
Abstract
To explore the general requirement of endothelial mTORC2 during embryonic and adolescent development, we knocked out the essential mTORC2 component Rictor in the mouse endothelium in the embryo, during adolescence and in endothelial cells in vitro. During embryonic development, Rictor knockout resulted in growth retardation and lethality around embryonic day 12. We detected reduced peripheral vascularization and delayed ossification of developing fingers, toes and vertebrae during this confined midgestational period. Rictor knockout did not affect viability, weight gain and vascular development during further adolescence. However during this period, Rictor knockout prevented skin capillaries to gain larger and heterogeneously sized diameters and remodeling into tortuous vessels in response to FGF2. Rictor knockout strongly reduced extensive FGF2-induced neovascularization and prevented hemorrhage in FGF2-loaded matrigel plugs. Rictor knockout also disabled the formation of capillary-like networks by FGF2-stimulated mouse aortic endothelial cells in vitro. Low RICTOR expression was detected in quiescent, confluent mouse aortic endothelial cells, whereas high doses of FGF2 induced high RICTOR expression that was associated with strong mTORC2-specific protein kinase Cα and AKT phosphorylation. We demonstrate that the endothelial FGF-RICTOR axis is not required during endothelial quiescence, but crucial for midgestational development and sustained and extensive neovascularization in the adult.
Similar content being viewed by others


Introduction
Controlled angiogenesis is required during early development to generate the peripheral vasculature and in the adult to regenerate damaged tissue and the endometrial lining. In contrast, uncontrolled angiogenesis, which is characterized by an unorganized, tortuous and hyper-permeable vascular network, is induced in many pathological processes, including tumor growth, metastasis, diabetic retinopathy and arthritis1,2. Angiogenic cascades are initiated by potent angiogenic molecules, such as fibroblast growth factor (FGF) or vascular endothelial growth factor (VEGF). FGF and VEGF both activate the protein kinase mammalian target of rapamycin (mTOR)3,4 via phosphatidylinositide 3-kinase (PI3K) activation. Pharmacological inhibition of mTOR-linked signaling has been shown to reduce tumor angiogenesis and tumor growth in various experimental models. The mTOR inhibitor, rapamycin, suppresses tumor angiogenesis in vivo by downregulating VEGF5,6. Furthermore, dual PI3K and mTOR inhibitors block VEGF-induced neovascularization in mice7,8,9. Similarly, dual mTOR inhibitors have been shown to considerably reduce angiogenesis and regrowth compared to rapamycin alone10.
Mammalian TOR acts as the core protein kinase in two different multi-protein complexes: mTOR complex (mTORC) 1 and 2. In most tissues, rapamycin is largely selective for mTORC111.
The complex of interest in this study is mTORC2, which contains the essential regulatory proteins RICTOR, mSIN1 and mLST8. mTORC2 integrates signals from growth factors to regulate cell survival or cytoskeleton organization. In addition, mTORC2 phosphorylates AGC kinase family members, such as AKT and protein kinase Cα (PKCα)11 and is implicated in the epithelial-mesenchymal transition (EMT)12,13. Embryos lacking Rictor or Mlst8 in the whole body are growth retarded and die at around midgestation14,15. We have previously shown that hypoxia, a main stimulus for angiogenesis, induces transient mTORC1 activity, whereas mTORC2-induced AKT activation is sustained and critical for endothelial proliferation16. This suggested a specific function of mTORC2 in angiogenesis in vivo. Recently, distinct mTORC2 signaling pathways have been reported to regulate endothelial cell proliferation and vascular assembly in response to VEGF17.
High doses of rapamycin or its prolonged delivery can also block mTORC2 in endothelial cells18. Thus, rapamycin-based inhibitors cannot differentiate between the functions of mTORC1 and mTORC2 and specific mTORC2 inhibitors are not available. Here we therefore deleted Rictor in the endothelium to study the general requirement of endothelial mTORC2 during embryonic and adolescent development. Our second main aim was to elucidate whether endothelial RICTOR participates in vascular changes upon wounding and extensive angiogenic stimulation in the existing capillary bed and during de novo angiogenesis.
Results
Loss of endothelial homozygous Rictor results in embryonic lethality around embryonic day (E) 11.5–12.5
Whole-body mTORC2 knockout mice are embryonically lethal. Guertin and colleagues suggested vascular defects as a potential reason for early embryonic death14,15. We further investigated the loss of Rictor in endothelial cells during embryogenesis by using a constitutive VE-Cadherin promoter-driven Cre and LacZ reporter containing19Rictor knockout. The analysis of 101 pups revealed two homozygous Rictor knockout mice, indicating predominant embryonic lethality. Heterozygous Rictor knockout and wildtype mice were born at expected Mendelian ratios (Fig. 1A). Interestingly, the two surviving RictorΔec mice were females, fertile and phenotypically normal. When further used for breeding, these two RictorΔec mice gave birth only to heterozygous and wildtype Rictor pups. We then analyzed 43 embryos received after terminated pregnancy on day E10.5. 11 out of these embryos were genotyped as homozygous knockouts. LacZ reporter-positive RictorΔec and wildtype embryos displayed endothelial cell-specific Cre recombination, as visualized by β-galactosidase staining in intersegmental vessels, intracranial arteries and the dorsal aorta. RictorΔec homozygous embryos generally appeared normal and were viable at this time point. LacZ activity in the dorsal longitudinal anastomotic vessel was clearly visible in wildtype embryos but was less prominent in 7 out of 11 RictorΔec embryos, indicating reduced or delayed angiogenesis into the periphery from intersegmental vessels (Fig. 1B, for additional pictures see supplementary Fig. S1). We found only one noticeable distinct vascular feature in 2 out of 11 RictorΔec embryos compared to controls: Vascular remodeling around the vitelline artery that usually occurs at this or earlier timepoints20 was characterized by the presence of numerous, thin parallel anastomosing vessels which are not observed in wild type mouse embryos (arrows in supplementary Fig. S1, lower panels). Thus, erythropoiesis and vasculogenesis of the primitive vascular plexus with further remodeling of arteries and veins, which is completed on E10.521, was not modulated by endothelial mTORC2. To precisely determine the time point of lethality, overlapping tamoxifen-injection schemes in pregnant mice with homozygous loxed Rictor gene and inducible VE-Cadherin CreERT2 recombinase were used (Fig. 2A). Injecting tamoxifen three times in pregnant mice, starting at E7.5, produced marked and significant reductions in litter size (Fig. 2B). However, injecting tamoxifen twice at E7.5 did not result in any differences in litter size (Fig. 2B). Rictor knockout by 60% is achieved with two injections of tamoxifen, whereas nearly homozygous (92%) knockout is achieved with three injections of tamoxifen every second day22. Thus, with three injections starting on E7.5, knockout of Rictor was likely to be maximal starting from E11.5–E12.5. On E17.5, one third of the embryos were growth retarded and the remaining embryos were absorbed (Fig. 2C). In addition, more than 90% of analyzed embryos were growth retarded after tamoxifen injections began on E6.5 and E8.5 (Fig. 2C). Interestingly, tamoxifen injections that began on E12.5 and E14.5 had no influence on viability and growth (Fig. 2C).

Constitutive homozygous endothelial Rictor knockout during embryonic development is generally lethal.
(A) Rictorflox/flox females were mated with Rictorflox/−; VE-Cadherin-Cre+/−; LacZ reporter+/+ males to generate homozygous deletion of Rictor in the endothelium. Litter genotypes were determined by qPCR and are displayed as total distribution and average number of pups per genotype (ntotal pups = 101, ****P < 0.0001, ***P < 0.001, 1-way ANOVA with Bonferroni multiple comparison) (B). The abovementioned breeding scheme was used to isolate embryonic day (E) 10.5 wildtype and endothelial Rictor knockout embryos. Representative β-galactosidase staining (blue) of E10.5 embryos shows the active sites of VE-Cadherin-Cre recombination. Arrows on the right indicate reduced peripheral LacZ staining in Rictor knockout embryos (n = 7 of 11).

Lethality and growth retardation of induced endothelial Rictor knockout mice peaks around E12.
(A) Tamoxifen (Tx) injection scheme: Three doses of Tx were administered to pregnant females every 48 hours, beginning on E6.5, E7.5, E8.5, E10.5, E12.5 and E14.5. Pregnant females were sacrificed and embryos were harvested on E17.5 for further histological analysis. Grey area symbolically depicts stepwise Rictor knockout. Control females were injected with corn oil. (B)RictoriΔec mice exhibit decreased litter size. Statistical analysis of the litter size of individual breedings (nEday) after three and two Tx injections at different time points (nE6.5 = 3, nE7.5 = 3, nE8.5 = 4, nE10.5 = 3, nE12.4 = 4, nE14.5 = 4,) compared to controls (n = 7). *P < 0.05, Student’s t-test. (C) Statistical analysis of normal, growth retarded and absorbed embryos per breeding after Tx-injection at different time points. Total litter size for breeding at each time point was set to 100%, nbreedings = 3. (D) Statistical analysis of the length of surviving embryos per litter at different starting time points of Tx injections. n = 4 (embryos per time point, length was measured in both extremities), *P < 0.01 compared to controls. Student’s t-test. (E) Representative picture of a E17.5 RictoriΔec embryo that was induced by Tx on E8.5 in comparison to a wildtype embryo. (F) RictoriΔec embryos displayed distinct vascular deficits in the eye when Tx injections started on E7.5 and embryos harvested at E12.5 and stained with the vessel-specific antibody, endomucin. Scale bar = 100 μm. Arrows indicate angiogenic sprouts. (G)RictoriΔec embryos display a delay in ossification. Representative pictures of the upper limbs of embryos stained with alizarin red (bone) and alcian blue (cartilage). Arrows: ossification centers. Below, quantification with number of ossification centers in fingers upon knockout of Rictor at indicated starting time points of Tx injections. *P < 0.05, **P < 0.01, compared to E14.5; nembryo = 4 (number of centers was measured in both extremities and averaged for each embryo), Mann-Whitney Rank Sum Test.
Embryos that were injected with tamoxifen on E8.5 had a body length of approximately 14 mm, whereas embryos that were injected on E14.5 had a body length of 19.5 mm. Wildtype embryos at embryonic day 17.5 displayed a body length of 18–22 mm (Fig. 2D). Furthermore, growth-retarded embryos did not display wrinkled skin; instead, the skin was rather thin and subcutaneous veins were visible (Fig. 2E). To investigate whether endothelial-specific Rictor knockout causes a delay in vascularization, embryos received three injections of tamoxifen that started on E7.5, after which they were sacrificed at E12.5. The majority of endomucin-stained vascular plexi were present in both RictoriΔec and control embryos. Due to mortality at this time point, we detected only 5 knockout embryos out of three terminated pregnancies with induced knockout. We detected reduced sprouting angiogenesis in subdermal vessels in the ventral region and of RictoriΔec embryos (2 out of 5 RictoriΔec embryos) (Fig. 2F). Also, the temporary hyaloid vessels in the eye had formed incompletely (3 out of 5 RictoriΔec embryos) (Fig. 2F). In growth-retarded RictoriΔec mice that were that were first injected with tamoxifen on E.6.5 and E8.5, ossification centers in the fingers were strongly reduced or missing when analyzed on E17.5 as compared to embryos that were first injected on E14.5 (Fig. 2G, alizarin red (bone) and alcian blue (cartilage) stainings and quantification of ossification centers below). Embryos that were first injected on E14.5 displayed an ossification progress comparable to wildtype mice as demonstrated earlier by Gollner et al.23 Similarly, the progress of ossification in the vertebrae was clearly delayed in embryos that were first injected with tamoxifen on E8.5 and the long bones of the upper and lower limbs were significantly shorter in embryos that received their first tamoxifen injections on E6.5 and E8.5 compared to embryos that received their first tamoxifen injections on E14.5 (supplementary Fig. S2). CD31 staining from skin, brain, skeletal muscle, lung and colon sections of growth-retarded RictoriΔec mice on the other hand were morphologically indistinguishable from sections of wildtype control mice (supplementary Fig. S3).
In summary, endothelial Rictor knockout resulted in lethality which peaked around E12 and growth retardation close to this time point in midgestation. Growth retardation was accompanied by delayed bone ossification in fingers, toes and vertebrae. Rictor knockout delayed peripheral angiogenesis, but did not generally affect vascular plexus formation.
Endothelial-specific Rictor knockout has no obvious effect on viability and weight gain during adolescence into adulthood
We proceeded to analyze the general requirement of endothelial mTORC2/RICTOR using the inducible VE-Cadherin-driven CreERT2 variant (RictoriΔec) during adolescence from the age of 4 weeks (at the time of tamoxifen-induced Rictor knockout) to adulthood, up to an age of 28 weeks. No statistically significant differences in weight gain were observed between genotypes and genders (Fig. 3A). Furthermore, all groups displayed normal health and viability. At the end of the weight study, the aorta from male control and RictoriΔec mice were removed, the endothelium was scraped and Rictor mRNA levels were determined by quantitative polymerase chain reaction (qPCR) to show stable, efficient, and significant knockdown of Rictor (Fig. 3B). Adolescent double transgenic mice (Cre+/+; Rictorlox/lox) also displayed specific CRE expression in capillaries from the mouse dermal skin muscle, as shown by estrogen receptor-specific staining in tissues that were used for intravital experiments (Fig. 3C). Although VE-Cadherin promoter-driven Cre recombination may also affect hematopoietic development22, we found similar hematological profiles in 8-week-old control and RictoriΔec mice. These profiles were comparable to those of healthy C57/Bl6 mice (supplemental Fig. S4).

Rictor knockout does not affect weight gain and viability in adolescent mice.
(A) Body weights were followed in male and female mice over a period of 27 weeks after induction of knockout or control on week 4. n = 10 per genotype and gender, n.s., 2-way ANOVA with group-wise comparison. All mice displayed normal health, behavior, and viability. (B) At the end of the experiment, RNA was extracted from endothelial layer for quantitative polymerase chain reaction (PCR) analysis to test for efficient excision of Rictor (n = 3/2, *P < 0.05. 2-tailed T-test) and qualitatively for purity of endothelial tissue (endothelial marker Pecam1, smooth muscle marker αSMA). Total aorta mRNA was used as comparative control. Representative immunostainings for estrogen receptor 2 (red fluorescence) in histological sections of the skinfold from 10-week-old RictoriΔec mice demonstrates specific expression of CreERT2 recombinase associated with capillaries.
These experiments demonstrate that the stable ablation of endothelial mTORC2 in adolescent mice, which persisted until late adulthood, did not cause obvious general health problems, as supported by normal hematological profiles, weight gain and viability.
Rictor knockout in mouse aortic endothelial cells (MAEC) differentially disables the formation of capillary-like endothelial networks
Our results so far suggested, that knockout of Rictor in endothelial cells has no obvious effects on the development during adolescence in mice and therefore may not affect basic physiologic parameters of the endothelial cell such as survival and homeostatic functions in the existing and developing vasculature. Before continuing investigations about the role of mTORC2 in activated endothelium in vivo, we used an in vitro assay to determine the angiogenic response of control and Rictor knockout mouse aortic endothelial cells to the two major angiogenic molecules FGF and VEGF (Characteristic endothelial markers of these cells are shown in supplementary Fig. S5). After plating on a basement membrane matrix gel, endothelial cells build capillary-like tubes with a lumen within a short time. Cells initially attach to the matrix and then migrate towards each other, after which they align and form tubes24. We found that control MAEC formed connected master segments in the presence of the diluent (1% fetal calf serum [FCS]), FGF2 and VEGFA (Fig. 4A). Rictor knockout was induced by CRE recombinase expression after adenoviral transfection (Fig. 4, lower left panel). We found, that the ability of MAEC with Rictor knockout (Rictor ko MAEC) to build stable contacts and connecting tubes was substantially disabled: Knockout of Rictor prevented MAEC from forming capillary-like tubes in unstimulated conditions. Upon FGF2 stimulation, Rictor ko MAEC arranged into star-like shapes, with sprouts extending from cell clusters, but did not establish contacts to other cell clusters and formed significantly less master segments compared to control (Fig. 4). In contrast to the study by Wang et al.17, VEGFA stimulation partially rescued endothelial network formation in Rictor ko MAEC. Rictor ko MAEC were able to form some substantial networks and organized tubes, albeit in in numbers that were small and similar to those of control MAEC (Fig. 4).

Rictor knockout in mouse aortic endothelial cells (MAEC) decreases endothelial network formation.
(A) Representative micrographs show endothelial network formation after 18 hours of seeding. Quantification (total number of master segments that connect to at least two other segments) of endothelial tube formation from three experiments is shown below. (Bars; mean and SEM, nexp = 3, *P < 0.05 versus control. Paired T-Test). FGF-treated Rictor ko MAEC typically formed star-shaped centers with omni-directional sprouting and no connection to neighboring centers. Efficient Rictor knockout displayed by RICTOR and CRE protein expression from control (AdCre–) and Rictor ko (AdCre+) MAEC (n = 3) is displayed on the lower right.
As we found that the angiogenic response to FGF2 was significantly more affected by Rictor knockout than that to VEGF, we further focused on FGF2-mediated responses in endothelial cells and angiogenesis assays in vivo in this study.
FGF2 amplifies RICTOR protein and RICTOR-dependent phosphorylation of AKT on serine 473 and PKCa on serine 657
Interestingly, we found a consistently low expression of RICTOR in starved, unstimulated and confluent MAEC isolates, whereas FGF2 amplified RICTOR protein levels. Quantification demonstrated a significant increase in RICTOR protein at 5 ng/ml of FGF2 peaking at an 8-fold expression compared to diluent at 50 ng/ml of FGF2. (Fig. 5A, upper left panels). We assessed how FGF2-induced signaling is altered in mTORC2-deficient endothelial cells and focused on one of the main mTORC2 downstream targets, PKCα and AKT25. In control MAEC, FGF2 induced the dose-dependent phosphorylation of the hydrophobic motif (HM) of PKCα on serine 657 (PSer657PKCα). Interestingly, deletion of Rictor strongly decreased PKCα protein levels and accordingly blunted the phosphorylation of the HM of PKCα in response to FGF2, FCS and insulin (Fig. 5A). PKCα signaling was strongly disabled in Rictor ko MAEC, and this was most likely a result of total PKCα protein destabilization and degradation due to absent phosphorylation25. We then assessed the impact of Rictor deletion in MAEC on mTORC downstream target protein kinase AKT (also known as PKB). The phosphorylation of the activation loop (A-loop) on threonine 308 (PThr308AKT) by phosphoinositide-dependent kinase 1 (PDK1) and of the HM at serine 473 of AKT (PSer473AKT) by mTORC2 results in AKT activation11,26. In control MAEC, FGF2 induced a marked and dose-dependent PSer473AKT that peaked at 25 ng/ml of FGF2. Rictor knockout efficiently and significantly blunted FGF2-, FCS-, and insulin-induced AKT phosphorylation (Fig. 5A, quantification to the right). The disruption of mTORC2 has been shown to decrease PThr308AKT in some cancer cell lines11. In control MAEC, PThr308AKT was robustly induced by FGF2, FCS, and insulin. After Rictor knockout, we did not detect a significant reduction in PThr308AKT (Fig. 5A). These results are consistent with reports in cells or tissues that were derived from mice with genetic deletions of mTORC2 components. In these mice, A-loop phosphorylation was not disrupted in the absence of PSer473AKT14,25,27.

FGF2 amplifies RICTOR protein and Rictor-dependent phosphorylation of AKT on serine 473 and PKCa on serine 657.
(A) Western blots show RICTOR and downstream targets of mTORC2 after 15 min stimulation of control and Rictor ko MAEC with 5–50 ng/ml of FGF2, 10% FCS or 1 μg/ml insulin (Ins). Significant increase in RICTOR protein at 5 ng/ml of FGF2 stimultion peaking at an 8-fold expression at 50 ng/ml of FGF2 compared to diluent in 3 repeated experiments in a MAEC isolate (control = white bars, nexp = 3, *P < 0.05, **P < 0.001, 1-way ANOVA with Bonferroni multiple comparison, upper left panels). PKCα protein was nearly absent in Rictor ko MAEC compared to control (middle panels). Densitometric quantification shows dose-dependent AKT Ser473 phosphorylation in response to FGF2, FCS and Ins (right panels, nexp = 3, *P < 0.05, **P < 0.01, ***P < 0.001, repeated measures ANOVA). Lower middle blots show S6K1 phosphorylation on PThr389 compared to total S6K1 and phosphorylation of ERK1/2 on PThr202/Tyr204 compared to total ERK1/2 after Rictor knockout. (B) mRNA expression after 1–6 hours stimulation (25 ng/ml FGF2) of control and Rictor ko MAEC of VEGF receptor 1 (mFLT1, sFlt1), VEGF receptor 2 (Kdr), VEGFA (Vegfa), FGF receptor 1 (Fgfr1) and protein kinase Cα (PKCα) detected by quantitative real-time PCR (n = 3, ns knockout versus control, repeated measures ANOVA). (C) Absolute proliferation values (Absorption = A450 nm-A650 nm) in FGF2-stimulated control (open circles) or Rictor ko (filled squares) MAEC are presented. (nexp = 3, n.s., repeated measures ANOVA). In response to 10% FCS (right), proliferation of Rictor ko MAEC was significantly lower compared to that of control MAEC (n = 3, *P < 0.05, two-tailed T-test). (D) Migration of MAEC was measured after FGF2 (25 ng/ml) or diluent administration for 1, 3, 6 and 9 hours (wound completely closed = 1, n = 3, n.s., repeated measures ANOVA).
The depletion of AKT phosphorylation by Rictor knockout may influence mTORC1 signaling. AKT phosphorylates and inhibits tuberous sclerosis 2 (also known as tuberin), thus resulting in the activation of mTORC1- p70S6 kinase1 (S6K1)28. We found that FGF2 dose-dependently promoted S6K1 phosphorylation in control MAEC. After Rictor knockout, a minor but insignificant reduction in FGF2-induced S6K1 phosphorylation was detected (Fig. 5A). No differences in the phosphorylation of extracellular signal-regulated kinase 1 and 2 (ERK1/2) by the FGF2 gradient were observed in control MAEC and Rictor ko MAEC (Fig. 5A)29.
A close cross-talk exists among FGF2 and the different members of the VEGF family during angiogenesis and several studies have suggested that FGF2 induces neovascularization indirectly by activation of the VEGF/VEGFR system30. We confirmed that Vegfa mRNA substantially increased after FGF2 stimulation, yet Rictor knockout did not affect this induction. To investigate whether endothelial mTORC2 might modulate in vitro angiogenesis in response to FGF2 by regulation of VEGF receptors, we measured the mRNA-levels of the two main VEGF-receptors Flt1 (VEGFR1), which is characteristic for stalk cells and quiescent endothelium, and Kdr (VEGFR2), upregulated in tip cells31. Interestingly we found a strong FGF2-induced upregulation of the membrane form of Flt1 (mFlt1) and an even stronger induction of soluble Flt1 receptor (sFlt1) mRNA (Fig. 5B). Kdr message was present and decreased after FGF-stimulation (Fig. 5B). However, Rictor knockout cells induced mFlt1 and sFlt1 and reduced Kdr message congruent to control cells after FGF2 stimulation, which argues against endothelial tip-/stalk-cell modulation by mTORC2. FGF2 also stimulates VEGFA in endothelial cells of forming capillaries and cultured aortic endothelial cells32. Similarly, FGF receptor 1 (Fgfr1) message equally increased in control and Rictor ko MAEC. The decrease in PKCα protein after Rictor knockout was due to posttranscriptional mechanisms as PKCα gene expression was equal in control and Rictor ko MAEC (Fig. 5B).
Furthermore, we did not observe any significant differences in endothelial proliferation over a period of 72 hours in the dose response to FGF2 (Fig. 5C) or FGF2-induced-migration in a wound assay (Fig. 5D). However, a weak but significant (P < 0.05, Nexp = 4) reduction in proliferation was detected in the presence of FCS (Fig. 5C). Similarly, we found a significant reduction in VEGFA-induced proliferation by Rictor knockout (Supplemental Fig. S6).
In conclusion, FGF2 amplified RICTOR protein in control MAEC and Rictor deletion in FGF2-stimulated MAEC strongly blunted PKCα signaling and reduced AKT activity by depleting Ser473 phosphorylation. Rictor deletion had no effects on mTORC1 signaling and FGF-induced proliferation, migration nor did it interfere with FGF2-induced modulation of Flt1, Kdr and Vegfa expression levels.
The structure of the capillary bed of the striated skin muscle is not altered by endothelial-specific Rictor knockout
In preparation to study mTORC2-dependent vascular changes in response to wounding and FGF2 in vivo, we first recorded the capillary morphology in the existing striated skin muscle (Panniculus carnosus) vascular bed as baseline. To do so we surgically mounted a dorsal skinfold chamber33,34 to 8-week-old control and RictoriΔec mice. We observed that the baseline capillary bed, which was recorded 3 days post-chamber surgery, appeared very similar in 8-week-old control and RictoriΔec mice (Fig. 6A; supplementary video 1). Analysis of all capillary diameters measured at baseline did not reveal significant differences (Fig. 6A)). In both experimental groups, the vascular bed consisting of the panniculus carnosus with its capillary structures was organized in the typical parallel orientation and the subcutaneous layer with draining arterioles and venules exhibited the typical perfusion pattern of the dorsal skinfold chamber capillaries, as observed in previous studies33. Thus, ablation of endothelial Rictor did not interfere with the development or directly observable functionality of the intact, unstimulated dermal microvasculature in adolescent mice, which is in line our observation of normal growth, viability and weight gain of adolescent mice lacking endothelial Rictor.

Rictor knockout disables a sustained increase in skin capillary diameters and restricts extensive capillary remodeling in response to FGF2.
(A) Stacked frames of representative videos of the capillary vasculature were recorded through the dorsal skinfold chamber by intravital fluorescence microscopy. Baseline capillary bed of the skin muscle was assessed in 10-week-old mice (6 weeks after knockout induction) by intravital microscopy through the unmodified dorsal skinfold chamber. Upper (20 × magnification) and lower micrographs (10×) display representative skin capillary beds of control (left) and RictoriΔec (right) mice. Capillary diameters from 10 control and RictoriΔec mice were quantified and pooled for statistical analysis (ncapillaries = 253/206, no differences between groups, 2-tailed T-test and Whisker plot, indicating median, 25% percentile and total range to the right). Color scheme applies for whole Fig. 6 and is displayed in upper right corner. B. Wounding response of the capillary bed. Skin muscle capillary structure from day 1, 2, 4 and day 7 in control (left) and RictoriΔec mice (right) after wound sealing with heparin-containing matrigel (20 × magnification). Capillary diameters from 4 control and RictoriΔec mice were quantified and pooled for statistical analysis by 1-way ANOVA followed by Bonferroni multiple comparison (Scatter plot with medians on the below; number of capillaries (ncapillaries) are indicated below the X-axis, ****P < 0.0001). C. Heterogeneous capillary diameter increase in the FGF2-stimulated capillary bed. Skin muscle capillary structure from day 1, 2, 4 and day 7in control (left) and RictoriΔec mice (right) after wound sealing with FGF2 (1.5 μg/ml; heparin-containing matrigel, 20 × magnification). Capillary diameters from 7 control and RictoriΔec mice were quantified and pooled for statistical analysis by 1-way ANOVA followed by Bonferroni multiple comparison (Scatter plot and medians below; number of capillaries are indicated below the X-axis, ****P < 0.0001). D. Capillary remodeling in the FGF2-stimulated capillary bed. The line graph displays the normalized distribution of capillaries (nmax = 100) resolved in a 1-μm range after FGF2 stimulation for 7 days and illustrates differences in remodeling between groups. Micrograph to the right shows 10x magnification of the vascular bed of control and RictoriΔec mice after 7 days of FGF2 exposure.
Wounding-induced capillary diameter remodeling is not impaired by endothelial-specific Rictor knockout
We then modified this chamber by additionally removing cutis and subcutis on the opposite side of the observation window. This defect was sealed by growth factor-reduced matrigel (suppl. Fig. S7). In the first set of experiments, we included diluent (heparin) in matrigel to assess baseline alterations as a response to wound healing processes without exogenous growth factor stimulation. Capillaries were observed and recorded on a daily basis by intravital microscopy up to 7 days after matrigel sealing (Fig. 6B and supplementary video 2). Vascular morphological parameters were quantified in vessels where perfusion was observable. The vessels encompassed diameters ranging from 4 to 20 μm, thus capillaries and some small arterioles or venules. Capillary diameters significantly increased from day 1, 2, 4 and 7 compared to the baseline by about 2 μm in average for both control and RictoriΔec capillary diameters (Fig. 6B). Importantly, the microvasculature of the panniculus carnosus responded similarly to wounding and matrigel sealing in control and RictoriΔec mice as we did not detect significant differences compared to controls at days 1, 2, 3, and 7 (Fig. 6B).
Endothelial-specific Rictor knockout limits increases in the capillary diameter of existing skin vasculature in response to high doses of FGF2
To assess remodeling in terms of individual increases in capillary diameters in response to strong angiogenic stimulation, we exposed the existing capillary bed to a high dose of FGF2 (1.5 μg/ml). We focused on FGF2, since we found that Rictor deletion specifically disabled FGF2- but not VEGFA-dependent angiogenesis in vitro as shown earlier in this manuscript. Furthermore, FGF2 also promotes maturation of larger vessels such as arterioles35. Intravital recordings through the dorsal skinfold chamber in control and RictoriΔec revealed that diameters increased at day 1 and day 2 after FGF2 exposure (Fig. 6C, day 1 and 2). In both groups, diameters had increased for about 3.5 μm and we found no statistical difference between groups after testing by Bonferroni multiple comparisons (Fig. 6C). At days 4 and 7, capillary diameters further significantly increased in the control group when comparing to diameters at day 1. In addition, tortuous and bulbous vascular structures developed, which were observed in capillaries and small draining arterioles and venules (Fig. 6C, day 4 and 7). In RictoriΔec mice, however, FGF2-induced capillary enlargement stopped and rather regressed after day 2. Statistically, capillary diameters at days 4 were not different from those measured at day 1. Diameters measured on day 4 and 7 in RictoriΔec were significantly smaller compared to the control group on those days (Fig. 6C). We observed, that at the end of the recording the vascular bed in RictoriΔec mice underwent a far more restrained and different mode of remodeling. Thin-connecting anastomoses emerged between capillaries and draining arterioles in RictoriΔec mice and the orientation of capillaries remained largely parallel (Fig. 6C and D, day 7 and supplementary video 3 and 4). The dilated and tortuous capillary structures that developed in control mice after longer (4 and 7 days) exposure to FGF2 are also found in vascularized tumors, which in addition exhibit hyperpermeability36. We assessed vessel leakage of the FGF2-stimulated skin vascular bed on day 7 by intravenously injecting fluorescently labeled ricinus communis agglutinin I (RCA I)37. Preliminary data could however not support an obvious decrease in permeability by endothelial Rictor deletion as plasma leakage points were present in capillary structures from both control and RictoriΔec mice to a similar extent (suppl. Fig. S8).
Taken together, loss of endothelial Rictor normalized vascular structure after 4 days of exposure to FGF2 and prevented the formation of a heterogeneous, irregular microvascular bed as seen in controls (Fig. 6D). This suggests that endothelial mTORC2 is central to the FGF2-mediated remodeling response that creates larger and heterogeneously sized capillaries and small arterioles in the existing skin capillary bed beyond day 4.
Endothelial-specific Rictor knockout strongly reduces FGF2-induced neovascularization in matrigel plugs and prevents local hemorrhage
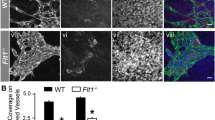
As technical limitations disabled us to monitor sprouting angiogenesis in the dorsal skinfold chamber, we used the matrigel plug angiogenesis assay to assess whether endothelial mTORC2 deficiency may alter de novo vascularization in adult mice in vivo in response to strong stimulation by FGF2 (1.5 μg/ml). Concentrations of FGF2 in this range have been used previously to achieve maximal neovascularization after 7 days38 and to achieve macroscopically a hemorrhagic appearance (see also supplementary Fig. S9). Knockout of Rictor in the endothelium was induced during adolescence in RictoriΔec mice. At 7 days after implantation, blood-containing microvessels and signs of hemorrhage were macroscopically evident in FGF2-containing plugs from control mice. In contrast, FGF2-containing plugs from RictoriΔec mice displayed homogeneous, unobtrusive vascularization. An estimation of blood content by optical densitometry demonstrated a significant decrease after Rictor knockout in FGF2 containing plugs compared to control plugs (Fig. 7A, 1st row). FGF2-induced vessels protruded to about 800 μm from the surface of the plug into the center in plugs from control mice, whereas microvessels protruded significantly less to only about 300 μm in plugs from RictoriΔec mice. FGF2-induced microvessel protrusion into the plugs from RictoriΔec mice was not completely abolished as we calculated a significant difference to diluent containing plugs (Fig. 7A, 2nd row). The presence of leaked erythrocytes in the FGF2-containing plugs from control mice from one set of experiments demonstrated local hemorrhage (Fig. 7B, left micrographs). In contrast, the microvasculature in FGF2-containing plugs from RictoriΔec mice was composed of homogenously small capillaries with no evidence of hemorrhage (Fig. 7B, right micrographs). Further micrographs showing hemorrhage in FGF2-containing plugs from 2 other sets of experiments are displayed in supplementary Fig. S10. During the 7 days of FGF2 administration, the matrigel plug was encapsulated by a thin layer of stromal tissue that also contained arterioles and venules. We found similar vessel densities in plugs from both experimental groups (data not shown), whereas the luminal diameter was significantly smaller in vessels of the stromal capsule from RictoriΔec mice compared with control mice. No stromal vessels were found in diluent containing plugs from both groups (Fig. 7A, 3rd row). Angiogenesis occurring during postnatal development is usually connected with inflammation39. Macrophages were demonstrated to promote angiogenesis via FGFs and placental growth factor signaling39 or by the release of pro-angiogenic molecules39,40. We therefore investigated by CD68 immunestaining whether the stromal halo around the plugs may contain varying amounts of macrophages that could influence de novo angiogenesis in our experimental setting. However, we found no significant differences in the ratio between CD68+ cells to total cell nuclei in the peripheral stroma when comparing diluent and FGF2-containing plugs from both groups (Fig. 7A, last row). The total amount of CD68+ cells per field counted was also not significantly modulated, however, we noticed a trend towards higher macrophage count in stromal halos around FGF2-containing matrigel plugs from RictoriΔec mice (Fig. 7A, last row). To further investigate the possibility of a mild FGF2-dependent proinflammatory state after endothelial Rictor knockout we measured the mRNA levels of monocyte-attracting protein 1 (Mcp1) and vascular- and inducible cell adhesion molecules (Vcam1, Icam1) in MAEC. FGF2 increased Vcam1 mRNA after 24 hours similarly in both control and Rictor knockout MAEC. A slight and insignificant increase for Icam1 mRNA was observed. FGF2 robustly (ca. 6.6 fold) induced Mcp1 mRNA in control and Rictor knockout MAEC. Interestingly, Mcp1 mRNA remained at high levels (ca. 5.3 fold induction) after 24 h of FGF2 stimulation and was significantly higher compared to controls (3.3 fold) at this timepoint (Fig. 7C). Thus, it is unlikely, that the strong reduction of FGF2-induced angiogenesis into matrigel plugs along with ‘normalized’ microvessel features by deletion of endothelial Rictor in vivo is caused by an altered inflammatory response.

Fibroblast growth factor 2(FGF2)-induced angiogenesis in matrigel plugs is reduced in RictoriΔec mice.
(A) 1st row: Representative matrigel plugs containing heparin (diluent) or 1.5 μg/ml FGF2 with heparin (FGF2) from 8-week-old male control (left) and RictoriΔec (right) mice removed 7 days after injection (scale bar = 1mm). Estimation of blood content by optical densitometry is shown on the right (nplugs = 4; **P < 0.01; 1-way ANOVA with Bonferroni multiple comparison). (A) 2nd row: Paraffin sections from corresponding plugs immunostained for CD31 (brown) and hematoxylin (blue/nuclei). Representative micrographs show 10 × magnification and display the depth of newly in grown microvessels from the surface towards the center of the matrigel plugs. Quantification to the right displays the significant reduction in the ingrowth (μm) of neovessels into plugs from RictoriΔec mice compared to control mice (nplugs = 4/7; **P < 0.01, ****P < 0.0001; 1-way ANOVA with Bonferroni multiple comparison). (A) 3rd row: Representative micrographs of peripheral stroma covering matrigel plugs. Identifiable inner microvessel diameters were measured (graph to the right; nplugs = 3; *P < 0.05;2-tailed T-test ). No vessels were found in peripheral stroma covering diluent-containing plugs. (A) 4th row: Representative micrographs of macrophage marker CD68-immunestainings of peripheral stroma and matrigel. Graphs to the right show ratio of CD68+/total cell nuclei in the stroma, and average of CD68 + cells per field counted in the stroma (nplugs = 4, no significant differences found after 1-way ANOVA/Bonferroni multiple comparison). (B) Representative micrographs of one set of experiments displaying hematoxylin and eosin stained (H&E) matrigel areas showing local leakage and hemorrhagic areas in FGF2 containing plugs from control mice compared to plugs from RictoriΔec mice (upper micrographs). Lower micrographs show higher magnification of CD31-stained matrigel areas. Arrowheads point to local spots of leaked erythrocytes in FGF2-containg control plugs. (C) Confluent and starved control and Rictor ko MAEC were stimulated for 1 and 24 hours with 25 ng/ml FGF2 or diluent. mRNA expression of monocyte attracting protein 1 (Mcp1), vascular and inducible cell adhesion molecules 1 (Vcam1, Icam1) was detected by quantitative real-time PCR (n = 3; *P < 0.05; 1-way ANOVA with Bonferroni multiple comparison test).
Taken together, we observe a dense, heterogeneous neovasculature with several patches of hemorrhage indicating disrupted or leaky capillaries that formed after 7 days of FGF2-exposure in control mice. In contrast endothelial Rictor knockout strongly and significantly reduced neovessel ingrowth in response to FGF2 with capillaries remaining homogenously small, with no evidence of hemorrhagic spots.
Discussion
In this study we deleted Rictor in the endothelium to study the general requirement of endothelial mTORC2 during embryonic and adolescent development. Our analysis of embryonic development using constitutive and inducible VE-Cadherin-Cre-specific Rictor knockout confirmed that homozygous Rictor deletion in endothelial cells is almost completely lethal and results in embryonic death around E12. Guertin et al. proposed that vascular defects may contribute to the lethality of whole-body Rictor knockout embryos on E10.514. Furthermore, Wang et al. demonstrated that the homozygous and Tie2-Cre-driven Rictor knockout is embryonically lethal17. Still, other than reductions in peripheral vascularization, we did not detect gross abnormalities of the normal vascular plexi on E10.5. Surviving embryos around this time frame showed rather distinct and rare deficits in vascularization at E10.5. Surviving Rictor knockout embryos, however, consistently displayed growth retardation and a delayed ossification of the vertebrae, toes, and fingers. Delayed ossification may explain growth retardation but may not categorically explain embryonic lethality of RictoriΔec in the confined midgestational timeframe. Thus, other essential functions that are regulated by mTORC2 during this short period in midgestation are probable and may also account for lethality.
Many signaling pathways that are involved in early embryonic development are also recapitulated during tumorigenesis41. As we found that endothelial mTORC2 was required during a confined, midgestational timeframe but had no apparent influence on viability before midgestation (E8.5), beyond midgestation (E14.5), physiological vascular development, vascular maintenance and growth from adolescence into adulthood, we hypothesized that endothelial Rictor might promote only ‘aberrant’ endothelial phenotype modulation such as in tumor angiogenesis or during a transition to an invasive mesenchymal phenotype. Guertin et al. demonstrated that Rictor has no significant role by itself in maintaining the integrity of a normal prostate epithelium in vivo but requires Rictor to be transformed into an invasive, malignant phenotype by Pten deletion, which results in strong PI3K activation42. Similarly, Drosophila embryos lacking mTORC2 activity are viable and display only minor growth defects42,43. However, PTEN loss-induced tissue overgrowth in the Drosophila eye requires dTORC243.
We found that particularly FGF2 and not VEGFA depended on the mTORC2 signaling hub to establish a capillary-like endothelial network on matrigel substrate when we tested these PI3K-activating angiogenic molecules in vitro. We found that FGF2 elicited a strong and fast increase in the diameter of existing skin capillaries in the dorsal skinfold chamber after two days of stimulation in both control and RictoriΔec mice. Over a period of 7 days, FGF2 induced the progressive and extensive remodeling of vessel structures characterized by heterogeneous and larger diameter sizes that also included larger arterioles and a tortuous vascular bed in control mice. RictoriΔec mice however, could not maintain heterogeneous capillary size remodeling beyond day 2of FGF2 exposure and returned to ‘normalized’ vascular features with homogenously and smaller sized capillary diameters and parallel-oriented capillaries. Tortuous and dilated capillaries are also found in vascularized tumors. Tumor blood vessels furthermore display hyperpermeability and do not mature into functional vasculature1,2. However, we could not detect fewer leakage points in FGF2-stimulated skin capillaries RictoriΔec in preliminary investigations.
However, the supply of FGF2 to the skin muscle over a period of seven days was limited by the volume of the matrigel carrier (16 μl) and may not have provided saturated FGF2-stimulation at all times. We therefore continued our investigations using the matrigel plug assay carrying pathologically high doses of FGF2 in a volume of 200 μl to induce aberrant, leaky and tumor-like angiogenesis as reported before38. Indeed, we observed a dense, heterogeneous neovasculature with several patches of hemorrhage that formed after 7 days in control mice. Endothelial Rictor knockout strongly and significantly reduced FGF2-mediated neovessel ingrowth and capillaries remained homogenously small with no microscopic signs of leakage. Our study thus demonstrates that endothelial mTORC2 acted as a central signaling hub for FGF2-induced persistent capillary diameter increases and remodeling into heterogeneous capillary structures and formation of a leaky, tumor-like neovasculature in the adult mouse.
FGF2, which is not secreted through vesicular pathways, can be exported from cells with unique extrusion pathways or large amounts can be released upon cell death44,38. FGF2 has been selectively determined as a crucial tumorigenic cytokine in prostate cancers in which both FGF2 and FGF2 receptor subtypes are co-expressed45,46. In addition many other tumor types express FGF2/FGFRs at high levels38,47,48,49,50,51. On the other hand, fibroblast growth factors are involved in the formation of skeletal elements within the developing limb52,53,54. Several FGFs are expressed in developing endochondral bone. FGF2 was the first FGF ligand to be isolated from growth plate chondrocytes52. FGF2 expression has also been observed in periosteal cells and in osteoblasts51,55. Targeted deletion of FGF2 causes a relatively subtle defect in osteoblastogenesis, leading to decreased bone growth and bone density51,56.
Mechanistically, we could not substantiate, that the extensive mTORC2-dependent FGF-induced angiogenic responses were executed by coupling to the VEGFA/VEGFR system, which also determines endothelial tip-/stalk phenotypes31. Also, FGF2-induced endothelial proliferation and migration were unaltered after Rictor knockout. In contrast, we found VEGFA-induced proliferation to be significantly decreased by Rictor knockout as previously reported17. Endothelial Rictor knockout resulted in dephosphorylation of typical mTORC2 downstream targets and blunted FGF2-induced phosphorylation of PSer473AKT and PSer657PKCα25. The results by Wang et al. suggest that mTORC2 is a critical signaling node required for VEGF-mediated angiogenesis through the regulation of AKT and PKCα in vascular endothelial cells17. AKT is activated by most endothelial growth factors (FGF2, VEGF and angiopoietin) and orchestrates a number of signaling pathways that are involved in angiogenesis1,57,58. For example, FGF2-induced capillary morphogenesis via FGFR1 is impaired in murine brain capillary endothelial cells expressing an inactive AKT57,59. FGF2-induced angiogenesis may partially depend on AKT signaling, potentially via transcription factors FoxO1/FoxO3a27,60.
Interestingly we found that quiescent and starved MAEC expressed low levels of RICTOR in control cells. High doses of FGF2 however induced RICTOR protein in correlation with high PSer473AKT. Comparable findings were reported during the epithelial to mesenchymal transition (EMT). Transforming growth factor (TGF-β), a strong inducer of EMT, increased RICTOR protein and thereby formation of mTORC2 in mouse mammary gland epithelial cells13. Without Rictor, the epithelial cells arrested in an intermediate stage between epithelial and mesenchymal differentiation, without the motile and invasive behavior of cells after EMT13. Interestingly, TGFβ-1 treatment also induces an interaction between RICTOR and integrin-linked kinase (ILK) and promotes ILK-dependent EMT. This complex was detected in cancer but not in normal cell types61 and overlaps with mTORC2 in the function as PSer473AKT kinase62. Thus, Rictor could promote EMT by forming different complexes.
In parallel to EMT, endothelial to mesenchymal transition (EndoMT) can be induced by transforming growth factor (TGF-β)63. EndoMT is a newly recognized type of cellular transdifferentiation that participates in development, but also in pathological conditions such as cancer and fibrosis63,64,65. New studies have shown that EndoMT represents a dedifferentiation of endothelial cells to a stem cell phenotype, which can redifferentiate into bone or cartilage cells63,64,66. Thus, hypothetically, endothelial Rictor may be required during midgestation to promote the transition of endothelial cells to a mesenchymal, osteogenic phenotyope to promote FGF2-directed ossification of limbs and vertebrae. During further development through normal adolescence where most vasculature is quiescent, with only 0.01% of endothelial cells undergoing division67, endothelial Rictor knockout may represent the characteristics of an unchallenged quiescent endothelial monolayer with low RICTOR expression and AKT activity. Recent studies suggest, during angiogenic sprouting, endothelial cells express many of EndoMT-driving genes and break down basement membrane. However, they retain intercellular junctions and migrate as a connected train of cells65. This process has been termed a partial EndoMT65. The permanently activated phenotype of tumor vasculature may well reflect the chronic activation of the EndoMT process, driven by persistent angiogenic cascades, leading to excessive sprouting and a failure to settle back into the mature, stable phenotype65. Thus, hypothetically, endothelial mTORC2, assembled through increased expression of FGF2-induced RICTOR may promote neovascularization by a sustained partial EndoMT.
In conclusion, we demonstrated that endothelial Rictor is crucial for progression through midgestation and for timely ossification. In adolescence, the FGF2-RICTOR axis promoted sustained, extensive and aberrant neovascularization. During adolescent vascular quiescence or moderate capillary remodeling, however, endothelial mTORC2/RICTOR was not required. Further studies are needed to uncover the exact molecular nature that may enable endothelial mTORC2/RICTOR to promote both embryonic development and extensive and aberrant FGF2-dependent angiogenesis in the adult.
Materials and Methods
Animal procedures
Mice with floxed Rictor exons68,69 were crossed with mice that express tamoxifen inducible Cre-ERT2 under the control of the endothelium-specific VE-cadherin promoter (VECad-Cre-ERT2)22 (kind gift of Dr. Iruela-Arispe, Department of Molecular, Cell & Developmental Biology, UCLA, USA) both on a congenic C57Bl/6J background. Offspring were genotyped for Cre-recombinase, Rictorfloxed and Rictorwt alleles using qPCR. Briefly, DNA was isolated from ear biopsies and amplified by qPCR with the following primer pairs (5′-3′): Forward: GCG GTC TGG CAG TAA AAA CTA TC; Reverse: GTG AAA CAG CAT TGC TGT CAC TT. The mice were bred, housed and handled according to the local animal ethics committee. All procedures with mice were approved by the Veterinary Office of the Canton of Zürich, Switzerland under licenses 77/2009 and 179/2012. We confirm that all procedures with mice were performed according the legislation of the Swiss Protection of Animals Act for vertebrates within the strict guidelines of the licenses 77/2009 and 179/2012 and additional local directives of the animal housing facilities.
Whole mount embryo staining
Embryos were stained with an antibody against endomucin according to Vieira et al.70. In brief, embryos were fixed for two hours on ice with PBS containing 4% PFA. After three washing steps with PBS containing 0.1% Triton X-100 and blocking of unspecific binding sites by incubation with PBS containing 10% FCS and 0.1% Triton X-100 for 30 minutes at room temperature, embryos were incubated with rat monoclonal antibody against endomucin (1:100, Santa Cruz Biotechnology) dissolved in PBS over night at 4 °C followed by five washing steps with PBS at room temperature. After incubation with secondary antibody anti-rat Alexa Fluor-555 (1:200, Molecular Probes) over night at 4 °C and three further washing steps with PBS, embryos were mounted and analyzed by laser scanning confocal microscopy (SP5; Leica, Wetzlar, Germany). Staining of whole embryos for LacZ was as described previously71.
Alizarin Red and Alcian Blue stainings
For skeletal staining, embryos were fixed in 95% ethanol for more than four days, after removal of fat and connective tissue, incubated in acetone for one day and stained with 0.15% alcian blue 8GS (Sigma-Aldrich)/0.05% alicarin red S (Sigma-Aldrich)/5% acetic acid in 70% ethanol at 37 °C for two days. After bleaching in 1% KOH for 12 to 48 hours embryos were destained with graded washes of glycerin (20% glycerin in 1% KOH, 50% glycerin in 1% KOH and 80% KOH in 1% glycerin). Embryos were stored in 100% glycerin.
Histological analysis
Embryos and dissected organs from adult mice were fixed in 4% PFA in PBS, transferred to ethanol and embedded in paraffin. Longitudinally-sectioned paraffin-embedded embryos and organs were stained with hematoxylin and eosin. For CD 31 staining paraffin sections were stained as described previously72. Sections from dorsal chambers were de-paraffinized and rehydrated in xylene and isopropanol and antigens retrieved by boiling in Na3 citrate. Rabbit polyclonal anti-mouse Estrogen Receptor α antibody (Millipore) was used for detection and visualized by secondary goat anti-rabbit Alexa-Fluor 555 (Invitrogen) using microscopes and cameras from Zeiss and Olympus.
Determination of Rictor mRNA expression levels in endothelial cells from thoracic aorta
Rictor deletion was induced in VECad-CreERT2+; Rictorfloxed/floxed mice at an age of four weeks with five consecutive intraperitoneal tamoxifen (Tx) injections according to the protocol of Monvoisin et al.22. (2 mg Tx/ml dissolved in corn oil at a concentration of 80 mg/kg bodyweight, T5648, Sigma-Aldrich). Littermate control mice were injected with corn oil alone. This injection protocol was used for all experiments in this study.
Aortae were excised from 6 month old Tx-injected (RictoriΔec) and control mice (Ctrl), cleaned from adhering tissue and opened longitudinally. Endothelial cells were carefully scraped directly in RLT buffer (Qiagen) and RNA was extracted using the RNAeasy micro kit (Qiagen) according to the recommendations of the manufacturer. Equal amounts of RNA were transcribed to cDNA by WT (Whole Transcript)-Ovation™ Pico RNA Amplification System (NuGEN). Quantitative real-time PCR was performed using a SYBR green-based standard protocol. Primer list is shown in section ‘Real-time quantitative reverse transcription polymerase chain reaction’. Specificity of the primers was tested by melt curve and agarose gel analysis and sequencing. Relative expression levels were calculated using the comparative ΔCt method73.
Isolation of endothelial cells
Mouse aortic endothelial cells (MAECs) were isolated from aortae of 8–10 weeks old Rictor floxed male mice as described earlier74,75. Briefly, fibrin gels were prepared by mixing 3 mg/ml of fibrinogen with serum-free DMEM complemented with non-essential amino acids, Sodium Pyruvate, Pen-Strep and thrombin on ice. 24-well plates were coated with the prepared fibrin gel allowed to polymerize at 37 °C. The excised aorta was cleaned, cut in small rings and placed on top of the gel and overlaid by fibrin gel. After gel had polymerized pre-warmed growth media (serum-free DMEM that contained 10% FCS, 200 μg/ml of ECGS, 10 ng/ml FGF2 and 0.1 IE/ml of heparin) was loaded to the wells. To protect the fibrin gel from degradation, 300 μg/ml of ε-amino caproic acid (Sigma A-7824) diluted in PBS was added to all wells every other day. After 10 days of culturing, capillary-like sprouts were observed under the microscope. The outgrown cells were harvested by pipetting up and down the fibrin gel. The gel-cell mixture was transferred to a six-well plate which had previously been coated with 0.1% gelatin gold and 1 ml/well of growth media w/o heparin was added. The next day, cells were washed once with warm PBS and new EC growth media was added. Confluent cells were split 1:2 by trypsinization using TrypLETM-Express and characterized with endothelial cell specific immuno-fluorescent marker von Willebrand Factor (VWF, LabForce AG).
Cell culture and treatments
For all experiments using MAECs, cell culture dishes were coated with 0.1% gelatin gold (Carl Roth GmbH 4274.1) for 20 minutes at 37 °C. MAECs were maintained in DMEM (Biochrom FG435), complemented with 10% or 1% (complete or starvation medium) FCS (Biochrom S0615), 1% sodium pyruvate (GIBCO 15140), 1% non-essential amino acids (GIBCO 11140) and 1% penicillin-streptomycin (GIBCO 15140). Stimulation with growth factors always included addition of heparin at a fixed ratio (1 IU heparin per 1.5 μg/ml FGF).
Generation of Rictor ko cells
Rictor knockout was induced by adenoviral transfection of Cre-Recombinase on Rictor floxed MAEC. 2.5 × 105Rictor floxed MAECs were seeded on a 6 cm culture dish. The next day, the media was removed and a virus (100 MOI) that contained either Ade-CRE-GFP (Vector Biolabs 1045) or Ade-CMV-GFP (Vector Biolabs 1060) was added in 1 ml of growth media to the cells. After 7 hours, the media was removed, replaced by 4 ml of normal growth media and incubated at 37 °C. Endothelial cells expressed GFP the following day. Down-regulation of Rictor and disruption of mTORC2 signaling was assessed by using qRT-PCR and western blotting 3 days after transfection (see also Fig. 4 lower left panel). Steps that involved handling viruses or virus-transfected cells were performed in a level-2 biosafety hood (Skan VSB 90) in a certified cell culture laboratory.
Endothelial network-formation assay in vitro
Angiogenesis in vitro was assessed on the basis of a tube formation assay. Twenty-four-well culture plates (Costar; Corning) were coated with growth factor-reduced matrigel (BD Biosciences) in a total volume of 150 μL and allowed to solidify for 30 min at 37 °C. Tracking dye green (CytoPainter, abcam ab138891) was dissolved in 100 μl DMSO (=stock solution (1000×)). 20 μl of stock solution was mixed with 5 ml assay buffer. Cells were washed once with PBS, and 5 ml tracking dye green working solution added to the cells and incubated for 45 min at 37 °C in an CO2 incubator. Labeled MAEC were trypsinized and resuspended to a concentration of 105/mL in DMEM/1% FCS. 500 μl of the cell suspension were added into each well and complemented with diluent (heparin) and growth factors (FGF2; VEGFA 25ng/ml with heparin). Then the cells were incubated at 37 °C for eighteen hours. The appearance of endothelial network was observed under an inverted microscope by fluorescence (FITC; Ex/Em = 490/520 nm) with a 4x objective (IX70, Olympus) and photographed. Number of master segments were quantified automatically using the Macro ‘Angiogenesis Analyzer’ by Gilles Carpentier (Gilles Carpentier. Contribution: Angiogenesis Analyzer, ImageJ News, 5 October 2012) for NIH Image J 1.47v Program.
Immunoblotting
To extract protein, cells were washed twice with ice cold PBS and then harvested by scraping with a cell scraper (BD Falcon 353089) into 1 ml of ice cold PBS. The cell suspension was collected with a 1 ml pipette, transferred to a 2 ml Eppendorf tube and then centrifuged for 5 minutes at 14,000 rpm at 4 °C. The pellet was resuspended in RIPA buffer (50 mM TrisHCl (pH7.4), 150 mM NaCl, 1 mM EDTA(pH8), 1% Triton X-100, 0.1% SDS, 0.25% Na-deoxycholate) that contained a complete mini protease inhibitor (Roche 11836153001) and a phosphatase inhibitor cocktail 2 (Sigma P5726). The sample was centrifuged for 15 minutes at 14,000 rpm at 4 °C. The resulting supernatant was transferred to a new 1.5-ml Eppendorf tube and the protein concentration measured by using a BCA protein assay kit (Thermo scientific 23223). Equal amounts of protein were loaded onto a 8% acrylamide-SDS gel. Separated proteins were transferred to a nitrocellulose membrane (Whatman BA85) using a semidry blotting procedure, blocked for 1 hour at room temperature (RT) in 5% Bovine Serum Albumine (BSA, Sigma-Aldrich A7906) in Tris buffered saline complemented with 0.1% Tween (TBS-T). Membranes were washed once with TBS-T and incubated overnight at 4 °C with one of the following primary antibodies against Rictor (#2140), Phospho-AKT (Ser473) (#9271), Phospho-AKT(Thr308) (#9275), AKT(#9272), PKCα, Phospho-PKCα (Ser657)(Santa Cruz Biotechnology, #12356), Phospho-ERK1/2 (Thr202/Tyr204) (#9101), ERK1/2 (#9102), Phospho-S6K1 (Thr389) (#9205), S6K1 (#9202) and ) and Phospho-S6 RP (Ser235/236) (#4858), all from Cell signaling (BioConcept, Switzerland) in a dilution of usually 1:1000. Mouse anti β-Actin 1:10,000 (Sigma A-5441) was used to control equal loading. The next day, the membrane was washed three times in TBS-0.1% tween and incubated with a secondary antibody goat anti rabbit HRP 1:5,000 (Cell Signaling 7074) or anti mouse HRP 1:50,000 (Cell Signaling 7076) for 1 hour at room temperature (RT). After another wash step, a chemiluminescent substrate that is used for the detection of HRP (Thermo Scientific 34080) was applied to the membrane and then the membrane was incubated for 1 minute. The signal was detected with a CL-XPosure film (Thermo Scientific 34088) and the resulting bands were quantified by using ImageJ (Wayne Rasband, NIH, MD, USA).
Real-time quantitative reverse transcription polymerase chain reaction
qRT-PCR was performed as described previously76,77. RNA was isolated using the RNAeasy Mini kit (Qiagen, Hilden, Germany), followed by an on column DNA digestion (Qiagen, Hilden, Germany). cDNA was transcribed from total RNA using Omniscript RT kit (Qiagen, Hilden, Germany) and random primers (Roche, Basel, Switzerland). To control for DNA contamination in the qRT-PCR, for each sample a control reaction missing reverse transcriptase was additionally amplified. Primers used are listed (5′ → 3′) below (Table 1). Primers were tested by cDNA dilution series to obtain optimal reaction conditions. qRT-PCR was performed using an iCycler iQ Real Time PCR Detection System (Biorad, Reinach, Switzerland) and iQ™ SYBR® Green Supermix (Biorad). Melting curve of each representative reaction was analyzed. The qRT-PCR was quantified using the formula: 2−ΔCT = CT gene of interest – CT Tubulin73.
Proliferation assay
4500 cells with 100 μl of growth media were seeded in a 96-well plate in 10 replicates. After serum-starvation (0.5% FCS) for 26 hours, cells were stimulated with 5–25 ng/ml of FGF2 or VEGFA, 10% FCS or 100μg/ml of insulin. After 72 hours, 10 μl of WST-1 solution (Roche Molecular Diagnostics) was added for 2 hours. Absorption was measured (A450nm-A690nm) by using a Spectramax M2 reader (Molecular Devices).
Migration assay (wound healing)
MAECs were grown to confluency, starved (0.5% FCS) for 24 hours. A straight scratch using a 200 μl sterile tip was applied to the confluent monolayer (Star Lab, #S1120-8810). The monolayer was stimulated with diluent and FGF2 (25 ng/ml). The migration of the cells was photographed with an inverted microscope (Olympus IX71) at different time points (0, 1, 3, 6 and 9 hours after the stimulation) and measured with the software T-Scratch® (Tobias Gebäck and Martin Schulz, ETH Zürich, 2008).
Mouse dorsal skin fold chamber and matrigel sealing
To study the capillary remodeling and angiogenesis in vivo we used the dorsal skin fold chamber as described previously33 with a novel modification in order to deliver growth factors via matrigel to the tissue. For chamber implantation, two symmetrical titanium frames were mounted on the dorsal skin fold of the animal. One skin layer and the underlying fat were then completely removed in a circular area of 15 mm in diameter and the remaining layers (consisting of striated skin muscle, subcutaneous tissue and skin) were covered with NaCl2 0.9% and a glass cover slip incorporated into one of the titanium frames33. The animals were allowed to recover for two days. Skin was detached from the underlying muscle and removed in a circular area of 7 mm in diameter from the back of the chamber. Growth-factor reduced matrigel was mixed with FGF2 (1.5 μg/ml) and Heparin (5 IU units) or Heparin alone. The defect on the back of the chamber was sealed with 16μl of the matrigel mixtures, allowed to polymerize and covered with a glass cover slip.
Intravital microscopy
Repetitive intravital microscopic analyses of skin microvasculature were carried out daily over a time period of 7 days. Microscopic images were taken at 8 different areas within the center and the periphery of the wound. After injection of 0.2 ml FITC-labeled dextran (2%; MW 70000, Sigma-Aldrich, Munich, Germany) the microcirculation was visualized by intravital fluorescence microscopy. (Leica DM/LM; Leica Microsystems, Wetzlar, Germany). Microscopic images were captured by a CCD television camera (Kappa Messtechnik, Gleichen, Germany) and recorded on video (50Hz; Panasonic AG-7350-SVHS, Tokyo, Japan) for subsequent off-line analysis. Using ×10 (N-Plan ×10/0.25 LD, Leica), ×20 (HCX Apo ×20/0.50W, Leica) objectives blood flow was monitored in capillaries of the superficial and deep dermal plexus of the skin muscle. The epi-illumination setup included a mercury lamp with a blue filter (450–490 nm/>520 nm excitation/emission wavelength) and a green filter (530–560 nm/>580 nm).
Matrigel plug assay
The matrigel plug assay was performed as previously described78. In brief, 8-week-old C57BL/6 mice were injected subcutaneously with 0.2 ml of matrigel containing 1.5 μg/ml bFGF with 1 μl (5 IU) Heparin. The injected matrigel rapidly formed a single, solid gel plug. After 7 days, mice were euthanized, the skin was pulled back to expose the matrigel plug. The matrigel plug was removed, photographed and fixed in formalin and paraffin embedded. Hemoglobin content was estimated by measuring mean pixel densities from five random 200 × 200 pixel areas from the magenta channel in CMKY converted images from each plug by the NIH ImageJ program.
The Matrigel plugs were removed and fixed in 4% buffered formalin. After 48 h fixation, the plugs were trimmed, dehydrated in graded alcohol and routinely paraffin wax embedded. Sections (3–5 μm thick) were prepared, mounted on glass slides, de-paraffinized in xylene, rehydrated through graded alcohols and stained with hematoxylin and eosin (HE) for the histological examination. For analysis of immunestainings, sections of one plug were photographed in three different areas (350 × 350 pixels), quantified and averaged. The n number refers to the mean of 3 areas of one plug. E.g. for microvessel invasion n = 4/7 refers to 4 groups of 3 averaged areas from control plugs compared to 7 groups of 3 averaged areas from 7 Rictor ko plugs. Immunohistology for CD31 antigen was employed to highlight endothelial cells. Anti-CD-31 immunohistochemical staining was performed according to manufacturer’s protocol (Lifespan Biosciences). Slides were photographed using a brightfield microscope (Zeiss Axioskop 2, Germany). Immunohistology for the CD68 antigen was employed to highlight cells of the monocyte/macrophage lineage79. Briefly, sections were deparaffinized in xylene (2 × 5 min) and rehydrated in decreasing concentrations of ethanol (2 × 3 min washes in 100% ethanol, followed by 1 × 3 min wash in 96% ethanol). Sections were washed twice with Tris-buffered saline (TBS, 0.1 M Tris–HCl with 0.9% NaCl, pH 7.4) and incubated for 1 h at 37 °C with the primary antisera (1:100, ab125212, Clone KP1, Abcam, Cambridge, United Kingdom), after heat pre-treatment in citrate acid (0.01M, pH 9.0) in a 97 °C water bath for 20 min. An anti-rabbit IgG DAB detection system was subsequently applied according to the manufacturer’s protocols (Discovery OmniMap anti-Rb HRP, Roche, Basel, Switzerland). Sections were then washed 3 × in TBS and 1 × in distilled water and counterstained for 1 min with hematoxylin, followed by rinsing for 5 min in tap water and dehydration in ascending alcohols, clearing in xylene, coverslipping and mounting. Sections of murine immune system organs were used as positive control. All slides were scanned using digital slide scanner NanoZoomer-XR C12000 (Hamamatsu, Japan) and images were taken using NDP.view2 viewing software (Hamamatsu).
Statistical analysis
Statistical tests were performed by GraphPad Prism 5.04 software (San Diego, CA, USA). On a general basis, two-way analysis of variance (ANOVA), followed by a Bonferroni-post test comparing all pairings was calculated whereby a P value of less than 0.05 was considered as statistically significant. For the comparison of two groups, the unpaired t-test was used. Datapoints in graphs represent average values ± standard error of the mean unless otherwise stated.
Additional Information
How to cite this article: Aimi, F. et al. Endothelial Rictor is crucial for midgestational development and sustained and extensive FGF2-induced neovascularization in the adult. Sci. Rep.5, 17705; doi: 10.1038/srep17705 (2015).
References
Munoz-Chapuli, R., Quesada, A. R. & Angel Medina, M. Angiogenesis and signal transduction in endothelial cells. Cell Mol Life Sci 61, 2224–2243 (2004).
Ziyad, S. & Iruela-Arispe, M. L. Molecular mechanisms of tumor angiogenesis. Genes Cancer 2, 1085–1096 (2011).
Lau, M. T., So, W. K. & Leung, P. C. Fibroblast growth factor 2 induces E-cadherin down-regulation via PI3K/Akt/mTOR and MAPK/ERK signaling in ovarian cancer cells. PloS one 8, e59083 (2013).
Sarbassov, D. D., Ali, S. M. & Sabatini, D. M. Growing roles for the mTOR pathway. Curr Opin Cell Biol 17, 596–603 (2005).
Guba, M. et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med 8, 128–135 (2002).
Seeliger, H. et al. Role of mTOR in solid tumor systems: a therapeutical target against primary tumor growth, metastases and angiogenesis. Cancer Metastasis Rev 26, 611–621 (2007).
Fokas, E. et al. Dual inhibition of the PI3K/mTOR pathway increases tumor radiosensitivity by normalizing tumor vasculature. Cancer Res 72, 239–248 (2012).
Maira, S. M. et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 7, 1851–1863 (2008).
Schnell, C. R. et al. Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 on the tumor vasculature: implications for clinical imaging. Cancer Res 68, 6598–6607 (2008).
Falcon, B. L. et al. Reduced VEGF production, angiogenesis and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res 71, 1573–1583 (2011).
Sarbassov, D. D., Guertin, D. A., Ali, S. M. & Sabatini, D. M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 (2005).
Tandon, M., Chen, Z. & Pratap, J. Runx2 activates PI3K/Akt signaling via mTORC2 regulation in invasive breast cancer cells. Breast Cancer Res 16, R16 (2014).
Lamouille, S. et al. TGF-beta-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J Cell Sci 125, 1259–1273 (2012).
Guertin, D. A. et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11, 859–871 (2006).
Shiota, C. et al. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev Cell 11, 583–589 (2006).
Li, W. et al. Hypoxia-induced endothelial proliferation requires both mTORC1 and mTORC2. Circ Res 100, 79–87 (2007).
Wang, S. et al. Regulation of Endothelial Cell Proliferation and Vascular Assembly through Distinct mTORC2 Signaling Pathways. Molecular and cellular biology 35, 1299–1313 (2015).
Sarbassov, D. D. et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22, 159–168 (2006).
Alva, J. A. et al. VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn 235, 759–767 (2006).
Zovein, A. C. et al. Vascular remodeling of the vitelline artery initiates extravascular emergence of hematopoietic clusters. Blood 116, 3435–3444 (2010).
McGrath, K. E., Koniski, A. D., Malik, J. & Palis, J. Circulation is established in a stepwise pattern in the mammalian embryo. Blood 101, 1669–1676 (2003).
Monvoisin, A. et al. VE-cadherin-CreERT2 transgenic mouse: a model for inducible recombination in the endothelium. Dev Dyn 235, 3413–3422 (2006).
Gollner, H. et al. Impaired ossification in mice lacking the transcription factor Sp3. Mech Dev 106, 77–83 (2001).
Arnaoutova, I. & Kleinman, H. K. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc 5, 628–635 (2010).
Ikenoue, T. et al. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J 27, 1919–1931 (2008).
Hiraoka, D., Okumura, E. & Kishimoto, T. Turn motif phosphorylation negatively regulates activation loop phosphorylation in Akt. Oncogene 30, 4487–4497 (2011).
Jacinto, E. et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137 (2006).
Inoki, K. et al. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4, 648–657 (2002).
Cross, M. J. & Claesson-Welsh L., FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci 22, 201–207 (2001).
Presta, M. et al. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev 16, 159–178 (2005).
De Smet, F. et al. Mechanisms of vessel branching: filopodia on endothelial tip cells lead the way. Arterioscler Thromb Vasc Biol 29, 639–649 (2009).
Seghezzi, G. et al. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol 141, 1659–1673 (1998).
Lindenblatt, N. et al. A new model for studying the revascularization of skin grafts in vivo: the role of angiogenesis. Plast Reconstr Surg 122, 1669–1680 (2008).
Calcagni, M. et al. In vivo visualization of the origination of skin graft vasculature in a wild-type/GFP crossover model. Microvasc Res 82, 237–245 (2011).
de Paula, E. V. et al. Dual gene transfer of fibroblast growth factor-2 and platelet derived growth factor-BB using plasmid deoxyribonucleic acid promotes effective angiogenesis and arteriogenesis in a rodent model of hindlimb ischemia. Transl Res 153, 232–239 (2009).
Goel, S. et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 91, 1071–1121 (2011).
Sounni, N. E. et al. Stromal regulation of vessel stability by MMP14 and TGFbeta. Dis Model Mech 3, 317–332 (2010).
Claffey, K. P. et al. Fibroblast growth factor 2 activation of stromal cell vascular endothelial growth factor expression and angiogenesis. Lab Invest 81, 61–75 (2001).
Szade, A. et al. Cellular and molecular mechanisms of inflammation-induced angiogenesis. IUBMB Life 67, 145–159 (2015).
Sunderkotter, C. et al. Macrophages and angiogenesis. J Leukoc Biol 55, 410–422 (1994).
Ma, Y. et al. The relationship between early embryo development and tumourigenesis. Journal of cellular and molecular medicine 14, 2697–2701 (2010).
Guertin, D. A. et al. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 15, 148–159 (2009).
Hietakangas, V., Cohen, S. M. & Re-evaluating, A. K. T. regulation: role of TOR complex 2 in tissue growth. Genes & development 21, 632–637 (2007).
Florkiewicz, R. Z., Majack, R. A., Buechler, R. D. & Florkiewicz, E. Quantitative export of FGF-2 occurs through an alternative, energy-dependent, non-ER/Golgi pathway. J Cell Physiol 162, 388–399 (1995).
Ittman, M. & Mansukhani, A. Expression of fibroblast growth factors (FGFs) and FGF receptors in human prostate. J Urol 157, 351–356 (1997).
Davol, P. A. & Frackelton, A. R., Jr. Targeting human prostatic carcinoma through basic fibroblast growth factor receptors in an animal model: characterizing and circumventing mechanisms of tumor resistance. Prostate 40, 178–191 (1999).
Berger, W. et al. Evidence for a role of FGF-2 and FGF receptors in the proliferation of non-small cell lung cancer cells. Int J Cancer 83, 415–423 (1999).
Sumitomo, S. et al. Immunohistochemical study of fibroblast growth factor-2 (FGF-2) and fibroblast growth factor receptor (FGF-R) in experimental squamous cell carcinoma of rat submandibular gland. Oral Oncol 35, 98–104 (1999).
Tamiya, S., Ueki, T. & Tsuneyoshi, M. Expressions of basic fibroblast growth factor and fibroblast growth factor receptor mRNA in soft tissue tumors by in situ hybridization. Mod Pathol 11, 533–536 (1998).
Ueki, T. et al. Expression of basic fibroblast growth factor and fibroblast growth factor receptor in advanced gastric carcinoma. J Pathol 177, 353–361 (1995).
Xerri, L. et al. Expression of FGF1 and FGFR1 in human melanoma tissues. Melanoma Res 6, 223–230 (1996).
Ornitz, D. M. & Itoh, N. Fibroblast growth factors. Genome Biol 2, REVIEWS3005 (2001).
Ornitz, D. M. & Marie, P. J. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev 16, 1446–1465 (2002).
Sullivan, R. & Klagsbrun, M. Purification of cartilage-derived growth factor by heparin affinity chromatography. J Biol Chem 260, 2399–2403 (1985).
Hurley, M. M. et al. Parathyroid hormone regulates the expression of fibroblast growth factor-2 mRNA and fibroblast growth factor receptor mRNA in osteoblastic cells. J Bone Miner Res 14, 776–783 (1999).
Moore, R., Ferretti, P., Copp, A. & Thorogood, P. Blocking endogenous FGF-2 activity prevents cranial osteogenesis. Dev Biol 243, 99–114 (2002).
Lee, C. et al. Signal transduction in endothelial cells by the angiogenesis inhibitor histidine-rich glycoprotein targets focal adhesions. Exp Cell Res 312, 2547–2556 (2006).
Hofer, E. & Schweighofer, B. Signal transduction induced in endothelial cells by growth factor receptors involved in angiogenesis. Thromb Haemost 97, 355–363 (2007).
Ruzinova, M. B. et al. Effect of angiogenesis inhibition by Id loss and the contribution of bone-marrow-derived endothelial cells in spontaneous murine tumors. Cancer Cell 4, 277–289 (2003).
Milkiewicz, M. et al. Identification of a mechanism underlying regulation of the anti-angiogenic forkhead transcription factor FoxO1 in cultured endothelial cells and ischemic muscle. Am J Pathol 178, 935–944 (2011).
Serrano, I., McDonald, P. C., Lock, F. E. & Dedhar, S. Role of the integrin-linked kinase (ILK)/Rictor complex in TGFbeta-1-induced epithelial-mesenchymal transition (EMT). Oncogene 32, 50–60 (2013).
McDonald, P. C. et al. Rictor and integrin-linked kinase interact and regulate Akt phosphorylation and cancer cell survival. Cancer Res 68, 1618–1624 (2008).
Medici, D. et al. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 16, 1400–1406 (2010).
Medici, D. & Olsen, B. R. The role of endothelial-mesenchymal transition in heterotopic ossification. J Bone Miner Res 27, 1619–1622 (2012).
Welch-Reardon, K. M., Wu, N. & Hughes, C. C. A role for partial endothelial-mesenchymal transitions in angiogenesis? Arterioscler Thromb Vasc Biol 35, 303–308 (2015).
Medici, D. & Kalluri, R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin Cancer Biol 22, 379–384 (2012).
Carmeliet, P. & Jain, R. K. Angiogenesis in cancer and other diseases. Nature 407, 249–257 (2000).
Bentzinger, C. F. et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab 8, 411–424 (2008).
Polak, P. et al. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab 8, 399–410 (2008).
Vieira, J. M., Schwarz, Q. & Ruhrberg, C. Selective requirements for NRP1 ligands during neurovascular patterning. Development 134, 1833–1843 (2007).
Nagy, A., Gertsenstein, M., Vintersten, K. & Behringer, R. Staining Whole Mouse Embryos for {beta}-Galactosidase (lacZ) Activity. CSH protocols2007, pdb prot4725 (2007).
Mihic-Probst, D. et al. Tumor cell plasticity and angiogenesis in human melanomas. PLoS One 7, e33571 (2012).
Schmittgen, T. D. et al. Real-time PCR quantification of precursor and mature microRNA. Methods 44, 31–38 (2008).
Peier, M. et al. Sprouty2 expression controls endothelial monolayer integrity and quiescence. Angiogenesis 16, 455–468 (2013).
Humar, R. et al. Hypoxia enhances vascular cell proliferation and angiogenesis In vitro via rapamycin (mTOR)-dependent signaling. FASEB J 16, 771–780 (2002).
Bhattacharya, I. et al. Rictor in perivascular adipose tissue controls vascular function by regulating inflammatory molecule expression. Arterioscler Thromb Vasc Biol 33, 2105–2111 (2013).
Walpen, T. et al. Loss of pim1 imposes a hyperadhesive phenotype on endothelial cells. Cell Physiol Biochem 30, 1083–1096 (2012).
Akhtar, N., Dickerson, E. B. & Auerbach, R. The sponge/Matrigel angiogenesis assay. Angiogenesis 5, 75–80 (2002).
Rehg, J. E., Bush, D. & Ward, J. M. The utility of immunohistochemistry for the identification of hematopoietic and lymphoid cells in normal tissues and interpretation of proliferative and inflammatory lesions of mice and rats. Toxicol Pathol 40, 345–374 (2012).
Acknowledgements
We thank Dr. Martin Peier and Dr. Thomas Walpen for valuable scientific discussions and for establishing molecular techniques used in this study. We thank Ana Isabel Perez-Dominguez for providing extensive technical assistance and Dr. Giovanni Pellegrini for immunohistochemistry. This study was supported by research grants from the Swiss National Science Foundation (No. 118349; EJB), the Foundation for Scientific Research at the University of Zürich (F-84401-10-01; EJB), the SwissLife Jubiläumsstiftung (F-84401-11-01; IK), the Zurich Center for Integrative Human Physiology (cooperative research project grant; RH) and the Novartis Foundation for Medical-Biological Research (RH).
Author information
Authors and Affiliations
Contributions
R.H. and E.J.B. produced the original idea, designed experiments and wrote the manuscript. F.A. performed molecular biology experiments and contributed writing the manuscript. R.H., F.A. and V.G.L. performed matrigel plug experiments. N.L. and A.H. provided rationale, equipment and training for dorsal skinfold experiments and intravital microscopy. S.G., I.K. and A.H. performed dorsal skinfold experiments and intravital microscopy. S.G. analyzed intravital videos and contributed writing the manuscript. I.K. assessed embryonic development and contributed writing the manuscript. F.L. performed endothelial network-formation assays. P.L. performed endothelial cell isolation. M.N.H. and M.R. generated RictorPlox mice. E.H. detected Rictor knockout in aortic samples and revised the manuscript. All authors have reviewed, discussed and approved the results and conclusions of this manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Aimi, F., Georgiopoulou, S., Kalus, I. et al. Endothelial Rictor is crucial for midgestational development and sustained and extensive FGF2-induced neovascularization in the adult. Sci Rep 5, 17705 (2016). https://doi.org/10.1038/srep17705
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep17705
- Springer Nature Limited
This article is cited by
-
The role of CD146 in renal disease: from experimental nephropathy to clinics
Journal of Molecular Medicine (2024)
-
CD146, from a melanoma cell adhesion molecule to a signaling receptor
Signal Transduction and Targeted Therapy (2020)
-
UV-blocking, transparent and hazy cellulose nanopaper with superior strength based on varied components of poplar mechanical pulp
Cellulose (2020)
-
Morphology regulation in vascular endothelial cells
Inflammation and Regeneration (2018)
-
Up-regulation of FGFBP1 signaling contributes to miR-146a-induced angiogenesis in human umbilical vein endothelial cells
Scientific Reports (2016)