
Abstract
The limitations of TB treatment are the long duration and immune-dampening effects of anti-tuberculosis therapy. The Cell wall plays a crucial role in survival and virulence; hence, enzymes involved in its biosynthesis are good therapeutic targets. Here, we identify Mycobacterium tuberculosis (Mtb) GlmM, (GlmMMtb) engaged in the UDP-GlcNAc synthesis pathway as an essential enzyme. We generated a conditional knockdown strain, Rv-glmMkD using the CRISPR interference-mediated gene silencing approach. Depletion of GlmMMtb affects the morphology and thickness of the cell wall. The Rv-glmMkD strain attenuated Mtb survival in vitro, in the host macrophages (ex vivo), and in a murine mice infection model (in vivo). Results suggest that the depletion of GlmMMtb induces M1 macrophage polarization, prompting a pro-inflammatory cytokine response, apparent from the upregulation of activation markers, including IFNɣ and IL-17 that resists the growth of Mtb. These observations provide a rationale for exploring GlmMMtb as a potential therapeutic target.
Similar content being viewed by others


Introduction
Tuberculosis (TB) is a deadly disease caused by one of the most successful and terrifying human pathogens, Mtb that silently encompasses most of the human population. Even though anti-tuberculosis therapy (ATT) can eradicate drug-sensitive strains in 6-8 months of treatment with a directly observed treatment short course (DOTS). The failure in the implementation of the full course becomes the reason for emerging DR (drug-resistant) strains1. The major drawback of ATT is that it does not have an immune modulator as suggested by the WHO to control contrary effects on the host (WHO. 2007). Therefore, innovative therapeutic strategies are urgently needed involving the identification of new drug targets and the impact of their inhibition on the host immune system to achieve the END TB target by 2030 (WHO. 2021). Post-infection Mtb is phagocytosed by antigen-presenting cells (APCs), which activate T lymphocytes to upregulate protective pro-inflammatory cytokines2,3. A complex immunological response is involved in the case of TB that decides the fate of infection predominantly governed by subsets of T lymphocytes4. The pro-inflammatory cytokine response primarily mediated by IFNγ and IL17 produced from CD4+ T cell subsets has been implicated in mediating its protective effect on macrophages and phagosomes by enhancing their defense mechanisms and antigenic stimulation5. While the immunological response during TB is entangled, Th1 and Th17 cells contribute to long-term immunity against TB. Conversely, Th2 and Treg cells critically impede the host’s protective immune responses and aid in disease progression. These host protective responses regulate a bacteria-free environment within the host6,7.
Pathogens have developed various mechanisms to survive stressful conditions offered by the host. Mycobacterial species have a unique cell wall structure that plays a key role in their growth, virulence, survival inside the host, and escape from immune responses8,9. The impermeability of cell walls is one major concern that affects the efficacy of existing antibiotics10. Therefore, identifying targets that can affect the strength of the cell wall or make it more pores could be an important strategy to deal with the pathogen. Hence, the enzymes involved in cell wall synthesis offer potential targets for new anti-tuberculosis drugs. It is composed of three layers of outer mycolic acid (MA), which is connected to the lower peptidoglycan (PG) layer via middle arabinogalactan (AG). PG is a repeat disaccharide unit (N-acetylmuramic acid-N-acetylglucosamine) attached to AG through a disaccharide linker (a-L-rhamnosyl-a-D-N-acetylglucosaminosyl-1-phosphate). UDP-N-acetylglucosamine (UDP-GlcNAc) is a direct glycosyl donor of the disaccharide linker and a precursor for PG synthesis, and it is also involved in various reactions8. Hence, its biosynthesis is crucial which starts from a glycolytic intermediate D-fructose-6-phosphate11. First, Glucosamine-6-phosphate synthase (GlmS) converts fructose-6-phosphate into glucosamine-6-phosphate, which becomes glucosamine-1-phosphate by Phosphoglucosamine mutase (GlmM) activity during the second reaction. Subsequently, glucosamine-1-phosphate acetyltransferase/N-acetylglucosamine-1-phosphate uridyltransferase (GlmU) converts glucosamine-1-phosphate into N-acetylglucosamine-1-phosphate by acetyltransferase activity followed by its uridyltransferase activity and the final product UDP-GlcNAc forms. However, in eukaryotes, glucosamine-6-phosphate converts into N-acetylglucosamine-6-phosphate by the activity of GAT (acetyl-CoA: D-glucosamine-6-phosphate N-acetyltransferase) which further converts into N-acetylglucosamine-1-phosphate by GNA1 (glucose-6-phosphate acetyltransferase; UAP1, UDP-N-Acetylglucosamine Pyrophosphorylase)12. The reactions catalyzed by GlmM and the first reaction (acetyltransferase activity) of GlmU are unique to prokaryotes, making them attractive therapeutic targets. GlmUMtb has been studied thoroughly, and effective inhibitors have been designed, which are under further investigation13,14,15.
The importance of GlmM was identified in E. coli, where its inactivation resulted in disturbed morphology and cell lysis16. Subsequently, it was studied that ureC from Helicobacter pylori restored the lethal effects of glmM mutant in E. coli17. A mutation in glmM affects cell growth, morphology, biofilm formation, and sensitivity to penicillin with increased polymorphonuclear leukocyte (PMN)-dependent killing in Streptococcus gordonii (2008, FEMS, 2009 FEMS). In Bacillus anthracis, crystal structure revealed key residues that play a role in catalysis and specificity18. GlmM was identified as a modulator of c-di-AMP levels in Lactococcus lactis and Staphylococcus aureus, where C-di-AMP synthesis enzyme CdaA and DacA was shown to be inhibited by direct binding of GlmM19,20. GlmM from M. smegmatis, MSMEG_1556, and Mtb, rv3441c have been identified, and the effects of mutation on biofilm formation and antimicrobial susceptibilities have been studied in M. smegmatis21,22.
In this study, we set out to answer if glmMMtb is essential for bacterial survival and virulence and to what extent. We have performed in vitro, ex vivo, and in vivo experiments to examine the importance of this gene in the growth of bacteria outside and inside the host and found that GlmM is essential in all three conditions. However, in the later part of the manuscript, we sought to evaluate the impact of GlmM depletion on the host immune system. Interestingly, we have observed increased pro-inflammatory immune response from macrophages and enhanced Th1 and Th17 cell activation in the lung and spleen of mice in the case of mutants compared to controls. It suggests that GlmM plays a crucial role in mycobacterial virulence, and suppressed expression of GlmM causes a better immune response. Hence, the study’s general objective was to establish GlmM as a potential drug target from both the angles of pathogen and host. Together, the data presented here demonstrate that GlmMMtb is a viable and promising target for therapeutic intervention against TB.
Results
GlmMMtb is essential for bacterial growth
High-throughput transposon-based mutagenesis studies suggested glmMMtb to be an essential gene for the in vitro growth of the bacteria23,24 as it is involved in the synthesis of UDP-GlcNAc a central metabolite of cell wall biosynthesis (Fig. 1a). Thus to understand the requirement of GlmM, we generated knockdown strains of Mtb H37Rv by using the CRISPRi-based approach described earlier25 and the schematic of the approach is provided in Fig. S1. The strain Rv-glmMkD thus generated shows depletion of GlmM in an anhydrotetracycline (ATc)-dependent manner, wherein the addition of ATc results in a knockdown. We have also generated Rv_vector control to check the impact of vector background on the phenotype of Rv-glmMkD strain. To confirm the knockdown, we grew the Rv and Rv-glmMkD in the liquid cultures in the presence and absence of ATc, which showed the inability of the Rv-glmMkD strain to sustain growth in the presence of ATc (Fig. 1b). To validate that observed growth defects are due to the downregulation of GlmMMtb, we compared RNA and protein expression levels in the presence and absence of ATc. Data revealed ~50 and ~85% suppression at the RNA level in the presence of 25 and 50 (ng/ml) of ATc, respectively, while the level of glmMMtb in the absence of ATc was comparable to Rv (Fig. 1c). Western blot analysis using GlmM antibodies (Fig. S2) showed that in the presence of ATc, the protein levels were significantly lower by the 4th day, which became undetectable on 7th day (Fig. 1d), confirming the generation of glmMMtb knockdown strain. In all subsequent experiments, we used 50 ng/ml ATc.

a Schematic of the UDP-GlcNAc synthesis pathway. In vitro growth analysis of different strains of Mtb H37Rv. b Rv and Rv-glmMkD cultures were gan rown to log phase to seed fresh cultures at an initial A600 of 0.1 in the presence and absence of ATc, growth of the culture in the tubes was accessed on 6th day. c Validation of CRISPRi-mediated silencing of genes by quantitative RT-PCR. Quantitative RT-PCR analysis showing levels of glmM transcripts in the knockdown strains of Mtb H37Rv. d Rv and Rv-glmMkD cultures grown in the absence of ATc were seeded at A600 of 0.1 and the cultures were grown in the absence or presence of ATc. Whole cell lysates (WCL) were prepared second day onwards post ATc addition. The WCLs were resolved and probed with α-GlmM and α-GroEL antibodies. e Growth of Rv-glmMkD strains was monitored in the presence of ATc (50 ng/ml) relative to controls by measuring A600 of bacterial cultures, days-wise growth was monitored by observing A600 till the 7th day. f Rv and Rv-glmMkD cultures were seeded at A600 of 0.1 in 7H9-ADC and the in vitro growth was monitored by enumerating CFU on 0, 4, and 6th day. The data represent the mean CFU log10/ml ± standard deviation (SD) of three independent replicates. **p < 0.001. g Rv and Rv-glmMkD cultures were inoculated at an initial A600 of 0.1 and the growth was monitored every day for eight days. ATc was added on 2nd day (A), on 4th day (B), on 6th day (C) ATc was added to the Rv culture on day 0 and Rv-glmMkD cultures were either grown in the absence of ATc or were supplemented with ATc in the growth media on 0, 2nd, 4th, or 6th day. Statistical significance was determined using unpaired t-test.
Next, we analyzed the growth profile of the Rv-glmMkD strain in the presence and absence of ATc by absorbance (A600) and colony-forming unit (CFU) over seven days. As presented in Fig. 1b, control cultures achieved A600 of ~4.3, while depleted Rv-glmMkD cultures failed to grow beyond A600 of ~0.27 (Fig. 1e). CFU enumeration indicated >2 log10 folds (100 fold) compromised growth in the mutant strain on the 4th day and >4 log10 fold differences 6 days post-ATc addition (Fig. 1f). However, we have not observed any significant growth difference in Rv_vector control in the presence and absence of ATc till 6th days (Fig. S3a). An ideal target of therapeutic intervention should be essential at different stages of growth, and early intervention should result in pathogen clearance. To evaluate the impact of GlmM depletion at different growth stages, we added ATc on either the day 0, 2nd, 4th, or 6th day post-inoculation. We observed a substantial reduction in growth even when ATc was added 4 days post-inoculation (Fig. 1g). Collectively, results suggest the importance of GlmM at early and late stages of bacterial growth in the extracellular conditions.
GlmM is essential for growth in hypoxic conditions
Survival in hypoxic conditions is crucial for being a successful pathogen. It was previously observed that GlmM plays an important role in biofilm formation in M. smegmatis, whose core is hypoxic21,22. Hence, we examined the survival of the Rv-glmMkD strain in the modified Wayne’s model26. Hypoxia was first established (for 20 days), followed by depletion of GlmM by the addition of ATc for 10 and 20 days (Fig. 2a). The addition of ATc resulted in significantly reduced growth at both time points (Fig. 2b), indicating the importance of GlmM in hypoxic conditions.

a Schematic outline of the hypoxia experiment. b Rv and Rv-glmMkD cultures were seeded at an initial A600 of 0.1 in 1.5 ml HPLC tubes containing penetrable caps. The establishment of hypoxia was monitored with the help of methylene blue color change (blue to colorless). CFUs were enumerated on day 0, day 20, and day 40. c Scanning electron microscopy of Rv and Rv-glmMkD grown for 96 h with or without ATc as indicated. The experiment was repeated thrice. d Transmission electron micrographs in result at 50,000X of Rv and Rv-glmMkD cultures grown with or without ATc. Scale bar: 20 nm. e Cell wall thickness was measured in nm for ~18 cells for each sample. ***p < 0.0001. Statistical significance was determined using an unpaired t-test.
Previous studies showed that orthologs of GlmM are involved in cell wall synthesis, and its depletion distorts the morphology of the cell27,28,29. To determine the impact of GlmMMtb depletion on cellular morphology, we performed TEM (transmission electron microscopy) and SEM (scanning electron microscopy) imaging analysis of Rv and Rv-glmMkD cells grown for 96 h in the absence and presence of ATc. The addition of ATc did not impact the cell morphology in the case of Rv. On the other hand, in the absence of GlmMMtb (+ATc condition), cells were crumbled and fused (Fig. 2c). TEM analysis indicated that in the case of Rv and Rv-glmMkD-ATc, the cell wall thickness is comparable. However, there we observed a significant decrease in the cell wall thickness in Rv-glmMkD + ATc samples (Fig. 2d, e). Together data suggests that the absence of GlmM results in decreased cell wall thickness, eventually crumbling and death.
GlmM depletion induces protective immunity inside macrophages
Macrophages are the key cellular source of cytokines that impact the commencement of both innate and adaptive immune arms. It is established from the previous studies that for survival and pathogenesis, Mtb escapes this immune attack by modulating the macrophage defense in its own favor30. Macrophages can polarize in two ways M1 and M2; while M1 secretes pro-inflammatory cytokines like IL-1β, IL-6, IL-12, and TNF-α that promotes resistance against Mtb, M2 induces an anti-inflammatory response like IL-10 that favors Mtb growth31. Mtb is known to favor M2 polarization and suppress the M1 response32. To evaluate the impact of GlmM depletion on bacterial survival in macrophages, we infected peritoneal macrophages (PФ) isolated from C57BL6 mice with Rv and Rv-glmMkd. GlmM was depleted from one set of Rv-glmMkd infected cells by adding ATc in the culture media at 4 h post-infection (p.i) (Fig. 3a). The infected cells were analyzed at different time points p.i for different purposes such as CFU analysis, cytokine profiling and FACS, western blot, and qRT-PCR experiments (Fig. 3a). We observed compromised survival of knockdown strain in the presence of ATc compared to controls (Rv and Rv-glmMkD-ATc). Whereas Rv and Rv-glmMkD-ATc showed 4 and 4.2 log10 CFU values at 96 h p.i the log10 CFU value of 2 was observed in Rv-glmMkD + ATc infected cells (Fig. 3b). No significant change in CFU was observed in Rv_vector control in the presence and absence of ATc (Fig. S3b), suggesting that these effects are not due to vector background. It is specifically due to the depletion of glmM. Moreover, the presence of ATc does not make any significant impact on cell survival (Fig. S4).

a Schematic to show experiment layout of ex vivo experiment. b Peritoneal macrophages were infected with Rv and Rv-glmMkD strains at 1:10 MOI. 4 hpi cells were washed and RPMI with ATc were added in one set of cells. The intracellular bacillary load was checked by CFU enumeration at 24 and 96 hpi. The data represent the mean CFU log10/ml ± SD of three independent replicates. c RNA was isolated from infected macrophages and used in RT PCR analysis to check the expression of macrophage activation markers MHCII and CD86. d–f At 48 h pi Infected murine peritoneal macrophages were surface stained with antibodies against CD11b (APC/Cy7), MHCII (PE), and CD86 (FITC) followed by flow cytometry. d Expression of CD11b on the surface of infected macrophages. e Expression of co-stimulatory markers MHCII and f CD86 on CD11b+ infected macrophages. g RT PCR to check the levels of Pro (IL-1β, IL-12, IL-22, IL-6, and TNFα) and anti-(IL-10) inflammatory cytokines in infected macrophages at 48 hpi. h Whole-cell lysates were prepared for the western blot experiment. Immuno-blot analysis depicting the phosphorylation status of P38, ERK, and AKT in the infected macrophages. The data are representative of two independent experiments. Statistical significance was drawn in comparison with glmMKd-ATc using an unpaired t-test.
We subsequently examined macrophage activation markers in Rv and Rv-glmMkD strain using qRT-PCR and FACS analysis and found significant upregulation of CD86 and MHCII (major histocompatibility complex II) in Rv-glmMkD + ATc infected cells compared with Rv and Rv-glmMkD -ATc infected cells (Fig. 3c–f). Next, we wanted to determine if this effect can be generalized to macrophages we performed the same experiment in BMDM (bone marrow-derived macrophages) and observed better immune response in Rv-glmMkD + ATc infected cells compared to controls again strengthening our results that depletion of glmM indeed enhances the macrophage activation (Fig. S5) Moreover, Rv-glmMkD strain in the presence of ATc showed increased levels of M1-specific pro-inflammatory cytokines viz. IL-1β, IL-12, TNF-α, IL-6, IL-22 with simultaneous downregulation of IL-10 (Fig. 3g). To examine whether this immune response can protect macrophages against bacterial burden, we have used the Rv-gfp strain. We have infected the macrophages with Rv-gfp followed by treatment with supernatant collected from Rv, Rv-glmMkD - ATc and Rv-glmMkD + ATc infected macrophages. 48 h post-treatment we checked Rv-gfp burden through FACS analysis and observed reduced Rv-gfp burden in cells treated from supernatant collected from Rv-glmMkD + ATc infected macrophages (Fig. S6). Further to delineate the molecular mechanism behind this M1-macrophage response, we have investigated MAPK and ERK signaling pathways known to be involved in the production of pro-inflammatory cytokines and the modulation of macrophage polarization33,34. MAPK phosphorylation in macrophages is associated with Th1 cell activation and differentiation, which is crucial for protecting against Mtb infection35. Moreover, it has been confirmed that the p38 MAPK pathway promotes M1 polarization in macrophages and enhances host immunity against tuberculosis through the release of pro-inflammatory cytokines36. The correlation between the p38MAPK pathway and M1 polarization is widely recognized across other cell types37,38. By that, we find that activation of p38 and ERK1/2 pathways as demonstrated by higher levels of phosphorylated proteins in Rv-glmMkD + ATc infected cells compared with either Rv or Rv-glmMkD -ATc infected cells (Fig. 3h). However, decreased phosphorylation of AKT was observed upon GlmM depletion. Since mTORC and AKT signaling are tightly connected and AKT activation through phosphorylation activates mTORC1 responsible for reduced autophagy supports Mtb survival39. Therefore, less phosphorylation of AKT further strengthened the hypothesis that depletion of GlmM improved the anti-mycobacterial response. Collectively, this data suggested that reduction in GlmM expression causes compromised bacterial survival in macrophages, and it also stimulates pro-inflammatory responses, probably via MAPK signaling.
GlmMMtb is indispensable for Mtb survival and pathogenesis in vivo
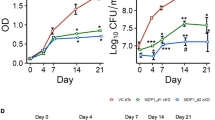
GlmM is an important enzyme involved in the synthesis of UDP-GlcNAc, a critical component of cell wall synthesis. Few pathogens are known to use GlcNAc from the host for the synthesis of UDP-GlcNAc, and others are dependent on cell wall recycling for the same in the absence of the UDP-GlcNAc biosynthesis pathway40,41. However, this information is not available in the case of Mtb. The essentiality of GlmU has been established in previous studies of the lab15. If Mtb can utilize the GlcNAc from the host, the activities of GlmS and GlmM may not be essential (Fig. S7). Hence, to investigate the possible presence of alternate pathways, we examined the in vivo survival and pathogenicity of Rv-glmMkD in a murine infection model. Mice were challenged with Rv and Rv-glmMkD strains through the aerosol route (Fig. 4a). CFU enumeration 24 h pi suggested efficient and equivalent deposition of both the strains Rv and Rv-glmMkD in the lungs of mice (Fig. 4b). Depletion of GlmM was initiated 1 day p.i by providing doxycycline (Dox) through drinking water, and CFUs were enumerated four weeks p.i. The bacillary load was significantly lower in the lungs and spleen of Rv-glmMkD infected mice treated with Dox compared with the absence of Dox treatment (Fig. 4b). Rv-infected mice treated with Dox were used as the reference control mice for CFU enumeration (Fig. 4b).

a GlmMMtb is essential for Mtb survival in vivo. a Schematic depicting the outline of in vivo murine infection experiment. Doxycycline (Dox) was introduced in the water on 1-day p.i. (In the schematic mice were created using MS Paint). b BALB/c mice were aerosolically challenged with 2 × 108 cfu of Rv or Rv-glmMkD and bacillary load was enumerated in the lung and spleen homogenates at day 1 and 28 days p.i. Each data point indicates CFU log10 /ml from the lung or spleen obtained from one mouse and the error bar represents as mean CFU log10/ml ± SD. *p < 0.01; **p < 0.001; ***p < 0.0001. c Schematic depicting the outline of the experiment. Dox was introduced after the establishment of the infection i.e., on day 14. BALB/c mice were infected with 100 CFU/mice of Rv or Rv-glmMkD, and the infection was established for 14 days. Dox was administrated to Rv and in one set of Rv-glmMkD (n = 5) infected mice, while the other set was left untreated. CFU was enumerated on days 1 and 2, 6, and 10 weeks’ p.i in the lung and spleen homogenates. d Data information: Each data point indicates CFU log10/lung or spleen obtained from one mouse and the error bar represents the mean CFU log10/lung or spleen ± SD. Statistical significance was drawn in comparison with glmMkd-ATc using an unpaired t-test. p < 0.01; **p < 0.001; ***p < 0.0001.
Subsequently, we sought to investigate the impact of GlmM depletion from an established infection. Mice were infected with Rv and Rv-glmMkD, and the infection was allowed to be established for two weeks (Fig. 4c). Subsequently, Rv-glmMkD mice were divided into two group’s one group was given Dox for 4 or 8 weeks to deplete GlmMMtb. CFUs were comparable for Rv and Rv-glmMkD at 1-day and 2 weeks p.i. CFUs enumerated 4- and 8-weeks post Dox treatment in Rv-glmMkD showed ~2.5 and 4 log10 fold decrease, respectively, compared with mice untreated with Dox (Fig. 4c). A similar trend was observed in the spleen as well. Gross pathology and histopathology data strengthened the observation wherein no significant granuloma formation was observed in the Rv-glmMkD +Dox samples (Fig. S8). Together, this data suggest that GlmM is critical for the survival of Mtb within the host at acute and chronic stages of infections.
Depletion of GlmM induces host protective immune response against Mtb
The host immune system and pathogen’s strength determine an infection’s eventual outcome. It is apparent from the data presented in Fig. 4 that the depletion of glmM significantly decreased bacillary load suggesting that the strength of the pathogen is lower, vis v vis host immune system. As shown in Fig. 2d, the depletion of GlmM reduces the thickness of the cell wall, which can also impact the permeability of the cell wall. Since the cell wall is the connection point between the pathogen and the host, any possible change in cell wall composition can affect the conversation with the host by impacting the secretion of virulence factors which plays an important role in modifying immune signals and protecting the bacteria inside the host. Thus, we set out to examine the host immune response in the mice infected with Rv or Rv-glmMkD in the absence and presence of Dox. Towards this, we infected C57BL6 mice with a low dose of Rv or Rv-glmMkD and glmM was depleted in one set for 60 days (Fig. 5a). As anticipated in Rv +Dox or Rv-glmMkD -Dox infected samples the bacillary load was comparable, while the Rv-glmMkD +Dox infected mice did not show any CFUs (Fig. 5b). Antigen-presenting cells and Th1 cells are the key players contributing to the immunological control of Mtb infection30. Hence, we profiled various immune cells in the spleen of infected mice.

a Schematic of the experiment showing C57BL/6 mice challenged via the aerosol route with a low-dose inoculum of ~110 CFU/mouse with H37Rv and RvGlmMkD strains, Dox treatment was started one day onwards for the next 60 days. Mice were sacrificed and lungs and spleen were harvested for estimation of bacterial burden and analysis of immune responses. (In the schematic mice were created using MS Paint). b CFU from lung and spleen homogenates at 60 days post-treatment. Each data point indicates CFU log10/lung or spleen obtained from one mouse and the error bar represents the mean CFU log10/lung or spleen ± SD. Statistical significance was drawn in comparison with glmMkd-ATc using an unpaired t-test. p < 0.01; **p < 0.001; ***p < 0.0001. c, d Activation profile of T cells (CD4+ and CD8+) in the spleen (c) and lung (d) of mice infected with H37Rv and glmMkD (-ATc and +ATc). e, f Cytokine profile (IFN-γ and IL-17) of CD4+ T cells in the spleen of different groups of mice. g, h Cytokine profile (IFN-γ and IL-17) of CD8+ T cells in the spleen of different groups of mice infected with H37Rv and glmMkD. i, j cytokine profile (IFN-γ and IL-17) of CD4+ T cells in the lung of different groups of mice. k, l cytokine profile (IFN-γ and IL-17) of CD8+ T cells in the lung of different groups of mice. m RT-PCR data to show expression of macrophage activation markers CD86 and MHCII at the transcriptional level in the spleen of different groups of mice infected with Rv and RvGlmMkD. n Profiling of intracellular cytokines (IL-1β, IL-12, IL-6, IL-10, and TNF-α) in splenocytes of different groups of mice infected with H37Rv and glmMkD (-ATc and +ATc). One-way ANOVA (Tukey analysis) was used to see the significance difference. Data is a representation of two independent experiments.
The gating strategy used in this study is depicted in Fig. S9. An increased percentage of CD11b+ cells with enhanced expression of co-stimulatory molecules CD86+ and MHCII+ was observed in the spleen of RvΔglmM +Dox infected mice compared to Rv +Dox or Rv-glmMkD -Dox (Fig. S10a–c). We also observed a significant increase in CD4+ and CD8+ T cells in the spleen of mutant-infected mice (Fig. S10d). The presence of increased CD69 surface expression suggests activation of both CD4+ and CD8+ T cells (Fig. 5c, d). Differentiation of CD4+ and CD8+ cells into protective Th1 and Th17 subsets was evident by the significant increase in gamma interferon (INFɣ) and Interleukin 17 (IL-17) levels in the splenic (Fig. 5e–h) and lung (Fig. 5i–l) T cells of Rv-glmMkD +Dox infected mice compared to Rv +Dox or Rv-glmMkD -Dox. We have also performed a similar experiment in GlmUMut15 another essential enzyme involved in the UDP-GlcNAc synthesis pathway shown in Fig.1a and observed a similar trend in the spleen (Fig. S11).
RT PCR analysis of spleenocytes demonstrated increased expression of macrophage activation markers CD86, MHCII, and pro-inflammatory cytokines IL-1β, IL-12, TNF-α, IL-6, while no significant change in IL-10 in Rv-glmMkD +Dox infected samples compared with Rv +Dox or Rv-glmMkD -Dox (Fig. 5m, n). Th17 cell responses play an important role in establishing protective immune responses against TB3 and do not majorly contribute to primary immune response42. We think that activation of Th17 response by targeting GlmMMtb can also help the host in recall responses. Together these results suggest a shift in the immune response in favor of host upon GlmM depletion.
Discussion
The mycobacterium cell wall is composed of a complex structure comprising highly impermeable mycolyl-arabinogalactan-peptidoglycan (mAGP) complex, which play an important role in survival and maintaining a cell shape. Due to the presence of such protective wall Mtb can replicate in the hostile environment of macrophages and resist the action of several therapeutic agents43. PG of this complex seems unexceptional but it contains various molecular subtleties that help Mtb to enter into the non-replicative dormant stage8. Most of the β-lactam antibiotics are ineffective in case of Mtb due to impermeability and highly active β-lactamase (BlaC) that efficiently hydrolyses many β-lactam drugs to render them ineffective44. Fosfomycin, a MurA inhibitor does not work in the case of Mtb due to alteration in a single amino acid in mycobacterial MurA45. D-cycloserine prevents L-alanine racemase (alr) and dipeptidyl synthetase (ddl) resulting in hindered formation of penta peptide side chains of PG but due to toxic effects on the central nervous system, its use is severely limited to MDR-TB46. A large proportion of the cell wall PG is cross-linked by non-classical l,d-transpeptidases, which are intrinsically impervious to these antibiotics47. Hence, the enzymes that catalyze the PG biosynthesis pathway are essential to bacterial cells and their restriction to the prokaryotes collectively makes them an attractive target for the development of new antibiotics.
Biosynthesis of UDP-GlcNAc, a central metabolite for both PG and AG synthesis involves three enzymes, GlmS, GlmM, and GlmU. In this study, we generated a knockdown of glmM in Mtb and examined its impact on growth and survival in in vitro, ex vivo, and in vivo. In compliance with the earlier studies reported for M. smegmatis, the closest nonpathogenic homolog of Mtb22 GlmMMtb was also found essential for the in vitro growth of bacteria (Fig. 1). GlmM is part of cell wall synthesis machinery and its role in morphology has been demonstrated in orthologs27,29. Here, SEM and TEM analysis revealed the crumpling of Mtb cells and reduced thickness of cell wall, respectively in the case of mutant (Fig. 2). Mycobacterial cell wall undergoes cell wall remodeling during hypoxia to adapt to the environment. Hence, we speculate that GlmM may play some role in hypoxic conditions since its role in biofilm formation has been shown earlier with orthologs21. In line with this hypothesis, bacterial survival was significantly reduced in hypoxic conditions upon GlmM depletion (Fig. 2).
In addition to the de novo pathway wherein fructose-6-phosphate an intermediate of glycolysis changed into UDP-GlcNAc, the salvage pathway is known to occur in some other bacteria and parasites48. In the salvage pathway, GlcNAc is taken from the host or available through cell wall recycling to the cytosol49. GlcNAc gets phosphorylated and converted into GlcNAc-6-phosphate and then GlcNAc-1-phosphate or directly to GlcNAc-1-phosphate which can enter in different parts of the synthesis. Therefore, the lack of information on such pathways in Mtb makes us interested in investigating the essentiality of GlmM for Mtb survival inside the host. Here we speculate, if a salvage pathway exists in Mtb for the synthesis of UDP-GlcNAc, in the absence of isomerase enzyme GlmM, bacteria should survive in the mutant. However, it was not reflected in ex vivo experiments performed using peritoneal macrophages, and Mtb survival was compromised in the absence of GlmM (Fig. 3). To further authenticate our ex vivo results we used the murine infection model for in vivo experiments and found that reduced GlmM expression result in less bacterial survival and pathogenicity (Fig. 4). Moreover, GlmMMtb was crucial for bacterial survival in all the stages of infection including before and after the establishment of infection (Fig. 4). Hence, it is conceivable that GlmMMtb is essential for bacterial survival in in vitro, ex vivo, and in vivo conditions.
During Mtb infection, the immune system plays a critical role in controlling the replication and survival of the mycobacteria inside the host, in the presence of a strong immune response Mtb enters into dormant a non-replicative stage. It is established that Mtb infection is more prevalent in the absence of an appropriate Th1 immune response5. The extent of Mtb infection correlates with the dynamics of pro- and anti-inflammatory responses which stimulate T helper cell differentiation. Interestingly, we found that downregulation of GlmM not only inhibits Mtb growth it simultaneously encourages the host immune system to fight better against infection. For that it stimulates macrophage towards the M1 response that secretes pro-inflammatory cytokines like IL-1β, IL-12, IL-6, IL-22, and TNFα (Fig. 3). However, the expression of anti-inflammatory cytokine IL-10 which promotes Mtb growth was less. To deepen more in the mechanism via which this pro-inflammatory response is occurring we observed the activation of p38 and ERK by phosphorylation (Fig. 3). P38 and ERK are MAPK which are known to stimulate the expression of pro-inflammatory cytokines and multiple effector molecules that will aid in protective host immunity during Mtb infection. The functional impact of M1 polarization through the p38 MAPK pathway not only reduced the bacterial load but also enhanced host immune action to combat the disease. Furthermore, the significant activation of IFNɣ and IL-17 was observed in the isolated lung and spleen of glmM mutant-infected mice, which is known to potentiate the host protective immunity against TB through Th1 and Th17 subsets of CD4+ T cells45 (Fig. 5).
The development of novel inhibitors for essential and conserved Mtb pathways is one potential strategy to shorten the duration of TB chemotherapy and eradicate drug-resistant TB. With the advancement of genome sequencing and molecular biology of mycobacteria, it is possible to identify essential bacterial pathways for drug development. Enzymes of UDP-GlcNAc pathway, especially GlmM due to its uniqueness to bacteria indeed offer an attractive target for new TB drug development. Here, in this study by giving a host immune angel we have suggested a distinct cause for the importance of GlmMMtb. The present study has given deep insight and knowledge about the potential of the GlmM enzyme for designing inhibitors, which will be further used together with current TB drugs for more effective TB treatment.
In the future, we will be testing the contribution of GlmM Mtb in mycobacterial survival and reprogramming of macrophages in the C3HeB/FeJ Kramnik’s mouse, which forms hypoxic, encapsulated granulomas with a caseous necrotic center. Further, since non-human primates such as Macaques are susceptible to infection with different strains of Mtb and produce the full spectrum of disease conditions very similar to human disease, it will be exciting to check the potential of GlmM in this model system to enhance the possibility of using GlmM as a future drug target.
Methods
Materials and growth conditions
Oligonucleotides (Sup. Table 1) were procured from Sigma. Doxycycline hydrochloride was purchased from Bio-chem Pharmaceutical. Antibodies used in this study; Anti-Mouse: CD3-Pacific Blue, CD4-QR, CD8-APCCy7, CD69-PE, IFNγ-BV510, IL17-BV650, CD11b-APCCy7, CD86-PerCPCy5.5, MHCII-PE from Biolegend, USA. Anti-human/anti-mouse: ERK, p-ERK, AKT, p-AKT, p38, P-p38, β-Actin were purchased from Cell Signaling Technologies. 7H9 medium supplemented with 10% ADC (NaCl, dextrose, bovine serum albumin, and catalase), 0.2% glycerol, and 0.1% Tween 80 was used for the liquid growth of Mtb strains. 7H11 agar with 10% OADC (ADC and oleic acid) and 0.2% glycerol were used for Mtb strain growth on plates. Mtb recombinants were selected on kanamycin (25 mg/mL). Medium components were from BD Difco, Sigma-Aldrich, and Hi-Media. Molecular grade reagents were procured from Merck, Ameresco, or Sigma; restriction-modification enzymes were from NEB; and SEM chemicals were from Electron Microscopy Sciences.
Expression, purification of GlmM, and generation of antibodies against it
glmM the gene of interest was PCR amplified and the amplicons were cloned into the pQE2 expression vector, which contains an N-terminal hexa-His tag. Temperature and induction conditions were optimized for the soluble expression of the protein. After optimizing the conditions, recombinant proteins was purified with the help of Ni2+-NTA (nickel-nitrilotriacetic acid) resin50. 50 µg of GlmM which was emulsified with Freund’s incomplete adjuvant was injected per mouse. After three booster doses, 1st bleed was collected to determine the antibody generation. The sensitivity of the antibody was evaluated using western blots with purified recombinant proteins. Finally, the ability of the antibody to recognize endogenous GlmM protein was evaluated by Mtb lysates using western blot analysis.
Construction of knockdown strains using CRISPRi
To achieve the repression of Rv3441c (glmM) gene, a pair of complementary oligonucleotides (Sup. Table 1) specific to the target ORFs near the 5’-end were synthesized, annealed, and cloned in pDCas9 + at AflII–AcII sites. This plasmid contains Dcas9, as described previously25. The Recombinants containing gene-specific guide sequence “GCCGCCGCGGCGCCCAGTGCC” (position on ORF from 72 to 92 nt from 5’) were electroporated in Mtb H37Rv to generate KanR knockdown strains. Since the plasmid contained a tetracycline-inducible promoter suppression was achieved by treatment of bacterial cultures with ATc.
Growth rate kinetics
Exponential-phase cultures of Rv, Rv-glmMkD -ATc, and Rv-glmMkD + ATc strains grown in Middlebrook 7H9-ADC medium were seeded at an optical density at 600 nm (OD600) of 0.05 in 7H9 medium. OD600 was monitored every 24 h, and the CFU were enumerated by serially diluting the cultures and plating them on 7H11 agar.
Western blot analysis
Rv, Rv-glmMkD strains grown in the absence of ATc were seeded at OD ~ 0.05 in the absence or presence of 50 ng/ml ATc. 1 Whole-cell lysates (WCLs) were prepared as described previously51 at different times points according to the Fig. 1. RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1.0% NP-40, 0.5% Sodium deoxycholate, 0.1% SDS) freshly supplemented with complete protease inhibitor and PhosSTOP purchased from Roche was used to prepare WCLs of peritoneal macrophages. Concentrations of WCLs were estimated using the Bradford protein estimation method, Samples were electrophoresed on SDS-PAGE and electro-blotted onto nitrocellulose membranes (Bio-Rad). After blocking with 5% BSA prepared in PBST (PBS and 0.05% Tween-20), blots were probed for different proteins using corresponding antibodies acquired from CST (Anti-human/anti-mouse: ERK, p-ERK, AKT, p-AKT, p38, P-p38, β-Actin) except α-GlmM, α-GroEL1 which were in-house generated. Blots were developed on autoradiograms using chemiluminescent HRP substrate (ECL, Millipore).
RNA isolation and qPCR analysis
For bacteria, total RNA was isolated from exponentially growing bacteria cells as described in ref. 52. Briefly, to isolate total RNA, cells were inoculated at 0.05 OD in the presence or absence of ATc and allowed to grow for three days. Cells were re-suspended in trizol reagent (Invitrogen), and total RNA was isolated from following mycobacterial cells followed by cDNA synthesis using iScript cDNA synthesis kit (Bio-Rad). qRT-PCRs was performed using the respective primers (Sup. Table 1). For peritoneal macrophages and splenocytes, total RNA was isolated using standard RNA isolation protocol followed by cDNA synthesis. Real-time PCR was performed using SYBR Green Master Mix (Bio-Rad). Bio-Rad Real-Time thermal cycler (BioRad, USA) was used for Real-time quantitative RT-PCR analysis. The list of Primers used in the study is provided in Sup. Table 1.
Hypoxia experiment
In vitro, hypoxia stress was assessed through a modified Wayne’s hypoxia model as described earlier15. Briefly, bacterial strains were inoculated in 7H9-ADC at an OD600 of 0.1; 1.5 mg/mL of methylene blue was added to a visual redox indicator. The experiment was carried out in tightly sealed glass tubes with 15% headspace at 37 °C without agitation. ATc (50 ng/ml) was injected into the cultures on the 20th day and the number of CFUs was determined at different time points. For that Bacterial cells were serially diluted and plated on 7H11-OADC agar, and after 15 days colonies appeared and counted.
Peritoneal macrophage (PM) infections
BALB/c and C57BL6 mice of 4–6 weeks were maintained in the Animal facility at ICGEB, New Delhi, India. Mice were accessed and obtained for experimental procedures from the facility. C57BL/6 mice were injected intra-peritoneally with 2 ml of 4% thioglycollate (Sigma). Five days later, ice-cold PBS was injected into the peritoneal cavity to extract macrophages. Cells were counted and seeded in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific Inc or Hyclone). Cells were washed with PBS to remove non-adherent cells after overnight incubation at 37 ˚C and 5% CO2.
For checking the cytotoxicity of ATc, mouse peritoneal macrophages were treated with 50 ng/ml ATc for 48 h followed by propidium iodide (PI) staining as described elsewhere53. For infection, single-cell suspensions were prepared from Log phase cultures of Rv and Rv-glmMkD strains by passing through a 26 gauge needle 5 times. Cells were infected at MOI of 1:10 (cell: bacteria). 4 h post-infection (p.i) media was removed, and cells were washed twice to remove extracellular bacteria. The cells were replenished with RPMI media containing 10% FBS with or without 50 ng/ml ATc. 24, 48, 72, or 96 h pi; cells were lysed in 100 μl of 0.02% SDS and CFUs were enumerated in different dilutions on OADC-containing 7H11 agar plates.
For Rvgfp infection, single-cell suspensions were prepared from Log phase cultures of Rvgfp strain and cells were infected at MOI of 1:1 (cell:bacteria). 4 hpi media was removed and supernatant from Rv, Rv-glmMkD-ATc and Rv-glmMkD + ATc were added along with fresh media in 1:1 ratio. 48 hpi Rvgfp burden was checked by FACS analysis in the FITC channel.
Bone marrow-derived macrophage (BMDM) infections
We have isolated the bone marrow cells from C57BL6 mice of 4–6 weeks. Post-RBC lysis cells were seeded into 6-well plates in DMEM complete media. Next day, media was changed and cells were supplemented with 20% M-CSF (macrophage-colony stimulating factor) and incubated at 37 ˚C and 5% CO2 for 3 days. On the third day, media was changed again complemented with MCSF again incubated for 2 days. On 3rd day cells were scraped, counted, and seeded in 12-well plates overnight in complete media. Next-day infection was done as described earlier in the case of PM followed by CFU and FACS analysis.
Murine infection experiment
Rv and Rv-glmMkD cultures were revived and grown up to exponential phase OD600 ~ 0.8, cultures were washed and re-suspended in the 1X sterile PBS, and single-cell suspensions were prepared by passing the culture five times through a 26 gauge needle. Bacterial cell suspension (1.5 × 106 cells per ml) total volume of 15 ml was placed in the nebulizer of the Madison aerosol chamber. Mice were grouped and labeled according to the experiment. First, bacterial deposition was checked in the lungs at 24 hpi. Doxycycline hydrochloride (1 mg/kg with 5% dextrose in drinking water) was provided as specified in the figures/legends, either from the time of the infection (day 1) or after the establishment of infection (21 days pi). 5–6 mice from each group were sacrificed at different time points and organs (lungs and spleen) were harvested, homogenized in 0.2 µm filtered PBS, and plated in different dilutions onto 7H11 Middlebrooks (Difco™) plates comprising 10% oleic acid, albumin, dextrose, and catalase (OADC) and incubated at 37 °C for 21–28 days. Mtb colonies were counted on different dilutions and CFUs were estimated as per dilution. Half the lung and spleen tissues were used for immune cell profiling. For histology1/4th portion of lung tissues were fixed in 10% neutral buffered formalin solution and coated with wax for sectioning followed by hematoxylin and eosin (H and E) staining and examined under a microscope. Granulomas for each animal in every group were screened in 5 different fields. Images in figures are illustrative of visualized section images.
Scanning electron microscopy and transmission electron microscopy
Rv and Rv-glmMkD strains were grown in the absence and presence of ATc at an OD600 of 0.6, harvested from 10-mL cultures. Scanning and Transmission electron microscopy (SEM & TEM) analysis of these samples were performed as described earlier15. For TEM, Cells were fixed in fixative containing 2.5% glutaraldehyde and 4% paraformaldehyde for 4 h at RT, followed by 24 h incubation at 4 °C. Cells were then washed with 0.1 M sodium cacodylate buffer to remove excess fixative and cells were embedded in 2% agar blocks before proceeding further. Samples were post-fixed in 2% osmium tetraoxide for 1 h, followed by dehydration in a graded series of ethanol (30%, 50%, 70%, and 100%) followed by 15 min of incubation in propylene oxide. Samples were infiltrated in Epson resin: propylene oxide (1:1 for 1 h at room temperature), and then infiltrated in pure Epson resin (overnight at room temperature) and polymerized at 60 °C for 72 h. Ultrathin sections (63 nm) were cut on Leica Ultramicrotome, placed on copper grids and stained with 5% uranyl acetate and 0.2% lead citrate. Sections were examined under a Tecnai G2 20 twin (FEI) transmission electron microscope. Cell length, width, and thickness were quantified with the help of Smart TIFF software and Carl Zeiss Tiff Annotation Editor.
Histology
Lung tissues were fixed in formalin solution and coated with wax for sectioning. Sections were stained with Hematoxylin and Eosin (H and E) dyes and slides were scored for granulomas by analyzing under a light microscope
Flow cytometry: surface and intracellular staining
For ex vivo experiments, infected macrophages were taken out 48 h pi and stained for different surface markers and intracellular cytokines as described in ref. 53. Briefly, macrophages were surface stained with CD11b (APC/Cy7), CD86 (PerCPCy5.5), and MHC-II (PE). For in vivo infections spleen and lungs from mice of different groups were isolated and macerated in ice-cold RPMI 1640 (Hyclone) media supplemented with 10% FBS using frosted glass slides to make single-cell suspension. RBC lyse was done using RBC lysis buffer, and cells were washed with 10% RPMI 1640. Cells were counted and 1 × 106 cells per well were seeded in 12-well plates for staining. Cells were activated using 10 µg/ml cell surface antigen (CSA) stimulation. Subsequently, 0.5 µg/ml Brefeldin and Monensin solutions (Bio Legend) were added during the last 4 h of culture. After treatment cells were washed with FACS buffer (PBS + 3% FBS) twice followed by antibody staining against surface markers and then 30 min fixation with 100 µl fixation buffer (Bio Legend). In the case of intracellular staining cells were first permeabilized using a permeabilizing buffer (Bio Legend) then staining was done with fluorescently labeled anti-cytokine antibodies. Anti-Mouse: CD3-Pacific Blue, CD4-QR, CD8-APCCy7, CD69-PE, IFNγ-BV510, IL17-BV650, from Biolegend, USA.
The intensity of fluorochromes was assessed by flow cytometry (BD LSRFortessa Cell Analyzer—Flow Cytometers, BD Biosciences) followed by data analysis via FlowJo (Tree Star, USA).
Statistics and reproducibility
All results were obtained from at least two or three independent experiments. Statistical significance was tested using an unpaired t-test and one-way ANOVA analysis unless specified otherwise. p < 0.05 was accepted as an indication of statistical significance. For Fig. 1, n = 3 biological replicates were used for two independent experiments. Statistical significance was determined using an unpaired t-test. For Fig. 2, statistical significance was determined using an unpaired t-test. n = ~20 were used for two independent experiments. For Fig. 3, n = 3 biological replicates were used for three independent experiments. Statistical significance was tested using an unpaired t-test and one-way ANOVA analysis. For Fig. 4, n = 6 biological replicates were used for two independent experiments. Statistical significance was determined using an unpaired t-test. For Fig. 5, two independent experiments were undertaken. Statistical significance was tested using an unpaired t-test.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
This published article and its supplementary information include all data generated or analyzed during this study. Unedited gel pictures of the western blot images are provided in the supplementary file as Supplementary Fig. 12. A complete source data file has been supplied as Supplementary Data 1. Mice in Figs. 4 and 5 and all the elements of Supplementary Fig. 1 were created by using PowerPoint and MS Paint.
References
Laurenzi, M., Ginsberg, A. & Spigelman, M. Challenges associated with current and future TB treatment. Infect. Disord. Drug Targets 7, 105–119 (2007).
Choi, H.-G. et al. Antigen-specific IFN-γ/IL-17-co-producing CD4+ T-cells are the determinants for protective efficacy of tuberculosis subunit vaccine. Vaccines 8, 300 (2020).
Khader, S. A. et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 8, 369–377 (2007).
Cadena, A. M., Flynn, J. L. & Fortune, S. M. The importance of first impressions: early events in Mycobacterium tuberculosis infection influence outcome. mBio 7, e00342–00316 (2016).
Chandra, P., Grigsby, S. J. & Philips, J. A. Immune evasion and provocation by Mycobacterium tuberculosis. Nat. Rev. Microbiol. 20, 750–766 (2022).
Jasenosky, L. D., Scriba, T. J., Hanekom, W. A. & Goldfeld, A. E. T cells and adaptive immunity to Mycobacterium tuberculosis in humans. Immunol. Rev. 264, 74–87 (2015).
Martino, M. de, Lodi, L., Galli, L. & Chiappini, E. Immune response to Mycobacterium tuberculosis: a narrative review. Front. Pediatr. 7, 350 (2019).
Alderwick, L. J., Harrison, J., Lloyd, G. S. & Birch, H. L. The mycobacterial cell wall-peptidoglycan and arabinogalactan. Cold Spring Harb. Perspect. Med. 5, a021113 (2015).
Brennan, P. J. & Nikaido, H. The envelope of mycobacteria. Annu. Rev. Biochem. 64, 29–63 (1995).
Jacobo-Delgado, Y. M., Rodríguez-Carlos, A., Serrano, C. J. & Rivas-Santiago, B. Mycobacterium tuberculosis cell-wall and antimicrobial peptides: a mission impossible? Front. Immunol. 14, 1194923 (2023).
Soni, V., Rosenn, E. H. & Venkataraman, R. Insights into the central role of N-acetyl-glucosamine-1-phosphate uridyltransferase (GlmU) in peptidoglycan metabolism and its potential as a therapeutic target. Biochem. J. 480, 1147–1164 (2023).
Milewski, S., Gabriel, I. & Olchowy, J. Enzymes of UDP-GlcNAc biosynthesis in yeast. Yeast Chichester Engl. 23, 1–14 (2006).
Han, X. et al. Action of dicumarol on glucosamine-1-phosphate acetyltransferase of GlmU and Mycobacterium tuberculosis. Front. Microbiol. 10, 1799 (2019).
Singh, M., Kempanna, P. & Bharatham, K. Identification of Mtb GlmU uridyltransferase domain inhibitors by ligand-based and structure-based drug design approaches. Mol. Basel Switz. 27, 2805 (2022).
Soni, V. et al. Depletion of M. tuberculosis GlmU from infected murine lungs effects the clearance of the pathogen. PLoS Pathog. 11, e1005235 (2015).
Mengin-Lecreulx, D. & van Heijenoort, J. Characterization of the essential gene glmM encoding phosphoglucosamine mutase in Escherichia coli. J. Biol. Chem. 271, 32–39 (1996).
De Reuse, H., Labigne, A. & Mengin-Lecreulx, D. The Helicobacter pylori ureC gene codes for a phosphoglucosamine mutase. J. Bacteriol. 179, 3488–3493 (1997).
Mehra-Chaudhary, R., Mick, J. & Beamer, L. J. Crystal structure of Bacillus anthracis phosphoglucosamine mutase, an enzyme in the peptidoglycan biosynthetic pathway. J. Bacteriol. 193, 4081–4087 (2011).
Tosi, T. et al. Inhibition of the Staphylococcus aureus c-di-AMP cyclase DacA by direct interaction with the phosphoglucosamine mutase GlmM. PLoS Pathog. 15, e1007537 (2019).
Zhu, Y. et al. Cyclic-di-AMP synthesis by the diadenylate cyclase CdaA is modulated by the peptidoglycan biosynthesis enzyme GlmM in Lactococcus lactis. Mol. Microbiol. 99, 1015–1027 (2016).
Kang, J. et al. Effect of phosphoglucosamine mutase on biofilm formation and antimicrobial susceptibilities in M. smegmatis glmM gene knockdown strain. PloS One 8, e61589 (2013).
Li, S. et al. Identification of M. tuberculosis Rv3441c and M. smegmatis MSMEG_1556 and essentiality of M. smegmatis MSMEG_1556. PloS One 7, e42769 (2012).
Griffin, J. E. et al. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLOS Pathog. 7, e1002251 (2011).
Levendosky, K., Janisch, N. & Quadri, L. E. N. Comprehensive essentiality analysis of the Mycobacterium kansasii genome by saturation transposon mutagenesis and deep sequencing. mBio 14, e0057323 (2023).
Choudhary, E., Thakur, P., Pareek, M. & Agarwal, N. Gene silencing by CRISPR interference in mycobacteria. Nat. Commun. 6, 6267 (2015).
Wayne, L. G. & Hayes, L. G. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 64, 2062–2069 (1996).
Shimazu, K. et al. Identification of the Streptococcus gordonii glmM gene encoding phosphoglucosamine mutase and its role in bacterial cell morphology, biofilm formation, and sensitivity to antibiotics. FEMS Immunol. Med. Microbiol. 53, 166–177 (2008).
Urano-Tashiro, Y., Saiki, K., Yamanaka, Y., Ishikawa, Y. & Takahashi, Y. Streptococcus gordonii DL1 evades polymorphonuclear leukocyte-mediated killing via resistance to lysozyme. PLoS ONE 16, e0261568 (2021).
Yajima, A. et al. Contribution of phosphoglucosamine mutase to the resistance of Streptococcus gordonii DL1 to polymorphonuclear leukocyte killing. FEMS Microbiol. Lett. 297, 196–202 (2009).
Flynn, J. L., Chan, J. & Lin, P. L. Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunol. 4, 271–278 (2011).
Saqib, U. et al. Phytochemicals as modulators of M1-M2 macrophages in inflammation. Oncotarget 9, 17937–17950 (2018).
Thiriot, J. D., Martinez-Martinez, Y. B., Endsley, J. J. & Torres, A. G. Hacking the host: exploitation of macrophage polarization by intracellular bacterial pathogens. Pathog. Dis. 78, ftaa009 (2020).
Cho, J.-E., Park, S., Cho, S.-N., Lee, H. & Kim, Y. S. c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38 MAPK) are involved in Mycobacterium tuberculosis-induced expression of Leukotactin-1. BMB Rep. 45, 583–588 (2012).
Pasquinelli, V. et al. Phosphorylation of mitogen-activated protein kinases contributes to interferon γ production in response to Mycobacterium tuberculosis. J. Infect. Dis. 207, 340–350 (2013).
Ashwell, J. D. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat. Rev. Immunol. 6, 532–540 (2006).
Singh, M. et al. The 1, 2-ethylenediamine SQ109 protects against tuberculosis by promoting M1 macrophage polarization through the p38 MAPK pathway. Commun. Biol. 5, 759 (2022).
Zhou, L. et al. DHZCP modulates microglial M1/M2 polarization via the p38 and TLR4/NF-κB SIgnaling Pathways in LPS-stimulated microglial cells. Front. Pharmacol. 11, 1126 (2020).
Zhang, B. et al. Targeting MAPK pathways by naringenin modulates microglia M1/M2 polarization in lipopolysaccharide-stimulated cultures. Front. Cell. Neurosci. 12, 531 (2018).
Covarrubias, A. J., Aksoylar, H. I. & Horng, T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin. Immunol. 27, 286–296 (2015).
Gisin, J., Schneider, A., Nägele, B., Borisova, M. & Mayer, C. A cell wall recycling shortcut that bypasses peptidoglycan de novo biosynthesis. Nat. Chem. Biol. 9, 491–493 (2013).
Sonnenburg, J. L. et al. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 307, 1955–1959 (2005).
Khader, S. A. & Cooper, A. M. IL-23 and IL-17 in tuberculosis. Cytokine 41, 79–83 (2008).
Batt, S. M., Burke, C. E., Moorey, A. R. & Besra, G. S. Antibiotics and resistance: the two-sided coin of the mycobacterial cell wall. Cell Surface 6, 100044 (2020).
Bush, K. & Bradford, P. A. β-Lactams and β-lactamase inhibitors: an overview. Cold Spring Harb. Perspect. Med. 6, a025247 (2016).
Silver, L. L. Fosfomycin: mechanism and resistance. Cold Spring Harb. Perspect. Med. 7, a025262 (2017).
de Chiara, C. et al. D-Cycloserine destruction by alanine racemase and the limit of irreversible inhibition. Nat. Chem. Biol. 16, 686–694 (2020).
Maitra, A. et al. Cell wall peptidoglycan in Mycobacterium tuberculosis: an Achilles’ heel for the TB-causing pathogen. FEMS Microbiol. Rev. 43, 548–575 (2019).
Biwi, J., Biot, C., Guerardel, Y., Vercoutter-Edouart, A.-S. & Lefebvre, T. The many ways by which O-GlcNAcylation may orchestrate the diversity of complex glycosylations. Mol. Basel Switz. 23, 2858 (2018).
Jankute, M., Cox, J. A. G., Harrison, J. & Besra, G. S. Assembly of the mycobacterial cell wall. Annu. Rev. Microbiol. 69, 405–423 (2015).
Sharma, K. et al. Transcriptional control of the mycobacterial embCAB operon by PknH through a regulatory protein, EmbR, in vivo. J. Bacteriol. 188, 2936–2944 (2006).
Agarwal, M., Soni, V., Kumar, S., Singha, B. & Nandicoori, V. K. Unique C-terminal extension and interactome of Mycobacterium tuberculosis GlmU impacts it’s in vivo function and the survival of pathogen. Biochem. J. 478, 2081–2099 (2021).
Singha, B. et al. The unique N-terminal region of Mycobacterium tuberculosis sigma factor A plays a dominant role in the essential function of this protein. J. Biol. Chem. 299, 102933 (2023).
Pahuja, I. et al. Berberine governs NOTCH3/AKT signaling to enrich lung-resident memory T cells during tuberculosis. PLOS Pathog. 19, e1011165 (2023).
Acknowledgements
We acknowledge the support of the DBT-supported Tuberculosis Aerosol Challenge Facility at the International Centre for Genetic Engineering and Biotechnology (ICGEB), New Delhi, India, and their staff in accomplishing this work. This work was supported by the funding provided by the Department of Biotechnology, Government of India (BT/PR13522/COE/34/27/2015) to V.K.N. M.A. acknowledges the DST-INSPIRE Faculty Fellowship, DST, and the Government of India (DST/INSPIRE/04/2019/002743). A.B. receives a DST-INSPIRE Faculty Fellowship from the Department of Science and Technology (DST), Government of India, and an HGK-IYBA Fellowship from the Department of Biotechnology, Government of India.
Author information
Authors and Affiliations
Contributions
Data curation: Meetu Agarwal, Biplab Singha, Suparba, Isha, Archna Singh, Shivam Chaturvedi, Ashima Bhaskar, Nisheeth Agarwal, Ved Prakash Dwivedi, Vinay Kumar Nandicoori. Formal analysis: Meetu Agarwal, Ashima Bhaskar, Biplab singha, Archna Singh, Ved Prakash Dwivedi, Vinay Kumar Nandicoori. Funding acquisition: Meetu Agarwal, Vinay Kumar Nandicoori. Investigation: Meetu Agarwal, Ashima Bhaskar, Ved Prakash Dwivedi. Methodology: Meetu Agarwal, Biplab, singha, Shivam Chaturvedi, Archna Singh, Ashima Bhaskar, Ved Prakash Dwivedi. Supervision: Meetu Agarwal, Vinay Kumar Nandicoori. Validation: Meetu Agarwal, Ved Prakash Dwivedi, Vinay Kumar Nandicoori. Visualization: Meetu Agarwal, Vinay Kumar Nandicoori. Writing—original draft: Meetu Agarwal. Writing—review & editing: Meetu Agarwal, Ved Prakash Dwivedi, Vinay Kumar Nandicoori.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
Animal experimentation: Animal experiments were carried out in accordance with the guidelines approved by the Animal Ethics Committee of National Institute of Immunology (NII, Approval ID: IAEC#462/18), New Delhi, India, International Centre for Genetic Engineering and Biotechnology (ICGEB, approval ICGEB/IAEC/18092021/IMB-19), New Delhi, India and the Department of Biotechnology (DBT) Government of India. Mice were ethically sacrificed according to institutional and DBT regulations.
Peer review
Peer review information
Communications Biology thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Tobias Goris.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Agarwal, M., Bhaskar, A., Singha, B. et al. Depletion of essential mycobacterial gene glmM reduces pathogen survival and induces host-protective immune responses against tuberculosis. Commun Biol 7, 949 (2024). https://doi.org/10.1038/s42003-024-06620-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06620-9
- Springer Nature Limited