
Abstract
The ability to finely tune the properties of magnetic topological insulators (TIs) is crucial for quantum electronics. We studied solid solutions with a general formula GexMn1-xBi2Te4 between two isostructural Z2 TIs, magnetic MnBi2Te4 and nonmagnetic GeBi2Te4 with Z2 invariants of 1;000 and 1;001, respectively. We observed linear x-dependent magnetic properties, composition-independent pairwise exchange interactions, and topological phase transitions (TPTs) between topologically nontrivial phases and the semimetal state. The TPTs are driven purely by the variation of orbital contributions. By tracing the x-dependent Bi 6p contribution to the states near the fundamental gap, the effective spin-orbit coupling variation is extracted. The gapless state observed at x = 0.42 closely resembles a Dirac semimetal above the Néel temperature and shows a magnetic gap below, which is clearly visible in raw photoemission data. The observed behavior demonstrates an ability to precisely control topological and magnetic properties of TIs.
Similar content being viewed by others


Introduction
Recently, magnetic topological insulators (MTIs) have become the focus of significant scientific interest. Among them, Z2 topological insulators (TIs) with ordered magnetic moments of embedded magnetic atoms have attracted special attention. One particularly intriguing example is the case of systems with a surface-normal magnetic easy axis coexisting with a topologically nontrivial surface state. Such materials demonstrate the quantum anomalous Hall effect, which manifests itself as chiral edge conduction channels that can be manipulated by switching the polarization of magnetic domains1.
To date, the most intensely studied MTIs are MnBi2Te4 (the so-called 124 phase), a layered compound composed of septuple layers (SLs) with an idealized layer sequence of Te–Bi–Te–Mn–Te–Bi–Te, along with its homological compounds 147 and 1610 etc. For them, partial intermixing of the Mn and Bi positions is observed in the cation sublattice, yielding MnBi and BiMn antisite defects. MnBi2Te4 is an antiferromagnetic (AFM) TI with TN = 24.5 K2. For such a system, gap opening is observed in the Dirac cone, although its width and origin are still under debate2. The system can be switched to the quantum anomalous Hall (QAH) regime in the case of ultra-thin flakes3.
Potential practical applications of MnBi2Te4 are restricted by the availability of only small crystals due to preparative issues and limited thermodynamic stability4. The MnBi2Te4 lattice can be stabilized by mixing with stable isostructural compounds such as GeBi2Te4, SnBi2Te4, and PbBi2Te4, which are believed to be Z2 TIs5,6,7. Among them, GeBi2Te4 is ideally suited and meets all requirements for its crystal structure (cation coordination, bond lengths) and the spin-orbit coupling (SOC) strength (related to core charge effect). It is known that in the related system MnTe-GeTe, a broad range of mutual solubility is observed for cubic MnxGe1−xTe, where Ge and Mn are also inside the Te octahedra8. One can expect similar behavior for the GeBi2Te4-MnBi2Te4 system which additionally holds the possibility to finely tune exchange interactions through dilution of the magnetic system. For instance, for MnxGe1−xTe, Mn substitution with Ge leads to a monotonic decrease of the Néel temperature in bulk crystals9. This behavior is further for the case of MnxGe1−xTe thin films, where the electronic band structure is modified by strain10. Moreover, GexMn1−xTe films have a Curie temperature as high as 200 K for MnTe concentration of 50%11.
Regarding chemical bonding in the 124 compound, the main difference between MnBi2Te4 and GeBi2Te4 is the orbital contributions in the electronic bands. Ge atoms in GeBi2Te4 are bonded through 4p electrons, while Mn atoms mainly contribute 4s electrons. Therefore, the effective SOC of the valence electrons can be varied with changing composition, thereby potentially driving a SOC-mediated gap closing across the GexMn1−xBi2Te4 series and producing a 3D Dirac/Weyl semimetal (3D DSM/WSM) state for mixed crystals12. Both 3D DSM and WSM phases host plenty of unique properties and phenomena, including high mobility of bulk carriers13, chiral anomaly and negative magnetoresistance14,15, Fermi arcs16, anomalous Hall effect17, charge-to-spin conversion18, spin Seebeck effect19. These effects have practical potential in topological electronics20, quantum computing, optoelectronics, spintronics and energy conversion21.
In this paper we report band structure and magnetic properties of GexMn1−xBi2Te4 crystals vs x, where the variation of p- and s- orbital contributions indirectly influences the Bi 6p contribution to the bands near the fundamental band gap and induces a band inversion and a bulk gapless state corresponding to x = 0.42. This semimetal state in turn demonstrates magnetic gap opening below Néel temperature. All observations were made for massive crystals of high structural perfection obtained by the original method developed in this work.
Results and discussion
Crystal growth and characterization
Initially, GexMn1−xBi2Te4 crystals were grown from the melt using the Bridgman method. Growth was performed from the melt of GexMn1−xBi2Te4 with x taken equal to the targeted composition. However, massive bulk crystals of high quality were obtained only for x ≥ 0.6. To grow crystals of lower x, we developed a growth protocol with the source initially including two phases L(Bi2Te3)+S(GexMn1−xTe). The solid phase was added as a compact tablet. Due to its lower density, the tablet floats over Bi2Te3 melt. After equilibration with the melt, the tablet dissolves gradually during growth. Preliminary nucleation temperature was determined by differential thermal analysis. Growth was performed in evacuated quartz cylindrical ampoules with conic tips. The ampoule’s inner surface was preliminarily graphitized. Each ampoule was placed into a furnace and equilibrated during the day at a temperature 10 °C higher than the nucleation temperature. Further, the ampoule was pulled in the temperature gradient at the rate of 0.2 mm/h. Growth conditions are summarized in Supplementary Tables 1 and 2. The result of this growth procedure produced good-quality, several-cm-sized crystals of the whole composition range of this system.
A typical crystal obtained is shown in Fig. 1a (inset). It should be noted that some crystals included in length several structural types of general formula nGexMn1−xTe*mBi2Te3 grown one by one. Most of the experiments show the general tendency of Y = (Ge+Mn)/(Ge+Mn+Bi) ≈ n/(n + 2 m) to decrease during crystal growth. Figure 1a exhibits GeBi2Te4 mole fraction along the longitudinal direction of 124 parts for all crystals according to X-ray fluorescent spectroscopy (XRF). X-Ray Diffraction (XRD) data are collected in Fig. 1c. They correspond well to \(R\overline{3}m\) structure (Fig. 1b). Both lattice constants, a and c, vary slightly with composition (less than one percent), as shown in Fig. 1d, e.

a Crystal composition longitudinal variation and general view of a typical crystal (cleaved), b crystal structure, c X-ray diffractograms, d, e lattice constants. Error bars shown correspond to deviation from mean composition and fit error of c and a.
Magnetic properties of GexMn1− xBi2Te4 crystals
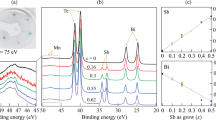
In order to unveil the magnetic properties, the Mn charge state and its possible variation for different compositions needs to be determined. To do this, X-ray photoemission spectroscopy (XPS) and absorption (NEXAFS) spectroscopy were employed. The corresponding Mn 2p spectra are presented in Fig. 2a and Supplementary Fig. 1. The absorption spectra show typical features I-IV which correspond to the transitions from 2p1/2 to split 3d levels, eg and t2g. Photoemission spectra demonstrate a multiplet splitting typical of Mn2+. No noticeable change of spectral shape is observed upon composition variation. All in all, both absorption and photoemission spectra and Mn 2p3/2 binding energies are similar to those of MnTe22, MnBi2Te44, and Mn0.08Bi1.92Se323, evidencing a 3d5 configuration. More detailed analysis of photoemission spectra is complicated by complex background shapes and limited intensity, especially for Ge-enriched samples.

a NEXAFS spectra for crystals with different Ge content. Temperature dependence of magnetic susceptibility (b), Néel temperature dependence on effective magnetic moment (c), and composition (d). Error bars shown correspond to deviation from mean composition and fit error of TN and μeff. GexMn1−xBi2Te4 (x = 0.6) (111) surface topography (e) and magnetic contrast (f), surface profile, and corresponding magnetic signal shown by curve color (f). The colorbar in (f) also implies (g), and arrows denote the magnetization of the layer. Crystal structure with indicated spin of Mn atoms (h).
Figure 2b shows temperature dependencies of magnetic susceptibility. Generally, all crystals are paramagnetic (PM) and demonstrate a transition to the AFM state with the easy magnetization axis, c, at the Néel temperature (additional data are available in Supplementary Fig. 2 and Supplementary Table 3). Materials demonstrate AFM interlayer order and ferromagnetic (FM) intralayer order as confirmed by magnetic force microscopy (MFM) results discussed below. Susceptibility curves for all compositions have a similar shape, and even the most dilute magnetic crystal of x = 0.77 ± 0.01 demonstrates behavior similar to that of pure MnBi2Te4. Upon dilution of the magnetic sublattice, the Néel temperature drops linearly from TN ≈ 20 K for x = 0.16 ± 0.03 to TN ≈ 6 K for x = 0.77 ± 0.01 (see Fig. 2d). Besides, Néel temperature is also a linear function of squared effective magnetic moment. Consequently, the effective exchange integral, in turn, is a linear function of dilution x; and a pairwise exchange coupling does not depend on x. This is true even for the most dilute crystals showing AFM properties. These observations demonstrate that the easy magnetization axis does not depend on composition, and magnetic dilution affects neither the choice of magnetically active sublattices nor the pairwise exchange interaction of ions.
To verify our supposition on magnetic structure, namely on interlayer and intralayer interactions, we studied a GexMn1−xBi2Te4 crystal with x = 0.6 by MFM. Figure 2e, f shows topography with terraces of septuple-layer (SL) height and the corresponding magnetic contrast below Néel temperature. One can see that the magnetic contrast appears at the steps of SL height (or odd number of SLs). There are also additional antiphase boundaries that are not related to the steps, an example of which is indicated with a violet line in Fig. 2f. Correspondence between the magnetic contrast and the step height is in line with AFM order between ferromagnetically ordered SLs as schematically shown in Fig. 2h. All in all, the behavior of GexMn1−xBi2Te4 enables precise control and fine-tuning of magnetic properties of the topological insulator by x variation.
Electronic band structure of GexMn1− xBi2Te4
Now we focus on the effect of Mn substitution with Ge on the electronic band structure and its evolution for mixed crystals of GexMn1−xBi2Te4 with x ranging from 0 to 1. Figure 3 shows angle-resolved photoemission spectroscopy (ARPES) results obtained with a photon energy of 21.2 eV at which the photoemission (PE) cross section for the topological surface states (TSS) of MnBi2Te4 is rather low24, and PE signal from the bulk bands dominates in the spectra. The upper row in Fig. 3 presents band dispersions along the K–Г–K direction of the surface Brillouin zone (BZ). In general, the ARPES data for all x values exhibit rather similar sets of bands. The bulk valence band (VB) has a nearly cone-like shape. Its constant-energy contours are represented by hexagonally warped circles with a decrease of warping close to the top of the band. The bulk character of this band is evidenced by its kz dispersion24,25. In the range of x = 0.2–0.8, this band demonstrates a monotonic shift towards the Fermi level that can be assigned to the gradual depopulation of the conduction band states. At x = 1, corresponding to GeBi2Te4, the material again becomes n-doped, most probably due to another type of dominant point defect. Generally, GeBi2Te4 crystals can have variable carrier concentration depending on nonstoichiometry26: Te excess provides a lower carrier concentration as shown in Supplementary Fig. 3 where ARPES data are presented for crystals with different composition.

The row (a) shows band dispersions measured along the high-symmetry \(\bar{{{{{{\rm{K}}}}}}}-\bar{\Gamma }-\bar{{{{{{\rm{K}}}}}}}\) direction of the surface-projected BZ. Spectral features SS (Surface state), CB (Conduction band), VB (Valence band), S(1,2), and TSS (Topological surface states) are marked by black arrows. Black dashed lines (Cut I, II) indicate the positions of constant-energy cuts. Black dotted lines define the edges of the bandgap. The row (b) displays Fermi-surface maps. The row (c) contains constant-energy maps plotted for different energies marked as “cut II”. The photon energy was 21.2 eV (He I radiation, 15 K).
The bulk conduction band (CB) for GexMn1−xBi2Te4 for low x is of a nearly conical shape and can be regarded as a counterpart of the VB (see Fig. 3). It should be noted that the gap between these states observed with ARPES strongly depends not only on the crystal composition but on the photon energy as well, and may be larger than the fundamental gap because of the kz dispersion of VB and CB states. In the range of x = 0–0.17 the observed gap is about 0.12 eV, while at x = 0.38 the gap shrinks. Due to the finite width of the PE spectral features, it is therefore unclear whether the gap becomes fully closed or not, but an upper limit of 0.05 eV can be placed with certainty. Upon further increase of x to 0.65, the ARPES data indicate no visible opening of the gap, while at x = 0.81, the VB apex moves above the Fermi level, and the gap becomes inaccessible for PE analysis.
Near the Fermi level, one can also see the band labeled as SS, which is barely visible at x = 0 but becomes more pronounced with increasing Ge content. As it was shown for MnBi2Te4, this band is formed by surface states that demonstrate Rashba-type splitting27 combined with magnetic exchange splitting28. Density functional theory (DFT) calculations predicted that these surface states should remain in the case of Mn0.5Ge0.5Bi2Te429 in full consistency with our ARPES data in Fig. 3. Upon the increase of x, the SS band appears closer to VB and reaches it at x = 0.65. Our additional calculations in slab geometry (Supplementary Note 1, Supplementary Fig. 9) reproduce well the Rashba band and TSS, although the calculated dispersion does not exactly correspond to the experimental data. In detail, the Rashba-band is of the same nature as in the case of pure MnBi2Te4 compound. In our calculations, the splitting of the minima of the Rashba band becomes more pronounced with increasing x. Also, TSS and the Rashba band have a strong hybridization, leading to the hybridization gap.
Our ARPES data for pure GeBi2Te4 (x = 1) demonstrate a characteristic TSS that appears in agreement with the previous studies of this compound30. The conduction band here is formed by the bands S1 and S2, which were assigned as bulk states on the basis of their photon energy dependence30. Their W-like form at the Г-point indicates an indirect character of the fundamental band gap that was estimated to be 0.12 eV wide30. In our data in Fig. 3, the apparent gap width (at hν = 21.2 eV) is about 0.17 eV. One can see that the TSS touches the VB so that its Dirac point appears below the VB maximum. Gradual energy shift of SS bands towards the VB upon the increase of Ge content suggests that the Rashba-type SS band, which is trivial at x = 027, evolves into the nontrivial TSS of GeBi2Te4. Taking into account that this transformation is accompanied by shrinkage and reopening of the bulk gap, we suggest that topological phase transitions (TPTs) occur at x value of 0.5 ± 0.1.
Now we consider the photon energy dependencies of the PE spectra. This type of measurement gives information on the dispersion of electronic states in the direction perpendicular to the sample surface (Г–Z direction in the Brillouin zone), thus allowing to distinguish bulk states from surface states which have no dispersion along Г–Z (kz direction). The analysis is based on the assumption that the electron is excited into the free-electron-like state inside the crystal with constant inner potential V0. In this case, the photoelectron energy is directly connected with its momentum in the ground state. Figure 4 shows the dependencies of the normal-emission spectra on the photon energy for different samples in the PM phase. In such a geometry, the in-plane components of momentum are fixed to zero, while the out-of-plane component kz is defined by the photon energy as
where φ is the work function, and EB is the binding energy. The experimentally observed periodicities in our kz dispersions are well described under the assumption that V0 = φ + EB. The respective relation between the photon energies and high-symmetry points is indicated above the bottom scales in Fig. 4. In our notations, Г0 corresponds to zero k. The kz dependence for MnBi2Te4 is presented in Ref. 25. indicates that the gap between the VB and CB varies between 0.13 eV at the Г point and 0.20 eV at Z. Our data for x = 0.16 (left panel of Fig. 4) show qualitatively similar gap variations, although the bottom of the CB is not well distinguished. The photon energy dependence changes significantly at x = 0.42. At this concentration, the bottom of the CB becomes well resolved, and its kz dependence closely resembles that of the top of the VB. Analysis of individual ARPES maps taken at different hν indicates that the CB minimum, which is observed at the Z point, is located at nearly the same energy as the VB maximum observed in the Г point. Thus, at x = 0.42, the system turns into a semimetal with zero indirect bandgap in the PM phase. At x = 0.56, the amplitude of the kz dispersion increases for the VB, while we do not detect any significant dispersion for the CB. Upon further increase of Ge content to x = 0.76, the VB dispersion does not change much in its shape, although it is shifted to the Fermi level; the system becomes p-doped, and the CB appears completely unoccupied. Finally, at x = 1, the system becomes n-doped again. In its kz dispersion, we recognize the dispersive VB and non-dispersive TSS marked with the dotted line in the right panel of Fig. 4. The non-dispersive character of the TSS confirms its localization at the surface. Additional data can be found in Supplementary Fig. 4 and 5.

The data taken for a x = 0.16 ± 0.02, white circles correspond to the VB position. b x = 0.42 ± 0.02, c x = 0.56 ± 0.01, d x = 0.76 ± 0.01, e x = 1.0, white dashed line indicates the position of TSS.
All in all, for mixed crystals of GexMn1−xBi2Te4, upon composition variation, we observed experimentally a bulk band gap closure around x = 0.4 and a further opening at x > 0.65 that may indicate TPTs between two topologically nontrivial materials. It is noticeable that this transition occurs neither due to strong SOC variation nor structural changes since both system components (MnBi2Te4 and GeBi2Te4) are rather similar in these properties. To shed light on the underlying mechanism, we performed a theoretical study of the electronic structure. First, we used a coherent potential approximation (CPA) in frames of the Korringa–Kohn–Rostoker (KKR) Green’s function method, which enables adequate modeling of mixed crystals by taking into account disorder induced by intermixing of Mn and Ge. Within this approach, crystal composition can vary smoothly without the construction of large supercells. To trace purely compositional effects in our calculations atomic positions for mixed crystals were fixed as they are known for MnBi2Te42. The results are collected in Fig. 5, where the KKR-calculated bulk band structure of MnBi2Te4 along the K–Г–Z direction in the BZ and its variations upon gradual substitution of magnetic Mn atoms with non-magnetic Ge atoms are shown, the spectral functions being with a minimal half-width of about 15 meV. According to these calculations, the bulk gap gradually decreases to zero for x variation from 0 to 0.4. For x > 0.6, a gap opens, and it increases as x approaches unity. The dependencies of the gap width in the Г and Z points on crystal composition are exhibited in Fig. 5i, j. These calculations reproduce our ARPES results despite both a slight difference in the structure of counterparts and the presence of Bi/Ge (and Bi/Mn) antisite defects, which were disregarded. The gapless states at x = 0.4–0.6 may pinpoint TPTs from a topologically nontrivial to a trivial phase or semimetal state with a zero bulk gap and further transition to TI above x = 0.6.

a–h Bulk band structure of GexMn1−xBi2Te4 in the AFM phase (except for nonmagnetic system with x = 1). i, j Gap width in Г (i) and Z (j) points of the BZ. Error bars shown correspond to twice the widths of the bands.
The underlying mechanism of TPTs may be related to the difference in chemical bonding of Mn and Ge atoms in 124 compounds. This difference is due to the fact that Mn atoms form bonds via 4s electrons while Ge uses 3p electrons. To evaluate orbital contributions into bands near the Fermi level, we performed additional DFT modeling in this part of the study. This approach does not allow simulation of randomly intermixed Ge and Mn atoms, for this reason we considered 2 × 2 supercells for certain x values of 0.25, 0.5, and 0.75. In these, calculations the atomic positions were also fixed, being equal to those of pristine MnBi2Te4, like in KKR calculations discussed above, with a single exception—Van-der-Waals (vdW) gap between SLs in MnBi2Te4 crystal was increased by 0.16 Å for better correspondence with ARPES data on kz dispersion. The DFT calculation results are illustrated in Fig. 6. Similarly to the KKR data discussed above, here bulk band gap becomes closed at the Г point at about x = 0.5 upon Mn substitution with Ge, followed by a gap reopening in Г when x approaches unity. However, in the DFT calculation for x = 1, the gap appears to be almost closed between the Г and Z points in contrast to the KKR and experimental results. One of the possible reasons is the strong sensitivity of the DFT calculated electronic structure of GeBi2Te4 to computational geometry. Indeed, the modeling of GeBi2Te4 with its own experimental crystal structure31 results in the gapped state, as it is clearly seen in Fig. 6f. Moreover, to ensure that gap closure at x = 0.5 is not the result of the artificially fixed structural parameters, we performed additional DFT calculations using the averaged cell parameters of MnBi2Te4 and GeBi2Te4. These calculations verified that the gap remains closed in this case (see Supplementary Note 1, Supplementary Fig. 6).

The gray color scale in a–f indicates the total band weight unfolded to a primitive 1 × 1 unit cell with a height of 1 SL. b Three exchange gaps are marked by Δ1–3. In f the bands of GeBi2Te4 were calculated for its own lattice parameters in contrast to (a–e), where the lattice parameters of MnBi2Te4 were used. The color scale in (g–k) illustrates the weight of Bi p-orbitals; in (h–j), the weight is unfolded to a 1 × 1 cell with the height of 2 SL; in (g) and (k), the weight is given without unfolding for the unit cells with the heights of 2 SL and 1 SL, respectively. l, m The weight of Bi p-orbitals in the Г and Z points. The lower and upper curves denote the states below and above the Fermi level.
It is worth noting that in the experimental ARPES data, the transition from the PM to the AFM state leads to minor changes in the spectral features, while in the calculation, the formal period of the unit cell changes from 1 SL to 2 SL. This leads to the doubling of all bands due to their folding. For this reason, in Fig. 6h, the VB of nonmagnetic GeBi2Te4 is represented with a single branch, while for the AFM systems in Fig. 6a–g, one can see two branches. To allow direct comparison with the ARPES data, we showed in Fig. 6a–d the bands unfolded to the unit cell of 1 SL height. In such a presentation, one can clearly see where the VB undergoes exchange splitting upon AFM order. Two examples of such exchange splitting are marked as Δ2 and Δ3 in Fig. 6b. They were observed in the ARPES data of MnBi2Te425,32, although our DFT calculations overestimate their experimental values (Δ2 ≈ 25 meV and Δ3 ≈ 45 meV at x = 032).
To disentangle the question of the gap closing and reopening upon variation of the Ge content, we trace the changes of the band orbital composition represented by the weight of Bi p-orbitals in Fig. 6g–k. Looking at the electronic states that form the bulk gap, it becomes evident that as x passes through the value of 0.5, the dominance of Bi-p at the VB maximum near the Г point is replaced by its dominance at the CB minimum. In other words, there is an inversion of the Bi p-orbitals contribution, indicating a possible transition from the TI phase into a semimetal state. Further increase of Ge concentration up to 100% leads to a new inversion of the Bi p-orbitals contribution, but near the Z point, which indicates the return of the system into the TI state. Summing all mentioned above, it is possible to suggest two possible phase transitions in GexMn1−xBi2Te4 as the value of x increases: (i) from TI to semimetal with increasing x value above 0.5 and (ii) from semimetal to TI when x approaches unity.
The analysis of changes in the Bi-p contributions in the bulk gap edges at the Г and Z points deserves special attention. It is evident from Fig. 6l, m that the inversion of the Bi-p contribution, occurring at some points of the intervals 0.5 < x < 0.75 at Г point and 0.75 < x < 1 at the Z point, is accompanied by a general decrease in the Bi-p contribution at the Г point and its increase at the Z point. Since Bi is the main source of SOC in the system, it can be assumed that the above-mentioned TPTs should be related to the modulation of the effective SOC strength at the Г and Z points.
It should be noted that TPTs cannot be revealed for materials of the same topological class. Generally speaking, a band inversion may occur in different time-reversal invariant momentum (TRIM) points that lead to distinct topological classes33. To clarify the topological properties of GeBi2Te4 and MnBi2Te4, we used a Wilson Loop technique and calculated Wannier Charge Centers34. The results show different topological classes, namely (1;000) for AFM MnBi2Te4 and (1;001) for GeBi2Te4 (Supplementary Note 1, Supplementary Fig. 7, 8). This is in accordance with previous calculations35. This explains the observed behavior in terms of topological band theory. In detail, upon x variation, two situations are possible: (i) transition to trivial state (0;000), (ii) transition to the weak TI state (0;001). Scenario (i) is realized when the gap is first closed in Г point and further in Z point. Contrary, case (ii) is implemented when the gap is closed in Z point first. Our KKR calculations fit better to the case (i). Unfortunately, due to data sensitivity to tiny variations of structural parameters, this conclusion is not rigorous.
Next, we discuss the nature of the gapless state of GexMn1−xBi2Te4. For this, let us now consider in detail the band structure of the system with x = 0.42 when GexMn1−xBi2Te4 becomes a semimetal. Our measurements of magnetic properties discussed above indicate that this crystal undergoes a transition from the PM to the AFM state at 13 K. Figure 7a shows a set of high-resolution temperature-dependent ARPES spectra that indicate the opening of a tiny bandgap around the Г point below Néel temperature, while at 25 K no traces of the gap are present in the data. This is clearly visible in the energy distribution curves (EDCs) in Fig. 7c, which clearly demonstrate the magnetic nature of the gap. The gap appears to be M-shaped and its width is about 20 meV. It is essential to note that the momentum distribution curves (MDCs) of the VB given in Fig. 7b cannot be described with a single band, but they show a multi-component structure that can be approximated by at least two bands (red and blue lines) differing in energy by ~30 meV. Such splitting is also observed for the CB, although this is not illustrated. We did not find any significant difference between the splitting values in the PM and AFM phases; therefore, it is not related to magnetism. The reason of such multi-component structure is related to the fact that the kz component of (quasi)momentum is not conserved in photoemission, therefore the ARPES data contain contributions from the electronic states with all possible kz, although with different intensities. Of course, the most intense contribution is expected from the kz value defined by Eq. 1. Nevertheless, the superposition of different kz leads to an apparent splitting with its value nearly equal to the amplitude of the kz dispersion.

a ARPES maps along the M–Г–M direction taken at hν = 12 eV. b MDCs of the VB along the dashed lines in the panel (a). The fine structure of the experimental MDSs can be approximated by at least two bands (red and blue curves). c Comparison of EDCs in the Г point at different temperatures. The bandgap in EDC taken at 1.4 K is marked by red spots and black lines.
A thorough analysis of the ARPES spectra taken at various photon energies and temperatures allowed us to propose the electronic structure of the semimetal phase at x = 0.42 schematically shown in Fig. 8, where the bands are presented along the in-plane direction at high-symmetry points and out-of-plane direction as a function of kz. As we already noted, in the PM phase, the VB and CB disperse synchronously and form a nearly zero indirect gap, whereas the local direct gap Δ0 is present at any kz, and its width is about 40 meV. Upon transition to the AFM phase the lattice period in the c (or z) direction gets doubled due to opposite directions of magnetic moments in neighbor SLs. This leads to shrinkage of the BZ and folding of bands. As a result, the bands originally located in the Z point are translated to the Г point, where the CB and VB may have touched each other. However, instead of touching, the bands demonstrate avoided-crossing behavior, and the 20 meV gap marked as Δ1 opens between them. The material, thus, becomes a semiconductor in the AFM phase. At the same time, the in-plane dispersion of the VB in the Г point gains an M-like shape. This seems to be a result of hybridization between the VB and CB states. Hybridization is evident from the photoemission intensity distribution. In particular, one can see that the VB is more intense than the CB (at hν = 12 eV), and the high intensity of the VB spreads slightly above the gap Δ1. This is a clear indication of the admixture of the orbital character of the VB to the bottom of the CB. In our scheme, we also admitted possible splitting of the VB and CB in the Z2SL points at the boundary of the AFM BZ (see Fig. 6); these gaps are marked as Δ2 and Δ3, respectively. The splitting is well seen in the AFM phase of MnBi2Te4 at the photon energy of 6.3 eV25,32. Since it has a magnetic nature, it is expected to be notably smaller in the GexMn1−xBi2Te4 crystals with large x. This is a probable reason why we do not resolve Δ2 and Δ3 in our ARPES data.

ARPES map (a). Scheme of the bulk bands in case of AFM (b) and PM (c) phase. Three exchange gaps are marked by Δ1–3.
The temperature behavior of the experimentally observed Δ1 gap size is illustrated in Supplementary Fig. 10. Generally, it obeys a power law Eg(T) = Eg0 ∙ (1-T/TN)β. According to the Ising model, the β value is expected to be β = 1/836 and β ≈ 0.337 in the case of 2D and 3D systems, respectively. In our case β = 1/8 corresponds better to the experimental observations. Unfortunately, the experimental statistics are insufficient to make any solid conclusions on this matter.
Taking into account our understanding of the bulk band structure of GexMn1−xBi2Te4 with x up to 0.42, the next question arises as to whether the TSS appears in this system. To clarify this point, we explored the systems with x = 0.16 and 0.42 with ARPES at hν = 6.3 eV, which is known to be suitable for efficient observation of TSS in MnBi2Te4. The data are displayed in Fig. 9. One can see that for x = 0.16, a quite intense TSS is present. The temperature of 15 K reached in this experiment is lower than the Néel temperature of about 20 K for this sample, but we did not observe clear traces of a magnetic gap opening in the TSS upon changing the temperature between 30 K and 15 K. For the sample with x = 0.42 we clearly see again that these bands have a multicomponent structure due to integration over kz-dimension. We also see where the bands overlay forming a diamond-shaped region from 0.24 eV to 0.19 eV (best seen in second derivative). As we have shown in Fig. 8, the VB and CB overlay at 0.20 ± 0.01 eV indicate that some extra states contribute to the higher BE part of the diamond-shaped region. Since the direct gap of at least Δ0 = 40 meV is present in the sample in the PM phase, it is natural to suppose that the extra contribution is due to the presence of TSS within the gap. Although the Rashba-type SS is clearly visible, the signal from TSS is weak in the system with x = 0.42. We suppose the following reason for this. As the fundamental gap shrinks upon the increase of Ge content, the spatial localization of the TSS shifts more and more inside the bulk. When the gap becomes as small as several dozens of meV, the TSS becomes localized in a rather extended region below the surface, so it becomes almost indistinguishable from the bulk states.

ARPES maps (a, b) were obtained with laser radiation of 6.3 eV and corresponding second derivatives of the PE intensity (c, d). Spectral features SS, TSS, VB, and CB are marked by black arrows.
Summing up, we have revealed and explained the general phenomenon of TPTs, also observed in similar systems, where Mn is substituted with group XIV elements, Sn38 and Pb39. In these systems, a decrease of the bulk gap down to zero was observed in some ranges of the x values. The authors of these works also suggested possible TPTs in these systems. In contrast to Sn and Pb, we consider an ideal model system in which the effect of geometry and atomic number on SOC is switched off, which enables the tracing of delicate SOC variation due to the change of the Bi 6p orbital contribution in the electronic bands near the fundamental gap. This change is caused indirectly by the substitution of the Mn 4s contribution with the Ge 4p contribution to chemical bonds.
Conclusions
We studied solid solutions between two Z2 TIs, magnetic MnBi2Te4 (Z2 invariants are 1;000) and nonmagnetic GeBi2Te4 (Z2 invariants are 1;001), which differ essentially neither in structural parameters nor in atomic core charges affecting SOC strength of the valent electrons. We have considered an ideal model system where geometry and atomic number effects are switched off, that enables us to trace chemical effects solely.
We observed a linear x-dependence of magnetic properties (Néel temperature), which points to composition-independent pairwise exchange interactions. Moreover, the easy axis does not depend on composition, and magnetic dilution affects neither the choice of magnetically active sublattices nor the pairwise exchange interaction of ions. MFM confirms antiferromagnetic order between ferromagnetically ordered SLs. The behavior of GexMn1−xBi2Te4 enables precise control and fine-tuning of magnetic properties of topological insulators by x variation.
In the electronic band structure probed by ARPES, and studied theoretically, the bulk band gap gradually decreases to zero for x variation from 0 to 0.4. For x > 0.6, a gap opens, and it increases while x approaches unity. This pinpoints two TPTs from a topologically nontrivial phase to a semimetal state that is possibly a Dirac semimetal state and back to the TI phase. The TPTs are driven solely by the variation of orbital contributions to chemical bonds (3p for Ge and 4s for Mn) and, in turn, indirectly to the states near the fundamental gap. Since Bi is the main source of SOC in the system, it can be assumed that the above-mentioned TPTs should be related to the modulation of the effective SOC strength. Using the Bi 6p contribution to the states near the fundamental gap, we traced effective SOC variation that causes band inversions at the Г and Z points at two different values of x, thus producing two TPTs upon x variation.
The gapless state observed at x = 0.42 closely resembles a Dirac semimetal state. It shows a magnetic gap below Néel temperature, which is clearly visible in raw ARPES data. We propose the detailed electronic structure of the semimetal phase. As the fundamental gap shrinks upon the increase of Ge content, the spatial localization of the TSS shifts more and more inside the bulk. When the gap becomes as small as several dozens of meV, the TSS becomes localized in a rather extended region below the surface, so it becomes almost indistinguishable from the bulk states.
Methods
Synthesis and characterization
All crystals were grown from the melt using the modified Bridgeman method in the four-zone vertical furnace from the mixture of binary compounds Bi2Te3 and Ge1−xMnxTe. Raw materials were synthesized by prolonged annealing of stochiometric mixtures of the elements. The obtained crystals were characterized by XRF and additionally by powder XRD. The XRF measurements were performed using a Bruker Mistral-M1 micro-focused system equipped with an XFlash 30 mm2 detector, the concentrations were determined using XSpect software by determining the area of individual peaks using the external standard model. In total, 4–7 measurements were taken for each crystal. For XRD measurements, the samples were powdered. XRD was performed using a Rigaku Smartlab SE diffractometer equipped with a PSD Pixel3D detector in the 2θ range of 10–80° with a step of 0.02° at room temperature. The data obtained were treated within the Jana 2006 software package.
Magnetic properties
Magnetic susceptibility of the GexMn1−xBi2Te4 crystals was studied in the temperature range of 1.77–300 K using a Quantum Design MPMS-XL and Quantum Design MPMS SQUID VSM magnetometer. Measurements were performed under magnetic fields applied along or transverse to the crystallographic c-axis. To determine the effective magnetic moment meff, the paramagnetic component of the magnetic susceptibility, χp(T), was analyzed using the Curie-Weiss expression cp(T) = Nameff2/3kB(T–θ). A part of the measurements was performed at the Center of Diagnostic of Functional Materials for Medicine, Pharmacology, and Nanoelectronics of the Research Park of St. Petersburg State University.
Magnetic force microscopy measurements were performed using AttoDry 1000 MFM system at the temperature T = 4.1 K using Bruker MESP-LN-V2 probes covered by CoCr coating.
Photoemission spectroscopy
ARPES data were recorded using several facilities. Laboratory-based measurements were made using a SPECS GmbH ProvenX-ARPES system located in ISP SB RAS equipped with ASTRAIOS 190 electron energy analyzer with 2D-CMOS electron detector and a non-monochromated He Iα light source with hν = 21.22 eV. Photon energy dependence of normal-emission PE spectra was measured at BaDElPh40 beamline of the Elettra synchrotron in Trieste. All measurements were carried out at T = 12 K using p-polarized photons. Temperature-dependent ARPES measurements were carried out at UE112-PGM2a undulator using the ARPES 13 endstation of the BESSY II synchrotron in Berlin. The temperature dependence was measured by sweeping down from 25 K to 1.4 K. We did not notice any aging effect during the time of the measurement at a single temperature (~40 min). We checked also the reproducibility of the effect by cycling the temperature. Laser-based data was collected using a laser ARPES endstation equipped with VG Scienta SES R4000 hemispherical analyzer at Hiroshima Synchrotron Radiation Center (HiSOR). Spin-resolved ARPES measurements were performed with linearly polarized light at the U125-2-PGM RGBL Undulator using the RGBL-2 endstation. The endstation was equipped with a hemispherical Scienta R4000 analyzer. The RGBL-2 endstation is equipped with a combined detector that comprises a 2D channel plate for ARPES and a Mott-type spin-detector41, which was operated at 25 kV. The energy and angular resolutions of spin-resolved ARPES measurements were 45 meV and 0.75°, respectively. XPS and NEXAFS measurements were performed at the Russian–German dipole beamline (RGBL) of the synchrotron light source BESSY-II at Helmholtz-Zentrum Berlin. The photoemission spectra were acquired with a hemispherical SPECS Phoibos 150 electron energy analyzer at normal emission geometry. NEXAFS spectra were recorded in the total-electron yield mode. The photon energy was calibrated using second-order reflection. All core-level spectra were fitted by Gaussian/Lorentzian convolution functions with simultaneous optimization of the background parameters using the UNIFIT 2014 software42. The background was modeled using a combination of Shirley- and Tougaard-type shapes. In all the cases, prior to measurements, the samples were cleaved in situ under ultrahigh vacuum at pressures below 1 × 10−9 Pa.
DFT calculations
The electronic structure supercell calculations with impurities were performed using the OpenMX code, which provides a fully relativistic DFT implementation with localized pseudoatomic orbitals43,44,45 and norm-conserving pseudopotentials46. The exchange-correlation energy in the PBE version of generalized gradient approximation (GGA) was employed47. Structural parameters from ref. 2 with modified vdW gap between SLs (2.89 Å) were used for MnBi2Te4 unit cell. For impurity calculations, 2 × 2 supercells of MnBi2Te4, which provide 4 non-equivalent positions of Mn atoms in each layer, were used. The Ge concentrations of 25, 50, and 75% were obtained by replacing one, two, and three Mn atoms with Ge atoms, respectively. The accuracy of the real-space numerical integration was specified by the cutoff energy of 450 Ry, and the total energy convergence criterion was 10−6 eV. The k-mesh for Brillouin zones were specified as follows: 5 × 5 × 5 mesh for pristine MnBi2Te4 slab and 3 × 3 × 1 mesh for 2 × 2 supercells. The basis functions were taken as Bi8.0—s3p2d2f1, Te7.0—s3p2d2f1, Mn6.0—s3p2d1, Ge7.0—s3p2d2 (the pseudopotential cutoff radius is followed by a basis set specification). The Mn 3d states were treated within the DFT + U approach48 within the Dudarev scheme49 where U parameter equals 5.4 eV2.
The band structure of (Mn1−xGex)Bi2Te4 with fully disordered impurities from first-principles calculations was obtained utilizing the spin-polarized relativistic Korringa–Kohn–Rostoker (SPR-KKR) code, version 7.750. The GGA in the PBE parametrization was used47 with U correction and the atomic-sphere approximation (ASA) in the fully relativistic approach. The coherent potential approximation (CPA)51 was used to simulate chemical disorder. We used the basic functions up to l = 3, a regular k-point grid with 250 points and 30 energy points.
For the topological classification of GeBi2Te4 and MnBi2Te4, the electronic structure was calculated using Quantum Espresso code52,53. The tight-binding Hamiltonian was obtained using Wannier90 code54. Calculations of Z2 invariants were done using WannierTools package55.
Data availability
All data related to this paper are available from the corresponding author upon reasonable request.
Code availability
The code used in this study is available from the corresponding author upon reasonable request.
References
Chang, C.-Z. et al. Experimental observation of the quantum anomalous Hall effect in a magnetic topological insulator. Science 340, 167–170 (2013).
Otrokov, M. M. et al. Prediction and observation of an antiferromagnetic topological insulator. Nature 576, 416–422 (2019).
Trang, C. X. et al. Crossover from 2D ferromagnetic insulator to wide band gap quantum anomalous Hall insulator in ultrathin MnBi2Te4. ACS Nano 15, 13444–13452 (2021).
Zeugner, A. et al. Chemical aspects of the candidate antiferromagnetic topological insulator MnBi2Te4. Chem. Mater. 31, 2795–2806 (2019).
Okamoto, K. et al. Observation of a highly spin-polarized topological surface state in GeBi2Te4. Phys. Rev. B 86, 195304 (2012).
Fragkos, S. et al. Topological surface states in epitaxial (SnBi2Te4)n(Bi2Te3)m natural van der Waals superlattices. Phys. Rev. Mater. 5, 014203 (2021).
Kuroda, K. et al. Experimental verification of PbBi2Te4 as a 3D topological insulator. Phys. Rev. Lett. 108, 206803 (2011).
Johnston, W. D. & Sestrich, D. E. The MnTe–GeTe phase diagram. J. Inorg. Nucl. Chem. 19, 229–236 (1961).
Zheng, Z. et al. Rhombohedral to cubic conversion of GeTe via MnTe alloying leads to ultralow thermal conductivity, electronic band convergence, and high thermoelectric performance. J. Am. Chem. Soc. 140, 2673–2686 (2018).
Chen, W. Q., Teo, K. L., Jalil, M. B. A. & Liew, T. Compositional dependencies of ferromagnetic Ge1−xMnxTe grown by solid-source molecular-beam epitaxy. J. Appl Phys. 99, 08D515 (2006).
Hassan, M. et al. Molecular beam epitaxy of single phase GeMnTe with high ferromagnetic transition temperature. J. Cryst. Growth 323, 363–367 (2011).
Yan, B. & Felser, C. Topological materials: Weyl semimetals. Annu. Rev. Condens. Matter Phys. 8, 337–354 (2016).
Liang, T. et al. Ultrahigh mobility and giant magnetoresistance in the Dirac semimetal Cd3As2. Nat. Mater. 14, 280–284 (2015).
Son, D. T. & Spivak, B. Z. Chiral anomaly and classical negative magnetoresistance of Weyl metals. Phys. Rev. B 88, 104412 (2013).
Nielsen, H. B. & Ninomiya, M. The Adler-Bell-Jackiw anomaly and Weyl fermions in a crystal. Phys. Lett. B 130, 389–396 (1983).
Xu, S.-Y. et al. Discovery of a Weyl fermion semimetal and topological Fermi arcs. Science 349, 613–617 (2015).
Li, P. et al. Giant room temperature anomalous Hall effect and tunable topology in a ferromagnetic topological semimetal Co2MnAl. Nat. Commun. 11, 3476 (2020).
Zhao, B. et al. Observation of charge to spin conversion in Weyl semimetal WTe2 at room temperature. Phys. Rev. Res 2, 013286 (2020).
Baltz, V. et al. Antiferromagnetic spintronics. Rev. Mod. Phys. 90, 015005 (2018).
Armitage, N. P., Mele, E. J. & Vishwanath, A. Weyl and Dirac semimetals in three-dimensional solids. Rev. Mod. Phys. 90, 015001 (2018).
Lundgren, R., Laurell, P. & Fiete, G. A. Thermoelectric properties of Weyl and Dirac semimetals. Phys. Rev. B 90, 165115 (2014).
Fukuma, Y. et al. Local environment of Mn atoms in IV-VI ferromagnetic semiconductor Ge1−xMnxTe. J. Appl. Phys. 99, 08D510 (2006).
Sánchez-Barriga, J. et al. Nonmagnetic band gap at the Dirac point of the magnetic topological insulator (Bi1−xMnx)2Se3. Nat. Commun. 7, 10559 (2016).
Chen, Y. J. et al. Topological electronic structure and its temperature evolution in antiferromagnetic topological insulator MnBi2Te4. Phys. Rev. X 9, 041040 (2019).
Hao, Y.-J. et al. Gapless surface Dirac cone in antiferromagnetic topological insulator MnBi2Te4. Phys. Rev. X 9, 041038 (2019).
Konstantinov, P. P., Shelimova, L. E., Avilov, E. S., Kretova, M. A. & Fleurial, J.-P. Transport phenomena in mixed layered tetradymite-like compounds in the GeTe–Bi2Te3 system. J. Solid State Chem. 146, 305–312 (1999).
Liang, A. et al. Approaching a minimal topological electronic structure in antiferromagnetic topological insulator MnBi2Te4 via surface modification. Nano Lett. 22, 4307–4314 (2022).
Estyunin, D. A. et al. Signatures of temperature driven antiferromagnetic transition in the electronic structure of topological insulator MnBi2Te4. Apl. Mater. 8, 021105 (2020).
Shikin, A. M. et al. Routes for the topological surface state energy gap modulation in antiferromagnetic MnBi2Te4. Phys. B Condens Matter 649, 414443 (2023).
Arita, M. et al. Angle resolved photoemission study of GeBi2Te4. Proc 12th Asia Pac. Phys. Conf. Appc.12 (2014) https://doi.org/10.7566/jpscp.1.012017.
Karpinsky, O. G., Shelimova, L. E., Kretova, M. A. & Fleurial, J.-P. X-ray study of the nGeTe·mBi2Te3 mixed layered tetradymite-like compounds. J. Alloy Compd. 265, 170–175 (1998).
Shikin, A. M. et al. Sample-dependent Dirac-point gap in MnBi2Te4 and its response to applied surface charge: a combined photoemission and ab initio study. Phys. Rev. B 104, 115168 (2021).
Fu, L., Kane, C. L. & Mele, E. J. Topological insulators in three dimensions. Phys. Rev. Lett. 98, 106803 (2007).
Soluyanov, A. A. & Vanderbilt, D. Computing topological invariants without inversion symmetry. Phys. Rev. B 83, 235401 (2011).
Neupane, M. et al. Topological surface states and Dirac point tuning in ternary topological insulators. Phys. Rev. B 85, 235406 (2012).
Kutlu, B. Critical exponents of the two-dimensional Ising model with next-nearest-neighbor and four-spin interactions on the Creutz cellular automaton. Phys. Stat. Mech. Appl. 243, 199–212 (1997).
Mailhot, A. Finite-size scaling of the frustrated Heisenberg model on a hexagonal lattice. Phys. Rev. B 50, 6854–6859 (1994).
Tarasov, A. V. et al. Topological phase transitions driven by Sn doping in (Mn1−xSnx)Bi2Te4. Symmetry 15, 469 (2023).
Qian, T. et al. Magnetic dilution effect and topological phase transitions in (Mn1−xPbx)Bi2Te4. Phys. Rev. B 106, 045121 (2022).
Petaccia, L. et al. BaD ElPh: A 4m normal-incidence monochromator beamline at Elettra. Nucl. Instrum. Methods Phys. Res. Sect. Accel. Spectrometers Detect Assoc. Equip. 606, 780–784 (2009).
Burnett, G. C., Monroe, T. J. & Dunning, F. B. High‐efficiency retarding‐potential Mott polarization analyzer. Rev. Sci. Instrum. 65, 1893–1896 (1994).
Unifit 2014. https://home.uni-leipzig.de/unifit/new.htm.
Ozaki, T. Variationally optimized atomic orbitals for large-scale electronic structures. Phys. Rev. B 67, 155108 (2003).
Ozaki, T. & Kino, H. Numerical atomic basis orbitals from H to Kr. Phys. Rev. B 69, 195113 (2003).
Ozaki, T. & Kino, H. Efficient projector expansion for the ab initio LCAO method. Phys. Rev. B 72, 045121 (2005).
Troullier, N. & Martins, J. L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 43, 1993–2006 (1991).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Han, M. J., Ozaki, T. & Yu, J. O(N) LDA + U electronic structure calculation method based on the nonorthogonal pseudoatomic orbital basis. Phys. Rev. B 73, 045110 (2006).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Ebert, H., Ködderitzsch, D. & Minár, J. Calculating condensed matter properties using the KKR-Green’s function method—recent developments and applications. Rep. Prog. Phys. 74, 096501 (2011).
Liechtenstein, A. I., Katsnelson, M. I. & Gubanov, V. A. Exchange interactions and spin-wave stiffness in ferromagnetic metals. J. Phys. F Met. Phys. 14, L125–L128 (1984).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens Matter 21, 395502 (2009).
Giannozzi, P. et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys.: Condens. Matter 29, 465901 (2017).
Pizzi, G. et al. Wannier90 as a community code: new features and applications. J. Phys.: Condens. Matter 32, 165902 (2020).
Wu, Q., Zhang, S., Song, H.-F., Troyer, M. & Soluyanov, A. A. WannierTools: an open-source software package for novel topological materials. Comput. Phys. Commun. 224, 405–416 (2018).
Acknowledgements
The Magnetic Force Microscopy at 4 K was partially supported by the Ministry of Science and Higher Education of the Russian Federation (No. FSMG-2023-0014). A.S.F. thank Russian Science Foundation (grant No. 22-72-10074 https://rscf.ru/en/project/22-72-10074/) for the support of ARPES study and synthesis. A.V.T. thank Saint Petersburg State University (Grant No. 95442847) for the supporting the theorethical modeling within the KKR approach. D.Yu.U. and V.S.S. thank Russian Science Foundation, Grant No. 23-72-30004 (https://rscf.ru/project/23-72-30004/) for supporting the DFT study. We thank Helmholtz-Zentrum Berlin and Elettra Sincrotrone Trieste for granting access to the beamlines UE112-PGM2a (13), RGBL, and BaDElPh, respectively, and for the provision of beamtimes. The project that gave rise to these results received the support of a fellowship from “la Caixa” Foundation (ID 100010434). We appreciate Dr. Anna Makarova, Dr. Vera Neudachina, and Elena Ushakova for their participation in spectra measurements. Calculations were performed using the facilities of the Joint Supercomputer Center of the Russian Academy of Sciences.
Author information
Authors and Affiliations
Contributions
A.S.F.: Conceptualization, investigation, formal analysis, data curation, writing-original draft, writing—review & editing, visualization, supervision. D.Yu.U, A.V.T., K.A.B: conceptualization, investigation, formal analysis, data curation, writing-original draft, writing—review & editing, visualization. I.I.K.: investigation, data curation, writing—review & editing. A.V.F., V.S.S., A.I.S., A.N.L., V.A.G., O.E.T., G.D.S., L.P., O.J.C.: investigation, data curation. J.S.-B.: investigation, data curation, conceptualization. L.V.Y.: Conceptualization, writing-original draft, writing—review & editing, supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Physics thanks Tyler A. Cochran and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Frolov, A.S., Usachov, D.Y., Tarasov, A.V. et al. Magnetic Dirac semimetal state of (Mn,Ge)Bi2Te4. Commun Phys 7, 180 (2024). https://doi.org/10.1038/s42005-024-01675-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42005-024-01675-w
- Springer Nature Limited