site (Dobu seep, Dobu control, Upa-Upasina seep, Upa-Upasina control) nested within reef system (Dobu, Upa-Upasina). In the environmental model, the genetic variance was solely predicted by the environment of the population pools (i.e., controls vs CO2 seeps) to test whether the environment has a significant effect on the endosymbiont genetic variance.
Microbiome taxonomic and functional characterization using a metatranscriptomic approach
Microbiome transcriptomic reads were taxonomically and functionally classified in order to study potential microbiome transcriptomic differences across environments. Taxonomic classification (indicative of transcriptionally active taxa) was performed on the unmapped reads (i.e., those that did not map the coral nor the photosymbiont genomes) with Kraken2 2.1.2102, using the NCBI taxonomic information combined with the RefSeq libraries “bacteria”, “archaea”, “fungi”, and “viral”. Kraken-biom 1.0.1103 was used to create a BIOM table from all Kraken reports that was then imported into R using the import_biom function in the phyloseq 1.40 R package104. Relative abundance of transcriptomic reads were plotted at the phylum level by site (following Morrow and coauthors22) using ggplot2 3.4.1105, and a non-metric multidimensional scaling (NMDS) plot was performed with the ordinate and plot_ordination functions in phyloseq using the NMDS method on the Bray-Curtis dissimilarities among colonies (method = “NMDS”, distance = “bray”).
Differences in microbiome functionality were assessed between environments following Gardiner and coauthors106. Functional classification was performed with a tree-shaped hierarchy of GO terms using the Molecular Function subset106. This GO tree-shaped hierarchy was combined with the RefSeq libraries “bacteria”, “archaea”, “fungi” and “viral” to build a database and to perform a direct functional classification of our unmapped clean reads with Kraken2 2.1.2. Kraken-biom 1.0.1 was used to create a BIOM table from all Kraken reports that was then imported into R using the import_biom function in phyloseq. An NMDS plot was performed on Bray-Curtis dissimilarities among colonies as detailed above. Additionally, in order to identify Molecular Functions that significantly differed between environments, a Welch t-test was run for every GO term independently using the oneway.test function in the stats 4.2.3 R package107, grouping samples by environment (seep vs control colonies). A Bonferroni correction was applied to the significance level to avoid false positives due to multiple testing.
Statistics and reproducibility
Four analyses were used to identify SNPs under natural selection or associated with pH in the coral host, and then those present in at least two of the four analyses were considered as under selection in order to avoid false positives. Subseqently, a Gene Set Enrichment Analysis was performed to detect over-represented GO terms in the candidate adaptive SNP annotations, using a false discovery rate approach to control for false positives. See more details in Methods section, subsection “Identification and annotation of loci under selection and genotype-environment association analyses in the coral host”.
For the coral microbiome, a Welch t-test was run for every GO term independently using the oneway.test function in the stats 4.2.3 R package107, in order to identify Molecular Functions that significantly differed between environments. The Welch t-test was chosen after checking for equal variances using the var.test in the stats 4.2.3 R package107, which indicated that there were significant differences in variances and hence the classic t-test could not be applied. A Bonferroni correction was applied to the significance level to avoid false positives due to multiple testing, lowering the significance level from 0.05 to 0.00027.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data for this study was obtained from public repositories: NCBI SRA BioProject PRJNA36265268, NCBI SRA BioProject PRJNA76766171, and the reefgenomics.org97 database. Numerical source data for graphs and charts can be found in Supplementary Data 3 and Supplementary Data 4. All other data are available from the corresponding author on reasonable request.
References
Doney, S. C. & Schimel, D. S. Carbon and climate system coupling on timescales from the Precambrian to the Anthropocene. Annu Rev. Env. Resour. 32, 31–66 (2007).
Khatiwala, S. et al. Global ocean storage of anthropogenic carbon. Biogeosciences 10, 2169–2191 (2013).
Friedlingstein, P. et al. Global carbon budget 2021. Earth Syst. Sci. Data 14, 1917–2005 (2022).
Doney, S. C., Fabry, V. J., Feely, R. A. & Kleypas, J. A. Ocean acidification: the other CO2 problem. Annu. Rev. Mar. Sci. 1, 169–192 (2009).
Knowlton, N. et al. Coral reef biodiversity. in Life in the World’s oceans: diversity, distribution, and abundance. (ed. Mcintyre, A. D.) 65–79 (Oxford, UK: Wiley-Blackwell, 2010).
Hoegh-Guldberg, O. et al. Coral reefs under rapid climate change and ocean acidification. Science 318, 1737–1742 (2007).
Cornwall, C. E. et al. Global declines in coral reef calcium carbonate production under ocean acidification and warming. Proc. Natl Acad. Sci. USA 118, e2015265118 (2021).
Pandolfi, J. M. & Kiessling, W. Gaining insights from past reefs to inform understanding of coral reef response to global climate change. Curr. Opin. Environ. Sustain. 7, 52–58 (2014).
Yamano, H., Sugihara, K. & Nomura, K. Rapid poleward range expansion of tropical reef corals in response to rising sea surface temperatures. Geophys. Res. Lett. 38, 4 (2011).
Torda, G. et al. Rapid adaptive responses to climate change in corals. Nat. Clim. Change 7, 627–636 (2017).
Barrett, R. D. & Schluter, D. Adaptation from standing genetic variation. Trends Ecol. Evol. 23, 38–44 (2008).
Bitter, M. C., Kapsenberg, L., Gattuso, J.-P. & Pfister, C. A. Standing genetic variation fuels rapid adaptation to ocean acidification. Nat. Commun. 10, 5821 (2019).
González-Delgado, S. & Hernández, J. C. The importance of natural acidified systems in the study of ocean acidification: what have we learned? Adv. Marine Biol. 80, 57–99 (2018).
Kang, J. et al. Rapid evolution fuels transcriptional plasticity to ocean acidification. Glob. Change Biol. 28, 3007–3022 (2022).
Bay, R. A. & Palumbi, S. R. Multilocus adaptation associated with heat resistance in reef-building corals. Curr. Biol. 24, 2952–2956 (2014).
Teixidó, N. et al. Ocean acidification causes variable trait‐shifts in a coral species. Glob. Change Biol. 26, 6813–6830 (2020).
Kenkel, C. D., Moya, A., Strahl, J., Humphrey, C. & Bay, L. K. Functional genomic analysis of corals from natural CO2‐seeps reveals core molecular responses involved in acclimatization to ocean acidification. Glob. Change Biol. 24, 158–171 (2018).
Fabricius, K. E. et al. Losers and winners in coral reefs acclimatized to elevated carbon dioxide concentrations. Nat. Clim. Change 1, 165–169 (2011).
Fabricius, K. E., De’ath, G., Noonan, S. & Uthicke, S. Ecological effects of ocean acidification and habitat complexity on reef-associated macroinvertebrate communities. Proc. R. Soc. B Biol. Sci. 281, 20132479 (2014).
Fuller, Z. L. et al. Population genetics of the coral Acropora millepora: Toward genomic prediction of bleaching. Science 369, eaba4674 (2020).
Noonan, S. H., Fabricius, K. E. & Humphrey, C. Symbiodinium community composition in scleractinian corals is not affected by life-long exposure to elevated carbon dioxide. PLoS One 8, e63985 (2013).
Morrow, K. M. et al. Natural volcanic CO2 seeps reveal future trajectories for host–microbial associations in corals and sponges. ISME J. 9, 894–908 (2015).
Goreau, T. F. The physiology of skeleton formation in corals. I. A method for measuring the rate of calcium deposition by corals under different conditions. Biol. Bull. 116, 59–75 (1959).
Bertucci, A. et al. Carbonic anhydrases in anthozoan corals—A review. Bioorg. Med. Chem. 21, 1437–1450 (2013).
Ramos-Silva, P. et al. The skeletal proteome of the coral Acropora millepora: the evolution of calcification by co-option and domain shuffling. Mol. Biol. Evol. 30, 2099–2112 (2013).
Peled, Y. et al. Optimization of skeletal protein preparation for LC–MS/MS sequencing yields additional coral skeletal proteins in Stylophora pistillata. BMC Mater. 2, 1–15 (2020).
Moya, A. et al. Whole transcriptome analysis of the coral Acropora millepora reveals complex responses to CO2‐driven acidification during the initiation of calcification. Mol. Ecol. 21, 2440–2454 (2012).
Kaniewska, P. et al. Major cellular and physiological impacts of ocean acidification on a reef building coral. PloS One 7, e34659 (2012).
Vidal-Dupiol, J. et al. Genes related to ion-transport and energy production are upregulated in response to CO2-driven pH decrease in corals: new insights from transcriptome analysis. PloS One 8, e58652 (2013).
Tambutté, S. et al. Coral biomineralization: From the gene to the environment. J. Exp. Mar. Biol. Ecol. 408, 58–78 (2011).
Tambutté, E. et al. Calcein labelling and electrophysiology: insights on coral tissue permeability and calcification. Proc. R. Soc. B Biol. Sci. 279, 19–27 (2012).
Tambutté, E., Ganot, P., Venn, A. A. & Tambutté, S. A role for primary cilia in coral calcification? Cell Tissue Res 383, 1093–1102 (2021).
Hemond, E. M., Kaluziak, S. T. & Vollmer, S. V. The genetics of colony form and function in Caribbean Acropora corals. BMC Genom.15, 1–21 (2014).
Karako-Lampert, S. et al. Transcriptome analysis of the scleractinian coral Stylophora pistillata. PLoS One 9, e88615 (2014).
Gutner-Hoch, E., Ben-Asher, H. W., Yam, R., Shemesh, A. & Levy, O. Identifying genes and regulatory pathways associated with the scleractinian coral calcification process. PeerJ 5, e3590 (2017).
Levene, H. Genetic equilibrium when more than one ecological niche is available. Am. Nat. 87, 331–333 (1953).
Yeaman, S. & Otto, S. P. Establishment and maintenance of adaptive genetic divergence under migration, selection, and drift. Evolution 65, 2123–2129 (2011).
Savolainen, O., Lascoux, M. & Merilä, J. Ecological genomics of local adaptation. Nat. Rev. Genet. 14, 807–820 (2013).
Leiva, C. et al. Population substructure and signals of divergent adaptive selection despite admixture in the sponge Dendrilla antarctica from shallow waters surrounding the Antarctic Peninsula. Mol. Ecol. 28, 3151–3170 (2019).
Barshis, D. J. et al. Genomic basis for coral resilience to climate change. Proc. Natl Acad. Sci. USA 110, 1387–1392 (2013).
Rose, N. H. et al. Genomic analysis of distinct bleaching tolerances among cryptic coral species. Proc. R. Soc. B 288, 20210678 (2021).
Smith, E. G. et al. Signatures of selection underpinning rapid coral adaptation to the world’s warmest reefs. Sci. Adv. 8, eabl7287 (2022).
Südhof, T. C. Synaptotagmins: why so many? J. Biol. Chem. 277, 7629–7632 (2002).
DeSalvo, M. K., Sunagawa, S., Voolstra, C. R. & Medina, M. Transcriptomic responses to heat stress and bleaching in the elkhorn coral Acropora palmata. Mar. Ecol. Prog. Ser. 402, 97–113 (2010).
DeSalvo, M. K., Estrada, A., Sunagawa, S. & Medina, M. Transcriptomic responses to darkness stress point to common coral bleaching mechanisms. Coral Reefs 31, 215–228 (2012).
Mao, Y. Structure, dynamics and function of the 26S proteasome. Subcell. Biochem. 96, 1–151 (2021).
Spriggs, K. A., Bushell, M. & Willis, A. E. Translational regulation of gene expression during conditions of cell stress. Mol. Cell 40, 228–237 (2010).
Zhang, X. et al. Translational control of the cytosolic stress response by mitochondrial ribosomal protein L18. Nat. Struct. Mol. Biol. 22, 404–410 (2015).
Kim, T.-H., Leslie, P. & Zhang, Y. Ribosomal proteins as unrevealed caretakers for cellular stress and genomic instability. Oncotarget 5, 860 (2014).
Matz, M. V. Fantastic beasts and how to sequence them: ecological genomics for obscure model organisms. Trends Genet 34, 121–132 (2018).
Cooke, I. et al. Genomic signatures in the coral holobiont reveal host adaptations driven by Holocene climate change and reef specific symbionts. Sci. Adv. 6, eabc6318 (2020).
Mattingsdal, M. et al. Demographic history has shaped the strongly differentiated corkwing wrasse populations in Northern Europe. Mol. Ecol. 29, 160–171 (2020).
Tanvet, C. et al. Corals adapted to extreme and fluctuating seawater pH increase calcification rates and have unique symbiont communities. Ecol. Evol. 13, e10099 (2023).
Mohamed, A. R. et al. The transcriptomic response of the coral Acropora digitifera to a competent Symbiodinium strain: the symbiosome as an arrested early phagosome. Mol. Ecol. 25, 3127–3141 (2016).
Yoshioka, Y. et al. Whole-genome transcriptome analyses of native symbionts reveal host coral genomic novelties for establishing coral–algae symbioses. Genome Biol. Evol. 13, evaa240 (2021).
Kuniya, N. et al. Possible involvement of Tachylectin-2-like lectin from Acropora tenuis in the process of Symbiodinium acquisition. Fish. Sci. 81, 473–483 (2015).
Oakley, C. A. et al. Symbiosis induces widespread changes in the proteome of the model cnidarian Aiptasia. Cell. Microbiol 18, 1009–1023 (2016).
Kvennefors, E. C. E. et al. Analysis of evolutionarily conserved innate immune components in coral links immunity and symbiosis. Dev. Comp. Immunol. 34, 1219–1229 (2010).
Neubauer, E.-F. et al. A diverse host thrombospondin-type-1 repeat protein repertoire promotes symbiont colonization during establishment of cnidarian-dinoflagellate symbiosis. Elife 6, e24494 (2017).
Quigley, K. M., Bay, L. K. & Willis, B. L. Leveraging new knowledge of Symbiodinium community regulation in corals for conservation and reef restoration. Mar. Ecol. Prog. Ser. 600, 245–253 (2018).
Daniels, C. A. et al. Metatranscriptome analysis of the reef-building coral Orbicella faveolata indicates holobiont response to coral disease. Front. Mar. Sci. 2, 62 (2015).
Sato, Y. et al. Unraveling the microbial processes of black band disease in corals through integrated genomics. Sci. Rep. 7, 40455 (2017).
Strahl, J. et al. Physiological and ecological performance differs in four coral taxa at a volcanic carbon dioxide seep. Comp. Biochem. Physiol. A. Mol. Integr. Physiol. 184, 179–186 (2015).
Madin, J. S. et al. A trait-based approach to advance coral reef science. Trends Ecol. Evol. 31, 419–428 (2016).
Rocker, M. M., Francis, D. S., Fabricius, K. E., Willis, B. L. & Bay, L. K. Variation in the health and biochemical condition of the coral Acropora tenuis along two water quality gradients on the Great Barrier Reef, Australia. Mar. Pollut. Bull. 119, 106–119 (2017).
Kleypas, J. et al. Designing a blueprint for coral reef survival. Biol. Conserv. 257, 109107 (2021).
Hoegh-Guldberg, O. Coral reef sustainability through adaptation: glimmer of hope or persistent mirage? Curr. Opin. Environ. Sustain. 7, 127–133 (2014).
NCBI BioProject Accession. Tag based RNA-seq of A. millepora from CO2-seeps in PNG. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA362652/ (2017).
Rogier, O. et al. Accuracy of RNAseq based SNP discovery and genotyping in Populus nigra. BMC Genom.19, 1–12 (2018).
Andrews, S. FastQC: a Quality Control Tool for High Throughput Sequence Data (Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom, 2010).
NCBI BioProject Accession. Acropora millepora RefSeq Genome sequencing and assembly. https://www.ncbi.nlm.nih.gov/assembly/GCF_013753865.1 (2019).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv Prepr. arXiv https://doi.org/10.48550/arXiv.1303.3997 (2013).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Van der Auwera, G. A. & O’Connor, B. D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (O’Reilly Media, 2020).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv https://doi.org/10.48550/arXiv.1207.3907 (2012).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Foll, M. & Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180, 977–993 (2008).
Gautier, M. Genome-wide scan for adaptive divergence and association with population-specific covariates. Genetics 201, 1555–1579 (2015).
Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930 (2003).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, s13742-015-0047–8 (2015).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. (Austin) 6, 80–92 (2012).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402 (1997).
Törönen, P., Medlar, A. & Holm, L. PANNZER2: a rapid functional annotation web server. Nucleic Acids Res 46, W84–W88 (2018).
Marble. PlantNGSTools: NGS Bioinformatics Tools for Plants. https://github.com/biomarble/PlantNGSTools/ (2023).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. 102, 15545–15550 (2005).
Supek, F., Bošnjak, M., Škunca, N. & Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PloS One 6, e21800 (2011).
Primmer, C. R., Papakostas, S., Leder, E. H., Davis, M. J. & Ragan, M. A. Annotated genes and nonannotated genomes: cross‐species use of Gene Ontology in ecology and evolution research. Mol. Ecol. 22, 3216–3241 (2013).
Bennett, K. L., McMillan, W. O. & Loaiza, J. R. The genomic signal of local environmental adaptation in Aedes aegypti mosquitoes. Evol. Appl. 14, 1301–1313 (2021).
Zou, T. et al. Uncovering the enigmatic evolution of bears in greater depth: the hybrid origin of the Asiatic black bear. Proc. Natl Acad. Sci. 119, e2120307119 (2022).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Jombart, T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Earl, D. A. & VonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Kopelman, N. M., Mayzel, J., Jakobsson, M., Rosenberg, N. A. & Mayrose, I. Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 15, 1179–1191 (2015).
Keenan, K., McGinnity, P., Cross, T. F., Crozier, W. W. & Prodöhl, P. A. diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 4, 782–788 (2013).
Goudet, J. Hierfstat, a package for R to compute and test hierarchical F‐statistics. Mol. Ecol. Notes 5, 184–186 (2005).
Liew, Y. J., Aranda, M. & Voolstra, C. R. Reefgenomics.Org—a repository for marine genomics data. Database 2016, baw152 (2016).
Vasimuddin, M., Misra, S., Li, H. & Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. in 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS) 314–324 (IEEE, 2019).
Gautier, M., Vitalis, R., Flori, L. & Estoup, A. f‐Statistics estimation and admixture graph construction with Pool‐Seq or allele count data using the R package poolfstat. Mol. Ecol. Resour. 22, 1394–1416 (2022).
Kolde, R. pheatmap: Pretty Heatmaps. https://rdrr.io/cran/pheatmap/ (2019).
Kamvar, Z. N., Tabima, J. F. & Grünwald, N. J. Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2, e281 (2014).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 1–13 (2019).
Dabdoub, S. Kraken-Biom Enabling Interoperative Format Convers. Kraken Results Version 12 https://github.com/smdabdoub/kraken-biom (2016).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8, e61217 (2013).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, 2026).
Gardiner, L.-J. et al. Re-purposing software for functional characterization of the microbiome. Microbiome 9, 1–12 (2021).
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2023).
Zoccola, D. et al. Specific expression of BMP2/4 ortholog in biomineralizing tissues of corals and action on mouse BMP receptor. Mar. Biotechnol. 11, 260–269 (2009).
Drake, J. L. et al. Proteomic analysis of skeletal organic matrix from the stony coral Stylophora pistillata. Proc. Natl Acad. Sci. USA 110, 3788–3793 (2013).
Acknowledgements
We thank Jeffrey Centino and Dr. Bastian Bentlage for their computing support. This work was supported by the National Science Foundation NSF-EPSCoR Grant No. OIA-1946352. Funding also came from the Spanish Government grants ADAPTIVE (PGC2018-100735-B-I00/MCIU/AEI/FEDER, UE) and ENVIOME (PID2021-128094NB-I00/MCIN/AEI/10.13039/501100011033/ and FEDER una manera de hacer Europa), a “Ramón y Cajal” contract to R.P.-P. (RYC2018-025070-I), and the project “DIVERGEN—Ayudas fundación BBVA a Proyectos Investigación Científica 2021”.
Author information
Authors and Affiliations
Contributions
C.L. and R.P.-P. conceived and designed the study; C.L. performed the analyses; C.L., R.P.-P. and S.L. interpreted and discussed the results; S.L. obtained funding; C.L. wrote the first version of the manuscript, and all coauthors contributed to the current version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Javier Rodriguez-Casariego and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Linn Hoffmann, George Inglis, and David Favero. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leiva, C., Pérez-Portela, R. & Lemer, S. Genomic signatures suggesting adaptation to ocean acidification in a coral holobiont from volcanic CO2 seeps. Commun Biol 6, 769 (2023). https://doi.org/10.1038/s42003-023-05103-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-05103-7
- Springer Nature Limited