
Abstract
The acquisition of ectopic fibroblast growth factor receptor 1 (FGFR1) expression is well documented in prostate cancer (PCa) progression, notably in conferring tumor growth advantage and facilitating metastasis. However, how FGFR1 contributes to PCa progression is not fully revealed. Here we report that ectopic FGFR1 in PCa cells promotes transferrin receptor 1 (TFR1) expression and expands the labile iron pool (LIP), and vice versa. We further demonstrate that FGFR1 stabilizes iron regulatory proteins 2 (IRP2) and therefore, upregulates TFR1 via promoting IRP2 binding to the IRE of TFR1. Deletion of FGFR1 in DU145 cells decreases the LIP, which potentiates the anticancer efficacy of iron chelator. Intriguingly, forced expression of IRP2 in FGFR1 depleted cells reinstates TFR1 expression and LIP, subsequently restoring the tumorigenicity of the cells. Together, our results here unravel a new mechanism by which FGFR1 drives PCa progression and suggest a potential novel target for PCa therapy.
Similar content being viewed by others


Introduction
Prostate cancer (PCa) is the most frequently diagnosed malignancy and the second leading cause of cancer death in men1. Organ-confined PCa can be effectively treated by androgen deprivation. However, such treatment inevitably leads to the recurrence of cancer, which generally is metastatic and androgen deprivation insensitive2. Ectopic expression of fibroblast growth factor (FGF) and FGF receptor (FGFR) is frequently found associated with a variety of human cancers, especially in prostate cancer3. Aberrant FGFR1 in prostate epithelial cells promotes PCa progression through increasing inflammation and Warburg effect, and deletion of FGFR1 impedes the initiation and progression of PCa both in vitro and in vivo4,5,6,7. However, the mechanism by which ectopically expressed FGFR1 contributes to PCa initiation and progression is not fully unraveled. Understanding the molecular mechanism by which FGFR1 contributes to PCa progression will facilitate the development of novel approaches for treating this deadly disease.
Iron, as an essential element necessary for the basic function of cells, is indispensable for cell growth and division8,9. Disorders of iron homeostasis such as iron acquisition and retention can trigger tumorigenesis and promote tumor growth10. Normally, iron binding to transferrin (TF) in the intracellular substance forms a complex with the transmembrane TF receptor 1 (TFR1) and then is internalized by endocytosis11,12. In endosomes, Fe3+ is reduced to Fe2+ and then transported from the endosome to the cytoplasm13,14. The free iron in the cytoplasm constitutes the labile iron pool (LIP), which is metabolically active especially in cancer. It has been observed that iron-rich supply helps cancer cell growth, while iron starvation retards their growth15,16. Tumor cells utilize iron for many important biochemical reactions such as DNA synthesis, mitochondrial metabolism and tumor proliferation17. Several clinical data indicate an increase of iron sequestration in PCa cells but low in normal cells adjacent to the tumor18,19. Malignant transformation of prostate epithelial cells is associated with increased intracellular iron content20,21. Iron is not only important for tumor survival but also for the activity of enzymes that control androgen receptor (AR) transcription22. A variety of strategies have been designed to target intracellular iron for antitumor therapies, however, whether and how ectopic FGFR1 contributes to dysregulated iron homeostasis also remains unknown.
Maintaining cellular iron content requires precise regulation of its uptake, storage and export. Among these process, iron uptake mediated by TFR1 is the most important way for iron absorption, and overexpression of TFR1 promotes tumorigenesis and cell proliferation through increasing LIP in tumor cells23,24,25,26. Therefore, targeting TFR1 could be a valuable pharmaceutical strategy for cancer intervention27. The primary regulators of TFR1 are iron regulatory proteins (IRPs), IRP1 and IRP2, which bind to a specific sequence of nucleotides termed iron responsive elements (IREs)28. However, the mechanisms of IRE binding activities of these two IRPs are different. In LIP abundant condition, IRP1 functions as an enzyme interconverting two intermediates of the TCA cycle, citrate and isocitrate. The deletion of IRP1 modestly affects cell growth through an iron-independent pathway. Unlike IRP1, IRP2 functions as an RNA binding protein, which is regulated at the post-translational level via the ubiquitin-proteasome system (UPS) in an iron-mediated manner. IRP2 when overexpressed in PCa cells facilitates iron retention and contributes to tumor progression29,30,31,32,33. Importantly, targeting iron dysmetabolism by reducing TFR1 expression significantly sensitizes PCa cells to small lead peptide treatment34. Therefore, it is critical to investigate the mechanism of TFR1 in tumorigenesis and the potential therapeutic effects of targeting TFR1 in PCa.
In the present study, we aimed to elucidate the mechanism of aberrant FGFR1 on regulating iron homeostasis to accelerate prostate cancer progression. To address these issues, we initially characterized the expression patterns of iron homeostasis-related molecules in human PCa tissues and PCa cells and then delineated how FGFR1 influenced the LIP via affecting regulators of iron homeostasis. Our results suggest that prostate tumor cells with greater progressive propensity exhibit a larger bioactive iron pool, which is closely associated with FGFR1 expression. Specifically, TFR1, as the transporter to allow cellular uptake of iron-loaded transferrin, is bound to IRP2 and functionally regulated by its degradation to promote PCa growth. These data suggest that there is a profound reciprocal relationship between liable iron and IRP2, and targeting the regulators of iron homeostasis could provide an effective therapeutic strategy for PCa.
Results
Ectopic expressed FGFR1 is associated with increased expression of TFR1 in PCa progression
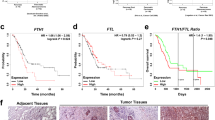
Iron dysregulation is closely related to cancer progression35,36,37. To determine whether iron content is the principal regulator of human PCa differentiation, we used Perl’s staining to detect the iron levels in well differentiated and poorly differentiated human PCa tissues (Fig. 1A). Then we analyzed the correlation of TFR1 expression with PCa progression in the TCGA database, from where told us the mRNA level of TFR1 was positively associated with the pathologic stages and Gleason scores (Fig. 1B, C). In addition, high TFR1 expression was closely related with short patient progression free survival in TCGA database and FGFR1 was increased in high TFR1 group than in low TFR1 group (Fig. 1D, E). Furthermore, the expression of FGFR1 at the mRNA level was positively associated with that of TFR1 (Fig. 1F).

A Representative Perl’s staining enhanced with DAB of well differentiated and poorly differentiated human PCa tissues. Scale bars: 50 μm. B The mRNA level of TFR1 in pathologic stage of T2C, T3A, and T3B in PCa patients of TCGA database. Other stages were excluded due to the inadequate number of cases. C Analysis between TFR1 mRNA level and Gleason score in PCa patients of TCGA. Gleason score ≥ 8 defined as high and <8 defined as low. D Kaplan–Meier survival analysis was performed in low and high TFR1 mRNA level in PCa patients. Cutoff sets 25%. E The mRNA level of FGFR1 based on the mRNA level of TFR1 in TCGA. Cutoff on TFR1 sets 25%. F Correlation analysis between the mRNA level of TFR1 and FGFR1 in PCa patients of TCGA database. G HE staining and double immunofluorescence staining with FGFR1 (green) and TFR1 (red) between well and poorly differentiated human PCa tissues and the quantitative analyses with Image J. Scale bars: 30 μm. WD well differentiated. PD poorly differentiated. TPM Transcripts Per Million.
We also found, as expected, poorly differentiated human PCa exhibited higher FGFR1 and TFR1 compared to the well differentiated PCa (Fig. 1G). Meanwhile, we used transgenic mouse PCa model TRAMP to investigate the correlation of FGFR1 and iron content in PCa. Normal prostate and PCa tissues from various week ages of mice were collected to test the detailed characterization. Although no apparent differences were observed in the morphology of prostate between wildtype and TRAMP group, either 12-week-old or 22-week-old, notable histological alterations could be detected in TRAMP tissues (Supplementary Fig. 1A, B). It was clear that the iron content, expression of FGFR1, and TFR1 were significantly increased in TRAMP prostate at the lesion areas, which were much higher during 22 weeks (Supplementary Fig. 1C–E). Collectively, these data show that ectopic expressed FGFR1 is associated with increased expression of TFR1 and LIP in PCa progression.
FGFR1 deletion decreases the LIP in PCa cells
To investigate whether ectopic FGFR1 expression increased the LIP in PCa, we generated FGFR1 null DU145 cells (DU145ΔR1 cells). The LIP size of DU145ΔR1 cells was significantly decreased compared with control cells (Fig. 2A), which prompted us to assess the expression of TFR1 and DMT1 responsible for iron uptake in PCa cells. The results indicated that the expression of TFR1 and DMT1 was reduced at both mRNA and protein levels in DU145ΔR1 cells, suggesting that deletion of FGFR1 reduced LIP via reducing iron intake in PCa (Fig. 2B, E). To confirm the function of FGFR1, we constructed the DU145ΔR1 cells exogenously transfected with FGFR1 (DU145ΔR1OE cells). The results showed FGFR1 restoration not only caused DU145ΔR1 cells to enhance the LIP, but also rescued the decreased TFR1 and DMT1 expression (Fig. 2C–E). In addition, exogenous expressed FGFR1 in LNCaP and C4-2 cells, both of which barely expressed FGFR1, significantly elevated TFR1 expression and LIP (Supplementary Fig. 2A, B). As expected, FGFR1 overexpression in DU145ΔR1 cells was necessary for the enhancement of FGF signaling pathway indicated as the phosphorylation of ERK (Fig. 2F). This was accompanied by the upregulation of IRP2 and TFR1 expression after FGF1 administration, which suggesting both FGFR1 expression and FGF signaling activity were correlated with the regulation of iron uptake (Fig. 2G). Taken together, these results further demonstrate that ectopic expressed FGFR1 promotes TFR1 expression, and therefore increase LIP in PCa (RT-PCR primers are listed in Table 1).

A Calcein AM was used to detect the labile iron pool. B Real-time qPCR was performed to analyze relative mRNA expression. C Calcein AM was used to detect the labile iron pool. D Real-time qPCR was performed to analyze relative mRNA expression. E Western blotting analysis of the indicated proteins and the quantitative analyses with Image J. β-actin was used as a loading control. F, G Western blotting analysis of the indicated proteins and the quantitative analyses with Image J. β-actin was used as a loading control (n = 4). Unless specified otherwise, data are represented as the mean ± SD of three independent experiments. DU145ΔR1, DU145 cells with FGFR1 knockout. DU145ΔR1OE, DU145ΔR1 with exogenous expression of FGFR1.
FGFR1 deletion potentiates the inhibitory effect of DFO in PCa cells
To determine whether ablation of FGFR1 sensitized the cells to iron deficiency, cells were treated with Deferoxamine (DFO), an iron-chelating agent, and then subjected to cell viability analysis. The CCK-8 analysis revealed that DFO inhibited cell viability more potently in DU145ΔR1 cells than in parental cells (Fig. 3A). Furthermore, EdU incorporation assays revealed that DFO showed more significant inhibitory effects in DU145ΔR1 cells than in parental cells (Fig. 3B). Consistently, expression of c-MYC was lower in DU145ΔR1 than in DU145 cells upon DFO treatment. Western blotting also confirmed that BCL2 expression in DU145ΔR1 cells was downregulated, and BAX was upregulated (Fig. 3C). To determine how ablation of ectopic FGFR1 contributed to DFO-induced apoptosis, SYTOX Green labeling was used to quantitate the percentage of apoptotic. Although there was no noticeable difference of apoptotic cells between DU145ΔR1 and parental cells, upon DFO treatment, DU145ΔR1 cells had significantly more apoptotic cells than the parental cells (Fig. 3D). Interestingly, LIP in DFO treated DU145ΔR1 cells was lower than in parental cells under the same treatment which was just on the contrary of TFR1 protein level, suggesting that depletion of ectopic FGFR1 signaling exhibited synergistic effect with DFO on iron content except the feedback increase of TFR1 (Fig. 3E, F). Together, these data document that FGFR1 deletion potentiates the inhibitory effect of DFO in PCa cells.

A CCK8 analysis was performed to analyze viability of DU145 and DU145ΔR1 cells treated with DFO at different concentration (0 μM, 20 μM, 50 μM, and 100 μM) for 24 h. B EdU incorporation assay was used to analyze the proliferation of cells treating with 20 μM DFO, the ratio of EdU positive cells were calculated in three random areas by Image J software, DAPI (blue) for the counterstaining of nuclear. Scale bars: 30 μm. C Western blotting analysis of the indicated proteins and the quantitative analyses with Image J. β-actin was used as a loading control (n = 4). D SYTOX Green was used to detect the survival of DU145 and DU145ΔR1cells treated with 20 μM DFO for 24 h, the dead cells were detected by FCM. E Labile iron pool of DU145 and DU145ΔR1cells treated with DFO were detected with Calcein AM. F Western blotting analysis of the indicated proteins and the intensity was quantitated with Image J. β-actin was used as a loading control (n = 4). Unless specified otherwise, data are represented as the mean ± SD of three independent experiments. DU145ΔR1, DU145 cells with FGFR1 knockout.
FGFR1 deletion disrupts ROS generation in PCa cells
At low to moderate levels, ROS acted as a signaling transducer to promote cancer cell proliferation and survival, which also regulated by iron content38,39,40. Thus, we were wondering whether FGFR1 deletion disturbed ROS production in DU145 cells. We detected the intracellular ROS level in both DU145 and DU145ΔR1 cells through DHE staining, which indicated FGFR1 deletion shrinking the ROS level apparently (Fig. 4A, B). Next, we tested the level of glutathione (GSH) and glutathione peroxidase (GPX) enzyme system which responsible for combating oxidative stress to maintain ROS homeostasis. Consistent with the DHE assay, cellular GSH and GPXs, especially GPX4, were upregulated after FGFR1 deletion benefiting for anti-oxidation (Fig. 4C, D). Furthermore, superoxide dismutase 2 (SOD-2), known as regulating reactive superoxide anions (O2•−) conversion, was markedly elevated after FGFR1 deletion (Fig. 4D). In contrast, malondialdehyde (MDA), a marker of oxidative stress, reduced markedly in DU145ΔR1 cells, as well as the number of mitochondria detected by transmission electron microscopy (Fig. 4C, E). In this condition, there was distinguished decrease of inflammatory proteins after FGFR1 deletion in DU145 cells, which indicated attenuation of oxidative stress (Fig. 4F). We conclude from these data that FGFR1 deletion disrupts mitochondrial function and ROS generation in PCa cells.

A The representative image of DHE staining in DU145 and DU145ΔR1 cells. Scale bars: 40 μm. B ROS level detected by FCM with DHE in DU145 and DU145ΔR1 cells (n = 8). C Intracellular GSH level, MDA level, and GPXs level detected in DU145 and DU145ΔR1 cells. D Relative mRNA and protein level of GPX4 and SOD-2 in DU145 and DU145ΔR1 cells. E The representative image of mitochondria in DU145 and DU145ΔR1 cells by TEM, and the number of mitochondria is counted in right panel (n = 6). Scale bars: 2 μm. F Western blotting analysis of the indicated proteins and the intensity was quantitated with Image J. β-actin was used as a loading control (n = 4). Unless specified otherwise, data are represented as the mean ± SD of three independent experiments. DU145ΔR1, DU145 cells with FGFR1 knockout.
FGFR1 deletion alters the main degradation pathway of IRP2
To determine how FGFR1 deletion affected TFR1 expression, quantitative RT-PCR analyses was employed to assess the expression of IRP1, IRP2, c-MYC, and P53, the main upstream regulatory factors of TFR141,42,43,44. The result showed that expression of IRP1 and c-MYC was reduced while that of IRP2 and P53 were increased in DU145ΔR1 cells compared with parental cells (Fig. 5A). Western blotting also showed that protein expression of IRP1, IRP2, and c-MYC was reduced while that of P53 was increased (Fig. 5B). Interestingly, the expression of IRP2 was increased at the mRNA level but decreased at the protein level, which suggested FGFR1 potentially regulating the degradation of IRP2. Consistent with this finding, exogenous expression of FGFR1 in DU145ΔR1 and C4-2 cells downregulated IRP2 at the mRNA level but upregulated IRP2 at protein level (Supplementary Fig 2C, D). To explore whether FGFR1 suppressed the degradation of IRP2 protein, the cells were treated with cycloheximide (CHX) to inhibit protein synthesis, and IRP2 proteins level was determined by Western blotting. The results clearly documented that the abundance of IRP2 decreased more rapidly in DU145ΔR1 cells than in parental cells (Fig. 5C). These results document that FGFR1 could be an important regulator to affect IRP2 degradation.

A Real-time qPCR was performed to analyze relative mRNA expression of IRP1/IRP2/c-MYC/P53 in DU145 and DU145ΔR1cells. B Western blotting analysis of the indicated proteins and the quantitative analyses of TFR1 with Image J. β-actin was used as a loading control. C Western blotting showing the expression of IRP2 in DU145 and DU145ΔR1 cells treated with CHX (100 μg/ml) for 0 h, 2 h, 4 h, 8 h, and 12 h. Data are shown as the mean ± SD (n = 5). Unless specified otherwise, data are represented as the mean ± SD of three independent experiments. DU145ΔR1, DU145 cells with FGFR1 knockout.
To further investigate whether the UPS or lysosome proteolysis pathway was responsible for IRP2 degradation, the cells were treated with the inhibitors of lysosome or proteasome and then subjected to the same analyses. The results showed that the degradation of IRP2 in parental cells was suppressed by proteasome inhibitor MG132, but not by lysosomal inhibitor chloroquine (CQ) (Fig. 6A). However, in DU145ΔR1 cells, the degradation of IRP2 was suppressed by CQ, but not by MG132, suggesting that IRP2 degradation in parental cells was largely mediated by the ubiquitination-proteosome pathway and in DU145ΔR1 cells was mainly mediated by lysosome proteolysis pathways (Fig. 6A). Consistently, exogenous expression of FGFR1 in C4-2 cells affected IRP2 degradation in the same pattern with DU145, both highly expressed FGFR1 (Supplementary Fig. 3A, B). To confirm whether the activation of autophagy accelerated the degradation of IRP2 after FGFR1 deletion, DU145 and DU145ΔR1 cells were treated with autophagy inducer metformin, rapamycin or ATG5 transfection (as no ATG5 expressed in DU145 (Supplementary Fig. 3C)), and then subjected to the same analyses. The results showed that the treatment of both metformin, rapamycin and ATG5 overexpression significantly increased IRP2 degradation in DU145ΔR1 cells but not in parental cells (Fig. 6B).

A DU145 and DU145ΔR1 cells treated with CHX (100 μg/ml) and CQ (50 μM) or MG132 (10 μM) for 0, 2, 4, 8 or 12 h, and then subjected to Western blotting with IRP2 antibody. B DU145 and DU145ΔR1 cells treated with Metformin (50 μM), Rapamycin (10 μM), or transfected with ATG5 for indicated time, followed by Western blotting showing the expression of IRP2. In A, B, panels were the quantitative analysis by Image J software and β-actin was used as a loading control. Data are shown as the mean ± SD (n = 3). CHX, Cycloheximide. CQ, Chloroquine. DU145ΔR1, DU145 cells with FGFR1 knockout.
F-box and leucine-rich repeats protein 5 (FBXL5) served as the ubiquitin ligase of IRP2 in the ubiquitin-proteosome degradation pathway45. Immunostaining demonstrated that IRP2 and FBXL5 colocalized in parental cells, whereas the colocalization was markedly reduced in DU145ΔR1 cells (Fig. 7A). Further analysis by Western blotting showed a significant decrease of FBXL5 protein level in DU145ΔR1 cells, while the ubiquitination of IRP2 was just a slight downregulation without statistical significance (Fig. 7B, C). Additionally, immunoprecipitation assay showed that FBXL5 pulled down more IRP2 in parental cells than in DU145ΔR1 cells, suggesting that more FBXL5-IRP2 complex existed in parental cells (Fig. 7D). The result showed that IRP2 degradation in DU145 cells was mediated primarily through the ubiquitination pathway, but deletion of FGFR1 could not compromise this pathway. Furthermore, RNA Immunoprecipitation was performed to figure out the correlation of IRP2 and TFR1 in DU145 cells affected by FGFR1. The result indicated that FGFR1 deletion markedly decreased the IRP2-binding complex, which was upregulated after FGFR1 overexpression (Fig. 7E). In addition, bound RNA is verified by qRT-PCR using TFR1 specific primers revealing mRNA level of TFR1 related with FGFR1 expression, which exhibited FGFR1 promoted IRP2 binding to IRE of TFR1 (Fig. 7F). Altogether, these data demonstrate that the degradation of IRP2 accelerated by FGFR1 deletion is mainly via lysosome in DU145ΔR1 cells.

A Immunofluorescence staining showing colocalization of IRP2 (green) and FBXL5 (red) in DU145 and DU145ΔR1 cells, DAPI for the counterstaining of nucleus. Middle/bottom panel was colocalization analysis and 3D view analysis of colocalization performed by image J software, intensity indication was in the upper left corner of 3D image. Scale bars: 20 μm. B Western blotting analysis of FBXL5 and the quantitative analyses with Image J (n = 3). C Western blotting analysis of the ubiquitination of IRP2 protein in DU145 and DU145ΔR1 cells. D The indicated cell lysates were prepared and immunoprecipitated with either agarose-conjugated anti-IRP2 or nonspecific anti-rabbit/mouse IgG antibodies as negative control. Immunoprecipitants and cell lysates were analyzed by western blotting. E, F RNA immunoprecipitation assay. Western blotting analysis of IRP2 after IRP2 specific antibody immunoprecipitation with cell lysate (n = 4). Input, cell lysate of DU145; IgG, DU145 cell lysate immunoprecipitated with IgG antibody as negative control (E). Quantitative RT-PCR analysis of the mRNA level of TFR1 after immunoprecipitation (n = 4) (F). Unless specified otherwise, data are represented as the mean ± SD of three independent experiments. DU145ΔR1, DU145 cells with FGFR1 knockout. DU145ΔR1/NC, DU145ΔR1 transfected with control plasmid. DU145ΔR1OE, DU145ΔR1 with exogenous expression of FGFR1.
Forced expression of IRP2 reinstates the LIP, increases cell proliferation, and restores tumorigenic activity in DU145ΔR1 cells
To elucidate whether reduced IRP2 expression underlie FGFR1 depletion deduced LIP content, we examined the IRP2 function in DU145 via transfected DU145ΔR1 cells with IRP2, designated DU145ΔR1/IRP2 or control vector, designated DU145ΔR1/NC. It was clear that the LIP in DU145ΔR1/IRP2, but not DU145ΔR1/NC cells, was significantly higher than that in DU145ΔR1 cells (Fig. 8A). EdU incorporation assay showed that forced expression of IRP2 reinstated cell proliferation to a level comparable to parental cells (Fig. 8B). Western blotting showed that IRP2 overexpression rescued the expression of TFR1 and FBXL5 (Fig. 8C). To determine whether forced expression of IRP2 restored the tumorigenic activity, parental, DU145ΔR1, DU145ΔR1/NC and DU145ΔR1/IRP2 cells implanted into nude mice for 6 weeks. The data indicated that the size of xenografts derived from DU145ΔR1/IRP2 cells were nearly similar to those derived from parental DU145 cells (Fig. 8D), suggesting that forced expression of IRP2 reinstated the tumorigenicity of DU145ΔR1 cells in nude mice. Furthermore, the LIP in DU145ΔR1/IRP2 cell derived xenografts was comparable to those in DU145 xenografts (Fig. 8E). Consistently, forced expression of IRP2 restores the expression of TFR1, FBXL5, and IRP2 in DU145ΔR1 cells (Fig. 8F). In addition, the DU145ΔR1/IRP2 xenografts exhibited a cell proliferation rate similar to that of DU145 xenografts (Fig. 8F). Altogether, these results demonstrate that forced expression of IRP2 reinstates the LIP, increases cell proliferation and restores tumorigenic activity in vivo and vitro.

A Statistical analysis of intracellular iron through Calcein AM for indicated cell groups (n = 3). B EdU incorporation assay was used to analyze the proliferation, the ratio of EdU positive cells were calculated in three random areas by Image J software, DAPI (blue) for the counterstaining of nuclear (n = 3). Scale bars: 30 μm. C Western blotting analysis of indicated proteins and the quantitative analyses with Image J (n = 3). D Tumor xenograft derived from the injection of DU145, DU145ΔR1, DU145ΔR1/NC, and DU145ΔR1/IRP2 cells into nude mice. Right panel was the statistical analysis of tumor volume and weight (n = 5). E The detection of labile iron pool in tumor xenograft tissues (n = 5). F Immunofluorescence staining showing FGFR1 (green) and TFR1 (red), FBXL5 (green) and IRP2 (red), and PCNA (red) in tumor xenograft. DAPI (blue) for the counterstaining of nuclear. Scale bars: 50 μm. Unless specified otherwise, data are represented as the mean ± SD of three independent experiments. DU145ΔR1, DU145 cells with FGFR1 knockout. DU145ΔR1/NC, DU145ΔR1 transfected with control plasmid. DU145ΔR1/IRP2, DU145ΔR1 transfected with IRP2 plasmid.
Discussion
Emerging evidence indicates that abnormal iron metabolism represents as a key metabolic hallmark closely associated with PCa initiation and progression46,47,48. Additionally, extensive investigation suggests that ectopic FGF/FGFR1 signaling plays a pivotal role in PCa development and progression6,7,49,50. It is well established that iron contents in PCa cells are higher compared to the adjacent non-cancerous prostate tissues19,51. However, to our knowledge, no study has yet elucidated the mechanism by which FGFR1 regulates iron homeostasis in PCa. In the present study, we have demonstrated that ectopic FGFR1 alters the LIP of PCa cells, which most possibly via inhibiting key regulators, TFR1 and DMT1, responsible for iron intake, as well as suppresses the activation of FGFR1 signaling. Recent studies have highlighted the elevated levels of iron metabolites and overexpression of TFR1 in prostate cancer52. TFR1 functions as the major receptor for cellular iron import, which significantly elevated in PCa has the potential to serve as a marker of PCa diagnosis and prognosis53,54. It has reported depleting TFR1 can potentially suppress tumor progression9. Consequently, it is possible that aberrant FGFR1 contributes to LIP increase through regulation of TFR1, which could be a potential target for PCa therapy.
DFO is the first iron chelator taken forward for clinical trial aimed at suppressing the growth of multiple types of cancers55,56,57. Herein, we have demonstrated that FGFR1 deletion synergistically enhances the effects of DFO on pro-apoptosis and anti-proliferation, and depletion of FGFR1 signaling combined with DFO significantly reduces LIP in PCa cells. Moreover, we have verified the expression of TFR1 after DFO administration and found that DFO significantly increases TFR1 protein levels either with or without FGFR1, which explicates as the feedback regulation. Given the reduction of LIP sensitizes cells to anticancer drugs, it is conceivable that the combination of FGFR1 inhibition and DFO could be used to enhance the efficacy of anticancer therapies for PCa.
Furthermore, in addition to the importance of iron levels in FGFR1 overexpressed tumor growth, we present the possibility that the intracellular ROS levels is apparently regulated by FGFR1 in PCa cells. Notably, the enzyme system GPX and GSH, which are responsible for combating oxidative stress, shows dramatically increase after FGFR1 depletion. It is well determined that iron content in an optimal range leads to the increase of ROS harmful for cell structures, while tumor cells have developed mechanisms protecting them against oxidative damage39. Therefore, only particularly high concentration of cellular iron can be detrimental for cancer cells, which make cells in extremely oxidative stress and may induce tumor death58. Accordingly, whether iron deprivation or iron overload in tumor cells can inhibit tumor growth and cause tumor cell death still needs further studies to uncover the precise mechanism.
IRP2 is a key regulator for iron homeostasis, which binds to the IREs in the non-coding region of TFR1 mRNA and prevents the mRNA degradation59,60. Recent reports show that IRP2 degradation is primarily mediated by ubiquitination45, which includes iron-dependent, oxygen-dependent and other redundant pathways61,62,63,64. Here we have uncovered that aberrant FGFR1 suppresses the degradation rate of IRP2 in PCa cells. Specifically, IRP2 degradation in FGFR1 overexpressed cells is largely mediated by ubiquitination proteosome pathway, whereas it shifts to lysosome proteolysis pathways after FGFR1 depletion. In addition, our findings reveal that FGFR1 deletion impairs the binding capacity of IRP2 to IRE of TFR1, which is subsequently restored by FGFR1 overexpression. As FBXL5 is an ubiquitin E3 ligase partnered for IRP2 contributing to iron-dependent degradation of IRP2 in iron-dependent pathway59, we further reveal that overexpressed IRP2 in FGFR1 deletion cells could reciprocally promote the expression of FBXL5, which with the reduction induced by iron decrease leading to FBXL5 instability64,65. Our data show that ectopic expression of FGFR1 enhances TFR1 expression through stabilizing IRP2 to increase intracellular iron content. Although we have demonstrated FGFR1 exhibits positive effect on FBXL5-IRP2 pathway to regulate TFR1 levels, further studies are required to excavate the incomplete mechanism of IRP2 on TFR1 expression under FGFR1 supervision.
In summary, we determine that ectopic FGFR1 regulates iron homeostasis by stabilizing IRP2, and therefore, upregulating TFR1 expression. Depletion of FGFR1 blocks the iron import via reducing TFR1 expression, which then lead to inhibition of cell proliferation and survival (Fig. 9). Accordingly, these findings implicate the new mechanism that FGFR1 influences the intracellular iron content contributing to PCa progression, then targeting the key regulators will facilitate the development of novel approaches to treat currently uncurable castrate-resistant PCa.

Iron bound to TF uptakes by TFR1 on the plasma membrane of cells is the most important way for cancer cells to absorb iron. TFR1 overexpression increases LIP in tumor cells to promote tumorigenesis and proliferation. FGFR1 positively regulates the cellular iron content via raising TFR1 expression through inhibiting the degradation of the master regulator IRP2, which is regulated in a reciprocal fashion to FBXL5, an iron-sensing E3 ubiquitin ligase. Under the condition of excess iron TFR1 expands, leading to accelerate PCa progression. TF, transferrin; TFR1, transferrin receptor 1; LIP, liable iron pool; IRP2, iron regulatory proteins 2; FBXL5, F-box and Leucine-rich repeat protein 5.
Methods
Animals and xenografts
Wildtype male C57BL/6 mice and female hemizygous for the transgenic adenocarcinoma of the mouse prostate (TRAMP) model were purchased from the Jackson Laboratory (Bar Harbor, ME, United States). The mice were housed at Laboratory Animal Resources Center of Wenzhou Medical University, maintained under 12 h light and dark cycles, and provided ad libitum access to food and water. The TRAMP transgenic construct was designed with simian virus 40 (SV40) early genes (large and small tumor antigens, Tag) under the control of the rat probasin promoter, which is a model for prostate cancer (PCa) that develops lesions ranging from preneoplastic to metastasis. The F1 offspring were genotyped by PCR. Male hemizygous TRAMP mice were used. After weaning, mice were randomly divided and housed in the animal facility. At the age of 12 and 22 weeks, mice were sacrificed, and prostate/prostate cancer tissues were collected for analysis. Male specific pathogen-free (SPF) BALB/c Nude mice were obtained from Vital River Laboratories (Beijng, China). For PCa xenograft implantation, 5×106 DU145 cells were mixed with phenol red free Matrigel (Corning, Unit States) and injected intraperitoneally into the nude mice. The tumor volume should not exceed 1200 cubic millimeters (1.2 cubic centimeters). Tumor harvest and volume assessment were performed 8 weeks after the injection. All animal experiments were performed according to the animal guidelines, and with prior approval from the Animal Experimentations Ethics Committee, Wenzhou Medical University. We have complied with all relevant ethical regulations for animal use.
Cell culture
DU145 cells (CVCL_0105, ATCC, VA, United States) were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Thermo Fisher Scientific, MA, United States), and LNCaP cells (CVCL_0395, ATCC) and C4-2 cells (CVCL_4782, ATCC) were cultured in Roswell Park Memorial Institute RPMI 1640 Medium (Gibco) both supplemented with 10% fetal bovine serum (FBS, Gibco), 100 units/ml penicillin and 100 μg/ml streptomycin in 5% CO2 incubators. All cell lines have recently been authenticated using STR profiling by Genetic Testing Biotechnology Corporation (Suzhou, China) and were performed with mycoplasma-free cells. For treatments of cells, iron-chelating agent Deferoxamine (DFO, Sigma-Aldrich, Louis, United States), protein synthesis inhibitor Cycloheximide (CHX, Sigma-Aldrich), Proteasome inhibitor (R)-MG132 (Merck, NJ, Germany), Lysosomal inhibitor Chloroquine (CQ, Sigma-Aldrich), autophagy inducer Metformin (Met, Aladdin, Shanghai, China), and autophagy inducer Rapamycin (Rap, Aladdin) were added to the medium at the indicated concentrations.
Hematoxylin and eosin (H&E) staining
Paraffin sections were dewaxed in xylene for 5–10 min, switched to fresh xylene and dewaxed for another 5–10 min, which were incubated in anhydrous ethanol for 5 min, 90% ethanol for 2 min, 80% ethanol for 2 min, 70% ethanol for 2 min, and distilled water for 2 min. Sections were stained by hematoxylin staining solution for 5 min, immersed in tap water and rinsed off excess staining solution for about 10 min, washed again with distilled water, and stained by eosin solution for 30 s. After dehydrated, cleared and mounted, sections were observed under a light microscope (ECLIPSE Ni-E, Nikon, Japan) to determine morphological changes.
Immunostaining assay
For tissue immunofluorescence staining, the sections were deparaffinized, rehydrated, and antigen retrieved. After blocking with 3% bovine serum albumin (BSA, beyotime, Shanghai, China) at room temperature for 30 min, the sections were incubated with mixture of primary antibodies overnight at 4 °C. Anti-FGFR1 antibody (1:200) was obtained from Cell Signaling Technology. Anti-TFR1 (1:200) were obtained from Santa Cruz Biotechnology (CA, United States). After a brief wash, the sections were incubated with ExtrAvidin peroxidase system (Sigma-Aldrich) to visualize specifically bound antibodies.
For cell immunofluorescence staining, cells were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.3% Triton X-100/PBS (v/v) for 15 min, blocked with 3% BSA at room temperature for 30 min and then incubated with primary antibody overnight at 4 °C. Anti-FGFR1 antibody (1:200) was obtained from Cell Signaling Technology. Anti-FBXL5 (1:1000) was obtained from Proteintech (Wuhan, China). Anti-TFR1 (1:200), anti-PCNA (1:200) and anti-IRP2 (1:200) were obtained from Santa Cruz Biotechnology (CA, United States). After a brief wash, the slides were incubated with mixture of fluorescence-conjugated secondary antibodies (Invitrogen) for 1 h and then counterstained with DAPI. Images were taken with a laser scanning confocal microscope (Leica TCS SP8, IL, United States) in three randomly selected areas.
Prussian blue staining
Prussian blue stain enhanced with DAB (Solarbio, Beijing, China) was used to detect the presence of iron. Briefly, 5-µm paraffin sections on slides (HistoCore, Leica Biosystems, Wetzlar, Germany) were immersed in xylene for 10 min. Repeated the step once again in fresh xylene for 10 min. Next, the sections were rehydrated by sequentially incubating with 100%, 95%, 80%, and 60% ethanol for 5 min each and then rinsed with distilled water three times for 3 min each. The sections were incubated for 20 min each with Prussian blue, incubation solution, and enhanced solution in sequence. Then the sections were incubated in hematoxylin solution for 40 s, followed by a wash with distilled water. Labeled cells were examined under a light microscope (ECLIPSE Ni-E, Nikon, Tokyo, Japan) to determine intracellular iron oxide distribution.
Gene ablation and overexpression
The lentivirus-based CRISPR-Cas9 system was used to ablate FGFR1 alleles in DU145 cells. The sequence of single guide RNA (sgRNA) was AACTTGTTCCGATGGTTATC. Two days after infection with the lentivirus, the virus-containing cells were selected by growing in medium containing 2 μg/ml puromycin. For transient transfections, cells cultured overnight (1 × 105 cells/well in 6-well plates) were transfected with plasmid and 5 μl Lipofectamine 2000 (Thermo Fisher Scientific, MA, United States). The cells were then incubated at 37 °C for 24 h before analysis.
Quantitative real-time RT-PCR
Total RNA was extracted from cells using the TRIzol RNA isolation reagents (Takara bio, Otsu, Japan). The first strand cDNA was reversely transcribed from the RNA templates using the GoScript Reverse Transcription system kit (Promega, WI, United States) and Oligo(dT)15 primers (Promega) according to the manufacturer’s protocols. Quantified real-time PCR (qPCR) analyses were carried out using the FastStart Essential DNA Green Master (Roche, Switzerland) as instructed by the manufacturer. The relative abundance of mRNA was calculated using the comparative threshold cycle method and normalized to β-actin as the internal control. Primer information can be found in Supplementary Data 2.
Western blotting analysis
Cells were lysed in radio-immunoprecipitation assay (RIPA) buffer, and the extracted proteins were harvested by centrifugation at 12,000 g. Samples containing 30 μg protein were separated on SDS-PAGE and blotted onto polyvinylidene difluoride membranes for Western blot analyses with the indicated antibodies. The dilutions and sources of the antibodies are as follows. Anti-TFR1 (1:500), anti-DMT1 (1:500), anti-IRP1 (1:500), anti-IRP2 (1:500), anti-c-MYC(1:500), anti-IL-6(1:500), anti-GPX4 (1:500), anti-SOD2 (1:500), and anti-β-actin (1:1,000) were all obtained from Santa Cruz Biotechnology (CA, United States). Anti-FGFR1 (1:1000), Anti-p-ERK (1:1000), Anti-ERK (1:1000), anti-BCL2 (1:1000), anti-BAX (1:1000), Anti-P65 (1:1000), Anti-NRF2 (1:1000), Anti- Ubiquitin (1:1000), and anti-P53 (1:1000), were obtained from Cell Signaling Technology (MA, United States). Anti-FBXL5 (1:1000) was obtained from Abcam (Cambs, United Kingdom). The membranes were washed with TBS with Tween 20 (TBST) buffer to remove nonspecific antibodies and then incubated with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (Jackson ImmunoResearch, PA, United States) at room temperature for 1 h. The specifically bound antibodies were then visualized using ECL-Plus chemiluminescent reagents.
Cell viability assays
DU145 cells were seeded in 96-well plates at a density of 5 × 103 cells per well. The next day, fresh media containing DFO (20 μM, 50 μM, 100 μM), or control (double distilled water) were added, and cells incubated for 24 h. Cell Counting Kit-8 from MedChemExpress (CCK-8, United States) was used to determine cell viability, as per the manufacturer’s instructions. Cell viability was normalized against the vehicle control, and the data expressed as a percentage of control from three independent experiments done in triplicate.
5-ethynyl-2’-deoxyuridine (EdU) incorporation assay
Cells were inoculated 1 × 104 cells per well in 24-well plates, then incubated for 24 h according to the test requirements by the addition of DFO. Before the termination of cell culture, 10 μM EdU (Beyotime, Shanghai, China) was added. The cells were further incubated at 37 °C for 40 min. The culture medium was then discarded, and the slide was washed three times with PBS. The cells were fixed with methanol for 10 min, and then air dried. After adding the reaction mixture, different fields were randomly chosen to calculate the total number of cells and the number of EdU-positive cells in each vision field.
Measurement of cell death by FCM
Cell death was measured by staining with SYTOX Green (MedChemExpress, NJ, United States) to detect the plasma membrane integrity through FCM (Berkman, United States) according to the manufacturer’s instructions. Briefly, SYTOX Green was excluded from live cells but penetrating dead cells and emitted a green fluorescence that can be quantified by FCM.
Measurement of cellular iron level
Cellular labile iron was measured according to the manufacturer’s instructions. Briefly, cells were seeded in 96-well plates (5 × 102 cells per well). Cells were incubated with 1 μM Calcein AM (Beyotime, Shanghai, China) for 30 min at 37 °C. Then, cells were washed twice with PBS. The samples were examined by microplate readers (Synergy NEO2, BioTek, VT, United States) followed by incubated with 10 μM Pyridoxal isonicotinoyl hydrazone (PIH, MedChemExpress, NJ, United States) for 5 min. The samples were examined by microplate readers again. The difference between the two fluorescence intensities is the intracellular LIP.
Correlation analysis of TCGA data
RNA-sequencing expression profiles and corresponding clinical Gleason score for prostate cancer were downloaded from the TCGA dataset (https://portal.gdc.com). The two-index correlation map is realized by correlation analysis and visualized. Pearson’s and Spearman’s correlation analysis to respectively describe the correlation between quantitative variables with or without normal distribution.
Co-immunoprecipitation assay
DU145 and DU145ΔR1 cell lysates containing 2 mg protein were subjected to immunoprecipitation assay overnight at 4 °C incubating with either anti-FBXL5/anti-IRP2 antibody or mouse IgG/rabbit IgG controls (CST, Beverly, Massachusetts). Then 40 μl protein A/G PLUS-Agarose (Santa Cruz, California) beads were added to each tube. After incubation for 3 h, the beads were collected and washed with PBS for five times at 4 °C. Immunoprecipitated proteins were examined by Western blotting with the indicated antibodies.
RNA immunoprecipitation (RIP) assay
The RNA Immunoprecipitation Kit (Cat#Bes5101, BersinBio, China) was rigorously employed according to the manufacturer’s guidelines. The indicated cells were lysed with a polysome lysis buffer fortified with both protein inhibitor and RNase inhibitor. Subsequently, the cell lysate was incubated with DNase at 37 °C for 10 min, followed by the addition of 0.5 M EDTA, 0.5 M EGTA, and DTT for stabilization. The mixture was then centrifuged at 16,100 g for 10 min at 4 °C to obtain the supernatant, which was further incubated with anti-IRP2 antibody and Protein A/G beads on a vertical mixer with gentle rotation at 4 °C for 16 h to immuoprecipitate the RNA-protein complexes. The Beads carrying the complexes were thoroughly washed with polysome washing buffers 1 and 2, each containing 5 µL DTT, to remove non-specifically bound materials. Afterward, Proteinase K was added to degrade proteins, thereby releasing the bound RNAs. The liberated RNAs were extracted using Trizol reagent and subjected to RT-qPCR for quantitative analysis. The relative enrichment was normalized to the input. Primer information can be found in Supplementary Data 2.
Statistics and reproducibility
All data were collected from three independent experiments unless otherwise stated. Statistical analysis was done using GraphPad Prism 8.0.2 (GraphPad Software, CA). The sample size refers to biological repeats, which means repeating using a different biological sample preparation. P Values among two groups were computed using Student’s t test. And P Values among exceeding three groups were computed using one-way ANOVA. The statistical P values of each experiment were shown above the bar. All statistical analysis data can be found in Supplementary Data 2.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available in Supplementary Data 1. The details that support the findings of this study are available from the corresponding author upon reasonable request.
References
Torre, L. A. et al. Global cancer statistics, 2012. CA Cancer J. Clin. 65, 87–108 (2015).
Isbarn, H. et al. Androgen deprivation therapy for the treatment of prostate cancer: consider both benefits and risks. Eur. Urol. 55, 62–75 (2009).
Wesche, J., Haglund, K. & Haugsten, E. M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 437, 199–213 (2011). p.
Corn, P. G., Wang, F., McKeehan, W. L. & Navone, N. Targeting fibroblast growth factor pathways in prostate cancer. Clin. Cancer Res. 19, 5856–5866 (2013).
Yang, F. et al. FGFR1 is essential for prostate cancer progression and metastasis. Cancer Res. 73, 3716–3724 (2013).
Wang, C. et al. Ectopic fibroblast growth factor receptor 1 promotes inflammation by promoting nuclear factor-kappaB signaling in prostate cancer cells. J. Biol. Chem. 293, 14839–14849 (2018).
Liu, J. et al. Aberrant FGFR tyrosine kinase signaling enhances the warburg effect by reprogramming LDH isoform expression and activity in prostate cancer. Cancer Res. 78, 4459–4470 (2018).
Xue, X. et al. Iron uptake via DMT1 integrates cell cycle with JAK-STAT3 signaling to promote colorectal tumorigenesis. Cell Metab. 24, 447–461 (2016).
Torti, S. V. & Torti, F. M. Iron and cancer: more ore to be mined. Nat. Rev. Cancer 13, 342–355 (2013).
Jung, M., Weigert, A., Mertens, C., Rehwald, C. & Brune, B. Iron Handling in Tumor-Associated Macrophages-Is There a New Role for Lipocalin-2? Front. Immunol. 8, 1171 (2017).
Daniels, T. R., Delgado, T., Helguera, G. & Penichet, M. L. The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clin. Immunol. 121, 159–176 (2006).
Hentze, M. W., Muckenthaler, M. U., Galy, B. & Camaschella, C. Two to tango: regulation of Mammalian iron metabolism. Cell 142, 24–38 (2010).
Brookes, M. J. et al. Modulation of iron transport proteins in human colorectal carcinogenesis. Gut 55, 1449–1460 (2006).
Zhang, C. & Zhang, F. Iron homeostasis and tumorigenesis: molecular mechanisms and therapeutic opportunities. Protein Cell 6, 88–100 (2015).
Fu, D. & Richardson, D. R. Iron chelation and regulation of the cell cycle: 2 mechanisms of posttranscriptional regulation of the universal cyclin-dependent kinase inhibitor p21CIP1/WAF1 by iron depletion. Blood 110, 752–761 (2007).
Khiroya, H. et al. IRP2 as a potential modulator of cell proliferation, apoptosis and prognosis in nonsmall cell lung cancer. Eur. Respir. J. 49, 1600711 (2017).
Richardson, D. R., Kalinowski, D. S., Lau, S., Jansson, P. J. & Lovejoy, D. B. Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents. Biochim. Biophys. Acta 1790, 702–717 (2009).
Guntupalli, J. N. et al. Trace elemental analysis of normal, benign hypertrophic and cancerous tissues of the prostate gland using the particle-induced X-ray emission technique. Eur. J. Cancer Prev. 16, 108–115 (2007).
Sarafanov, A. G. et al. Prostate cancer outcome and tissue levels of metal ions. Prostate 71, 1231–1238 (2011).
Ornstein, D. L. & Zacharski, L. R. Iron stimulates urokinase plasminogen activator expression and activates NF-kappa B in human prostate cancer cells. Nutr. Cancer 58, 115–126 (2007).
Choi, J.-Y. et al. Iron intake, oxidative stress-related genes (MnSOD and MPO) and prostate cancer risk in CARET cohort. Carcinogenesis 29, 964–970 (2008).
Xiang, Y. et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc. Natl Acad. Sci. USA 104, 19226–19231 (2007).
Xiao, C. et al. Transferrin receptor regulates malignancies and the stemness of hepatocellular carcinoma-derived cancer stem-like cells by affecting iron accumulation. PloS ONE 15, e0243812 (2020).
Jung, S. et al. Depletion of ST6GALNACIII retards A549 non-small cell lung cancer cell proliferation by downregulating transferrin receptor protein 1 expression. Biochem. Biophys. Res. Commun. 575, 78–84 (2021).
Lodhi, M. et al. Probing transferrin receptor overexpression in gastric cancer mice models. ACS Omega 6, 29893–29904 (2021).
Huang, N. et al. Iron metabolism protein transferrin receptor 1 involves in cervical cancer progression by affecting gene expression and alternative splicing in HeLa cells. Genes Genom. 44, 637–650 (2022).
Senyilmaz, D. et al. Regulation of mitochondrial morphology and function by stearoylation of TFR1. Nature 525, 124–128 (2015).
Pantopoulos, K. Iron metabolism and the IRE/IRP regulatory system: an update. Ann. N. Y. Acad. Sci. 1012, 1–13 (2004).
Philpott, C. C., Klausner, R. D. & Rouault, T. A. The bifunctional iron-responsive element binding protein/cytosolic aconitase: the role of active-site residues in ligand binding and regulation. Proc. Natl Acad. Sci. USA 91, 7321–7325 (1994).
Guo, B., Phillips, J. D., Yu, Y. & Leibold, E. A. Iron regulates the intracellular degradation of iron regulatory protein 2 by the proteasome. J. Biol. Chem. 270, 21645–21651 (1995).
Samaniego, F., Chin, J., Iwai, K., Rouault, T. A. & Klausner, R. D. Molecular characterization of a second iron-responsive element binding protein, iron regulatory protein 2. Structure, function, and post-translational regulation. J. Biol. Chem. 269, 30904–30910 (1994).
Iwai, K., Klausner, R. D. & Rouault, T. A. Requirements for iron-regulated degradation of the RNA binding protein, iron regulatory protein 2. EMBO J. 14, 5350–5357 (1995).
Deng, Z., Manz, D. H., Torti, S. V. & Torti, F. M. Iron-responsive element-binding protein 2 plays an essential role in regulating prostate cancer cell growth. Oncotarget 8, 82231–82243 (2017).
Currie, C., Bjerknes, C., Myklebust, T. A. & Framroze, B. Assessing the potential of small peptides for altering expression levels of the iron-regulatory genes FTH1 and TFRC and enhancing androgen receptor inhibitor activity in in vitro prostate cancer models. Int. J. Mol. Sci. 24, 15231 (2023).
Luscieti, S. et al. The actin-binding protein profilin 2 is a novel regulator of iron homeostasis. Blood 130, 1934–1945 (2017).
Wang, Y.-F. et al. G9a regulates breast cancer growth by modulating iron homeostasis through the repression of ferroxidase hephaestin. Nat. Commun. 8, 274 (2017).
Kerins, M. J. & Ooi, A. The Roles of NRF2 in modulating cellular iron homeostasis. Antioxid. Redox Signal. 29, 1756–1773 (2018).
Wu, K., El Zowalaty, A. E., Sayin, V. I. & Papagiannakopoulos, T. The pleiotropic functions of reactive oxygen species in cancer. Nat. Cancer 5, 384–399 (2024).
Nakamura, H. & Takada, K. Reactive oxygen species in cancer: current findings and future directions. Cancer Sci. 112, 3945–3952 (2021).
Jung, M., Mertens, C., Tomat, E. & Brune, B. Iron as a central player and promising target in cancer progression. Int. J. Mol. Sci. 20, 273 (2019).
Henderson, B. R., Menotti, E. & Kühn, L. C. Iron regulatory proteins 1 and 2 bind distinct sets of RNA target sequences. J. Biol. Chem. 271, 4900–4908 (1996).
O’Donnell, K. A. et al. Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Mol. Cell. Biol. 26, 2373–2386 (2006).
Zhang, Y. et al. Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev. 31, 1243–1256 (2017).
Liu, J., Zhang, C., Hu, W. & Feng, Z. Tumor suppressor p53 and metabolism. J. Mol. cell Biol. 11, 284–292 (2019).
Wang, H. et al. FBXL5 regulates IRP2 stability in iron homeostasis via an oxygen-responsive [2Fe2S] cluster. Mol. Cell 78, 31–41.e5 (2020).
Beshara, S. et al. Anemia associated with advanced prostatic adenocarcinoma: effects of recombinant human erythropoietin. Prostate 31, 153–160 (1997).
Lee, M. H. & Means, R. T. Extremely elevated serum ferritin levels in a university hospital: associated diseases and clinical significance. Am. J. Med. 98, 566–571 (1995).
Shen, Y. et al. Transferrin receptor 1 in cancer: a new sight for cancer therapy. Am. J. Cancer Res. 8, 916–931 (2018).
Taylor, B. S. et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22 (2010).
Fan, Z. et al. Crosstalk of FGFR1 signaling and choline metabolism promotes cell proliferation and survival in prostate cancer cells. Int. J. Cancer 150, 1525–1536 (2022).
Guntupalli, J. N. R. et al. Trace elemental analysis of normal, benign hypertrophic and cancerous tissues of the prostate gland using the particle-induced X-ray emission technique. Eur. J. Cancer Prev. 16, 108–115 (2007).
Johnson, I. R. D. et al. Altered endosome biogenesis in prostate cancer has biomarker potential. Mol. Cancer Res. 12, 1851–1862 (2014).
Keer, H. N. et al. Elevated transferrin receptor content in human prostate cancer cell lines assessed in vitro and in vivo. J. Urol. 143, 381–385 (1990).
Zapała, P. et al. E-Cadherin, Integrin Alpha2 (Cd49b), and Transferrin Receptor-1 (Tfr1) are promising immunohistochemical markers of selected adverse pathological features in patients treated with radical prostatectomy. J. Clin. Med. 10, 5587 (2021).
Lang, J. et al. Targeted Co-delivery of the iron chelator deferoxamine and a HIF1α inhibitor impairs pancreatic tumor growth. ACS Nano 13, 2176–2189 (2019).
Komoto, K. et al. Iron chelation cancer therapy using hydrophilic block copolymers conjugated with deferoxamine. Cancer Sci. 112, 410–421 (2021).
Sandoval-Acuña, C. et al. Targeting mitochondrial iron metabolism suppresses tumor growth and metastasis by inducing mitochondrial dysfunction and mitophagy. Cancer Res. 81, 2289–2303 (2021).
Li, Z., Tanaka, H., Galiano, F. & Glass, J. Anticancer activity of the iron facilitator LS081. J. Exp. Clin. Cancer Res. 30, 34 (2011).
Moroishi, T., Nishiyama, M., Takeda, Y., Iwai, K. & Nakayama, K. I. The FBXL5-IRP2 axis is integral to control of iron metabolism in vivo. Cell Metab. 14, 339–351 (2011).
Guo, B., Brown, F. M., Phillips, J. D., Yu, Y. & Leibold, E. A. Characterization and expression of iron regulatory protein 2 (IRP2). Presence of multiple IRP2 transcripts regulated by intracellular iron levels. J. Biol. Chem. 270, 16529–16535 (1995).
Wang, J. et al. Iron-mediated degradation of IRP2, an unexpected pathway involving a 2-oxoglutarate-dependent oxygenase activity. Mol. Cell. Biol. 24, 954–965 (2004).
Ishikawa, H. et al. Involvement of heme regulatory motif in heme-mediated ubiquitination and degradation of IRP2. Mol. Cell 19, 171–181 (2005).
Elton, L., Carpentier, I., Verhelst, K., Staal, J. & Beyaert, R. The multifaceted role of the E3 ubiquitin ligase HOIL-1: beyond linear ubiquitination. Immunol. Rev. 266, 208–221 (2015).
Salahudeen, A. A. et al. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 326, 722–726 (2009).
Vashisht, A. A. et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 326, 718–721 (2009).
Funding
This work was supported by the National Natural Science Foundation of China (82173013, 82372941, 81971894), the Natural Science Foundation of Zhejiang Province of China (Z23H160024, LR20H310001, LWY20H300001, LY17H150003), Project of Wenzhou Science & Technology Bureau (Y2023161, Y20210085).
Author information
Authors and Affiliations
Contributions
Wang Cong: Conceptualization, Investigation, Resources, Writing-original draft, Writing-review&editing, Project administration, Funding acquisition. Li Xiaokun: Conceptualization, Resources, Funding acquisition. Pan Xuebo: Conceptualization, Resources, Investigation, Project administration. Lin Hui: Methodology, Software, Validation, Formal analysis, Data curation, Writing-original draft. Shi Liuhong: Methodology, Validation, Data curation. Lin Shuaijun: Methodology, Validation, Data curation. Lin Manjie & Xiao Yunbei: Methodology, Software, Validation. Xu Guangsen: Validation. Li Shupeng & Li Zhiquan: Methodology, Formal analysis. Chen Jiale & Catherine Nakazibwe: Methodology, Formal analysis. The work reported in the paper has been performed by the authors, unless clearly specified in the text.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics Statement
Approval of the research protocol by an Institutional Reviewer Board: The experimental procedures for plasmid construction were approved by the Genetically Modified Organisms Safety Committee of Wenzhou Medical University. Animal Studies: The male BALB/c nude mice were housed under the Program of Animal Resources of the Wenzhou Medical University in accordance with the principles and procedures of the Guide for the Care and Use of Laboratory Animals.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Toshiro Moroishi and Christina Karlsson Rosenthal. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, H., Lin, S., Shi, L. et al. FGFR1 governs iron homeostasis via regulating intracellular protein degradation pathways of IRP2 in prostate cancer cells. Commun Biol 7, 1011 (2024). https://doi.org/10.1038/s42003-024-06704-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06704-6
- Springer Nature Limited