
Abstract
Bone homeostasis is maintained as a delicate balance between bone-resorption and bone-formation, which are coupled to maintain appropriate bone mass. A critical question is how bone-resorption is terminated to allow bone-formation to occur. Here, we show that TGFβs inhibit osteoclastogenesis and maintain bone-mass through Smad4 activity in osteoclasts. We found that latent-TGFβ1 was activated by osteoclasts to inhibit osteoclastogenesis. Osteoclast-specific Smad4 conditional knockout mice (Smad4-cKO) exhibited significantly reduced bone-mass and elevated osteoclast formation relative to controls. TGFβ1-activation induced expression of Irf8 and Bcl6, both of which encode factors inhibiting osteoclastogenesis, by blocking their negative regulator, Prdm1, in osteoclasts in a Smad4-dependent manner. Reduced bone-mass and accelerated osteoclastogenesis seen in Smad4-cKO were abrogated by Prdm1 deletion. Administration of latent-TGFβ1-Fc to wild-type mice antagonized LPS-induced bone destruction in a model of activated osteoclast-mediated bone destruction. Thus, latent-TGFβ1-Fc could serve as a promising new therapeutic agent in bone diseases marked by excessive resorption.
Similar content being viewed by others


Introduction
Bone is continuously resorbed and constructed, and bone volume is controlled as a balance of both activities, a process termed “coupling”1. The coupling system is also critical to determine regions for new bone production, which are often sites where resorption has occurred2. Coupling failure results in bone diseases such as osteoporosis, in which bone formation following resorption is less effective, reducing bone mass3. To date, drugs such as bisphosphonates have been launched as osteoporosis therapy; however, their use frequently causes multiple adverse effects, including osteonecrosis of the jaw due to excessive inhibition of osteoclastic activity beyond levels required for physiological bone turnover4. Thus, understanding the coupling system is required to better enable us to increase bone formation at required levels and at sites where resorption occurs.
Several factors have been proposed to function as coupling factors5. For example, osteoclast/osteoblast signaling is reportedly transduced by ephrin B2/EphB4, Ephrin A2/EphA2, and Semaphorin 4D/PlexinB1 interactions or by Semaphorin 3A secreted by osteoblasts6,7,8,9. In addition, mice deficient in genes encoding either dendritic cell-specific transmembrane protein or the v-ATPase V0 subunit d2 exhibit increased osteoblastic activity, reduced osteoclastic bone resorption, and increased bone bone mass10,11. Collagen triple helix repeat containing 1 (Cthrc1) produced by bone-resorbing mature osteoclasts is also reportedly required for bone formation12.
Transforming growth factor beta 1 (TGFβ1), a member of the TGFβ superfamily, also serves as a coupling factor. This ligand family consists of TGFβ1–5 as well as bone morphogenetic proteins (BMPs), however, TGFβ4 and TGFβ5 are expressed only in chick and Xenopus, respectively13,14,15,16,17. TGFβ ligands signal by binding to specific receptors and activating receptor-regulated transcription factors called Smads (R-Smads), namely Smad2/3 (for TGFβ1–5) and Smad1/5/8 (for BMPs), which form oligomers with the common mediator Smad4. Thus, loss of Smad4 results in loss of both TGFβs-Smad2/3 and BMPs-Smad1/5/8 signals18. Osteoblast-specific Smad4-deficient mice show altered bone formation19, while chondrocyte-specific Smad4-deficient mice reportedly exhibit dwarfism20; however, although bone tissues comprised of osteoclasts are rich in TGFβs and BMPs21,22, Smad4 function in those cells is largely unknown.
TGFβ1 and insulin like growth factor 1 (IGF1) reportedly accumulate in bone matrix and are released following osteoclastic bone resorption, stimulating osteoblastic bone formation23,24. TGFβ1 is initially produced in an inactive form (latent-TGFβ1), which is then activated extracellularly under highly acidic conditions25. Activated TGFβ1 reportedly promotes bone marrow stromal cell migration to enable bone formation25. Nonetheless, TGFβ1 function in osteoclasts is controversial: various investigators report that TGFβ1 exerts both stimulatory and inhibitory effects on osteoclast differentiation in vitro, the former of which was reported by majority as TGFβ1 action on osteoclasts26,27,28. Recently, osteoclast-specific ablation of TGFβ receptor 2 in mice was reportedly resulted in no significant impact on osteoclast numbers or activity in vivo29. BMP2 reportedly stimulates osteoclastogenesis30,31; however, osteoclast-specific conditional ablation of the gene encoding its receptor BMP receptor type 1A (BMPR1a) increases osteoclast differentiation in vivo32.
Osteoclast differentiation is regulated as a balance between stimulators and inhibitors33,34. RANKL-dependent factors that promote differentiation include Nuclear factor of activated T cells 1 (NFATc1), c-Fos and Blimp1, the latter encoded by the PR domain 1 (Prdm1)35,36,37,38. In contrast, interferon regulatory factor 8 (Irf8) and B cell lymphoma 6 (Bcl6) reportedly inhibit osteoclast differentiation, and are suppressed by RANKL stimulation in osteoclasts via Blimp1 activity38,39,40.
Here, we show that Smad4 is expressed in osteoclasts and report that osteoclast-specific Smad4 conditional knockout mice (Smad4-cKO: Cathepsin K (Ctsk)Cre/+/Smad4flox/flox) exhibit significantly reduced bone mass due to accelerated osteoclast formation. High TGFβ1 concentrations inhibited osteoclast differentiation of wild-type cells in vitro, and such inhibition was blocked in Smad4 cKO cells. We also show that TGFβ1 inhibits Prdm1 expression, which in turn upregulates Irf8 and Bcl6 expression, inhibiting osteoclast differentiation. Reduced bone mass and elevated osteoclastogenesis in Smad4-cKO were abrogated in Smad4/Blimp1 doubly mutant mice. Latent-TGFβ1 was converted to an active form by osteoclastic activity in cultured cells, and administration of latent-TGFβ1-Fc to wild-type mice blocked LPS-induced bone destruction. We conclude that following bone resorption, inhibition of osteoclastogenesis by activated TGFβ1 via Smad4 expressed in osteoclasts is crucial to maintain bone mass.
Results
Osteoclastogenesis is differentially regulated in osteoclast progenitor cells by high concentrations of TGFβ1 or TGFβ3 in vitro
To assess expression of TGFβ factors in bone, we undertook analysis of transcripts encoding these factors in bone tissues of wild-type mice and identified TGFβ1, 2, 3 and BMP2 mRNAs (Fig. 1a). We also found that osteoclastogenesis, as assessed by expression of osteoclastic genes such as Cathepsin K (Ctsk) and NFATc1 following RANKL treatment of cultured Raw264.7 cells, was enhanced by co-incubation of cells with RANKL plus either TGFβ1 or β3 (Supplementary Fig. 1). Osteoclast differentiation in bone marrow macrophages (BMMs) was also significantly inhibited by SB431542, a TGFβ inhibitor, in vitro (Supplementary Fig. 2a–c). Interestingly, in vitro osteoclastogenesis in wild-type BMMs was stimulated at a lower concentration (0.016 ng/ml) of either TGFβ1 or TGFβ3, while differentiation was significantly inhibited at higher concentrations (0.4, 2 or 10 ng/ml) of either TGFβ1 or TGFβ3 dose-dependently (Fig. 1b–d, Supplementary Fig. 3 and Supplementary Fig. 4). In contrast, osteoclast differentiation from wild-type BMMs was stimulated by high concentrations of either TGFβ2 (10 ng/ml) or BMP2 (200 ng/ml) (Fig. 1b,c). Osteoclastogenesis, as evidenced by appearance of multi-nuclear TRAP-positive cells, was stimulated by either 40 or 200 ng/ml BMP2 but inhibited by 1,000 ng/ml of BMP2 (Supplementary Fig. 5). These results suggest that osteoclastogenesis is regulated in a complex manner by TGFβ superfamily members in the bone microenvironment.

TGFβ1/β3 and BMP2/TGFβ2 have differential effects on osteoclastogenesis.
(a) Expression of TGFβ1–3 and BMP2 transcripts in mouse humerus bone was confirmed. Osteoclast progenitors from wild-type mice were cultured with recombinant BMP2 (200 ng/ml), TGFβ1 (10 ng/ml), TGFβ2 (10 ng/ml) or TGFβ3 (10 ng/ml) in the presence of M-CSF (50 ng/ml; M) and RANKL (25 ng/ml; R) for five days and then assessed for osteoclast formation by TRAP staining (b), by counting the number of multi-nuclear TRAP-positive cells (c) and by expression of the osteoclast markers Ctsk and NFATc1 based on realtime PCR (d). Data represent mean Ctsk or NFATc1 expression relative to β-actin ± SD (n = 3). Bar = 100 μm. *P < 0.05; ***P < 0.001; NS, not significant. Representative data of at least three independent experiments are shown.
Smad4 is required to inhibit osteoclastogenesis and maintain bone mass
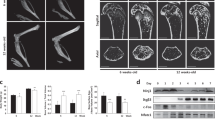
As noted, lack of Smad4 results in abrogation of both TGFβ and BMP signaling. We detected Smad4 expression in osteoclasts (Fig. 2a). To assess roles of Smad4 and downstream signaling in regulating osteoclastogenesis and bone mass in vivo, we generated osteoclast-specific Smad4 conditional knockout mice (Smad4 cKO) using Ctsk-Cre mice (Fig. 2b). Based on DEXA analysis, Smad4 cKO mice exhibited significantly reduced bone mass with accelerated osteoclastogenesis as analyzed by TRAP staining and bone morphometric analysis compared with controls in vivo (Fig. 2c–f). Osteoblastogenesis was normal in Smad4 cKO mice, while osteoclast formation was activated (Fig. 2d–f). Thus, reduced bone mass seen in Smad4 cKO mice is likely due to elevated osteoclastogenesis in vivo.

Smad4 is expressed in osteoclasts and required for osteoclast inhibition.
(a) Electrophoresis gel images of RT-PCR (left panels) or quantitative real-time PCR (right panel) analysis of Smad4 expression in macrophages (M), osteoclasts (OC) and osteoblasts (OB). β-actin served as an internal control. Data represent mean Smad4 expression relative to β-actin ± SD. (b) Western blotting in osteoclasts to assess Smad4 deletion efficiency in Smad4 cKO (cKO) compared with control (Smad4flox/flox) osteoclasts. (c) Bone mineral density (BMD) of femurs bisected equally longitudinally from control (Smad4flox/flox) and Smad4-cKO (cKO) mice. *P < 0.05; **P < 0.01; ***P < 0.001. (d) Bone histomorphometrical analysis of femurs from control and Smad4 cKO (cKO) female mice. Osteoblast surface per bone surface (Ob.S/BS) was determined. NS, not significant. Tibial sections from control (Smad4flox/flox or flox/+) and Smad4 cKO mice were stained with TRAP (e), and the number of TRAP-positive osteoclasts was scored (f). Bar = 100 μm. **P < 0.01. Representative data of at least two independent experiments are shown.
TGFβ1 and β3 inhibit osteoclast differentiation via Smad4
We next focused on identifying osteoclast-inhibiting signals mediated by Smad4 in vitro (Fig. 3). Osteoclastogenesis in wild-type BMMs as analyzed by formation of multi-nuclear TRAP-positive cells in vitro, was significantly inhibited in the presence of high concentrations of TGFβ1 or TGFβ3, and inhibition was significantly reversed in Smad4 cKO cells (Fig. 3a,b and Supplementary Fig. 6a,b). Expression of the osteoclast differentiation markers Ctsk and NFATc1 was significantly inhibited by treatment of wild-type osteoclasts with either TGFβ1 or TGFβ3 in vitro, an effect reversed in Smad4 cKO cells (Fig. 3c and Supplementary Fig. 6c).

High TGFβ1 concentrations inhibit osteoclastogenesis via Smad4 activity.
Osteoclast progenitors from control (Smad4flox/flox or flox/+) or Smad4-cKO (cKO) mice were cultured with or without 10 ng/ml TGFβ1 in the presence or absence of M-CSF (M) and RANKL (R). Osteoclast formation was evaluated by TRAP staining (a), by the number of multi-nuclear TRAP-positive cells (b) and by Ctsk and NFATc1 expression as analyzed by realtime PCR (c). Data represent mean Ctsk or NFATc1 expression relative to β-actin ± SD (n = 3). Bar = 100 μm. *P < 0.05; **P < 0.01; ***P < 0.001; NS, not significant. Representative data of at least three independent experiments are shown.
Smad4 regulates Bcl6 and Irf8 expression
To define molecular mechanisms underlying TGFβ inhibition of osteoclastogenesis through Smad4, we analyzed expression of potential inhibitory factors following treatment of wild-type osteoclasts with TGFβs. Candidates included Bcl6 and Irf8, both transcriptional repressors and reported inhibitors of osteoclastogenesis39,40. Bcl6 and Irf8 mRNA expression was upregulated following stimulation of wild-type osteoclasts with TGFβ1 (Fig. 4a). Interestingly, Bcl6 and Irf8 upregulation was significantly blocked in Smad4 cKO cells (Fig. 4b), suggesting that such upregulation is dependent on Smad4. Thus, next we treated Bcl6-deficient BMMs with TGFβ1 or β3 and found that their inhibition of osteoclast formation was abrogated relative to wild-type cells (Fig. 4c,d, and Supplementary Fig. 7a,b). Likewise, decreased expression of the osteoclastic genes Ctsk and NFATc1 seen following TGFβ1 or β3 treatment in wild-type osteoclasts was significantly rescued in Bcl6-deficient osteoclasts (Fig. 4e, Supplementary Fig. 7c). Similarly, Irf8-deficient cells were resistant to inhibition of osteoclastogenesis and suppression of osteoclastic gene expression by either TGFβ1 or β3 (Fig. 4f–h, Supplementary Fig. 7d–f).

TGFβ1 stimulates increases in Bcl6 and Irf8 expression in osteoclasts via Smad4 expressed.
(a) Osteoclast progenitors were isolated from wild-type mice and cultured in the presence or absence of M-CSF (M) and RANKL (R) with or without indicated concentrations of TGFβ1. Irf8 and Bcl6 expression was then determined by realtime PCR. Data represent mean Bcl6 or Irf8 expression relative to β-actin ± SD (n = 3). (b) Osteoclast progenitors were isolated from control (Smad4flox/flox or flox/+) or Smad4 cKO (cKO) mice and cultured in the presence or absence of RANKL with or without 10 ng/ml of TGFβ1. Irf8 and Bcl6 expression was determined by realtime PCR. Data represent mean Bcl6 or Irf8 expression relative to β-actin ± SD (n = 3). (c–h) Osteoclast progenitors were isolated from wild-type, Bcl6-deficient (c–e) or Irf8-deficient (f–h) mice and cultured in the presence or absence of M-CSF (M) and RANKL (R) with or without 10 ng/ml TGFβ1. Osteoclast formation was evaluated by TRAP staining (c,f), by the number of multi-nuclear TRAP-positive cells (d,g) and by Ctsk and NFATc1 expression as analyzed by realtime PCR (e,h). Irf8 and Bcl6 expression was determined in Bcl6 and Irf8-deficient mice, respectively, by realtime PCR (i). Data represent mean Ctsk or NFATc1 or expression relative to β-actin ± SD (n = 3). Bar = 100 μm. *P < 0.05; **P < 0.01; ***P < 0.001; NS, not significant. Representative data of at least two independent experiments are shown.
Interestingly, deficiency of either Bcl6 or Irf8 was sufficient to block TGFβ1- or β3-induced inhibition of osteoclastogenesis (Fig. 4a–h), and Irf8 or Bcl6 expression was significantly inhibited in Bcl6- or Irf8-deficient osteoclasts, respectively (Fig. 4i).
Blimp1 is a direct target of Smad4 in osteoclasts
Both Bcl6 and Irf8 expression in osteoclasts is reportedly negatively regulated by Blimp1, a transcriptional repressor encoded by Prdm139,40. Thus, we asked whether elevated Bcl6 and Irf8 expression seen following TGFβ1 or TGFβ3 treatment was accompanied by decreased Prdm1 expression. In accordance, Prdm1 mRNA expression was significantly inhibited by either TGFβ1 or TGFβ3 treatment of wild-type osteoclasts (Fig. 5a, Supplementary Fig. 8), but such Prdm1 inhibition was abrogated in Smad4 cKO cells (Fig. 5a, Supplementary Fig. 8). To assess whether Prdm1 is a direct target of Smad in osteoclasts, we employed chromatin immune precipitation sequencing (ChIP seq) analysis using anti-Smad2/3 antibodies, and observed that Smad2/3 bound to an upstream region of the Prdm1 gene in osteoclasts under TGFβ1 stimulation (Fig. 5b). When we generated osteoclast-specific Smad4/Prdm1 double knockout (DcKO: CtskCre/+/Smad4f/fPrdm1f/f) mice, in which Prdm1 is deleted from Smad4 cKO mice, we found that the significantly decreased bone mass seen in Smad4 cKO mice was reversed and rather increased in DcKO mice (Fig. 5c–e). These observations suggest that Smad4 is required for Prdm1 inhibition in osteoclasts and to maintain bone mass following stimulation with either TGFβ1 or TGFβ3.

Prdm1 deletion rescues bone loss in Smad4-cKO mice.
(a) Osteoclast progenitors from control (Smad4flox/flox or flox/+) or Smad4 cKO mice were cultured with or without 10 ng/ml TGFβ1 in the presence or absence of M-CSF and RANKL. Prdm1 expression was analyzed by realtime PCR. Data represent mean Prdm1 expression relative to β-actin ± SD (n = 3). (b) Osteclast progenitor cells from wild-type mice were cultured in the presence of M-CSF and RANKL with 10 ng/ml TGFβ1, and chromatin immune precipitation-sequencing analysis was preformed by using anti-Smad2/3 antibody. Smad2/3 bound to regions upstream of the Prdm1 gene. (c) Bone mineral density of femurs divided equally longitudinally from control, Smad4 cKO and Smad4/Prdm1 double-mutant (DcKO) female mice. (d,e) Tibial sections from control (Smad4flox/flox or flox/+), Smad4 cKO and DcKO mice were stained with TRAP (d), and the number of TRAP-positive osteoclasts was scored (e). Data represent mean number of TRAP-positive cells per section ± SD (n = 3). Bar = 100 μm. *P < 0.05; **P < 0.01; NS, not significant. Representative data of at least two independent experiments are shown.
We also found that Prdm1 expression was significantly upregulated by either TGFβ1 or TGFβ3 in Raw263.7 cells (Supplementary Fig. 9). Although, Bcl6 expression was rather upregulated, elevated Prdm1 expression may explain, at least in part, why osteoclastogenesis was stimulated in Raw264.7 cells by either TGFβ1 or TGFβ3. Osteoclast formation was stimulated by 200 ng/ml of BMP2, however, either Prdm1, Bcl6 or Irf8 expression level remained unchanged following BMP2 treatment of wild-type osteoclasts (Supplementary Fig. 10). Furthermore, Prdm1 expression was significantly inhibited, while Bcl6 and Irf8 expression was significantly upregulated in SB431542-treated wild-type osteoclasts (Supplementary Fig. 11).
Latent-TGFβ1 inhibits LPS-induced osteoclast formation and bone destruction
As reported, TGFβ1 is converted from a non-active, latent-TGFβ1 form to an activated form25. First, we established primary cultures of wild-type osteoclasts with or without latent-TGFβ1, and found that osteoclastogenesis in vitro was inhibited when wild-type BMMs were treated with active TGFβ1 but not by latent-TGFβ1 (Fig. 6a). Then, we treated wild-type BMMs with supernatants from primary cultures in the presence of M-CSF and RANKL (Fig. 6b). Latent-TGFβ1 is reportedly activated by osteoclastic bone-resorption25. In accordance, we found that osteoclastogenesis was inhibited in secondary cultures treated with supernatants from osteoclasts cultured with latent-TGFβ1 (Fig. 6b). Based on these results, we concluded that administered latent-TGFβ1 is converted to an active form by osteoclast to inhibit osteoclast formation. To test this hypothesis, we administered latent-TGFβ1-Fc or control CD4-Fc protein by injection in vivo in a mouse model of LPS-induced bone destruction, in which LPS was injected on wild-type mouse calvariae. We found that LPS-induced bone-resorption and osteoclast formation as analyzed by micro CT (μCT), and anti-Ctsk with anti-NFATc1 staining, respectively, were significantly inhibited by latent-TGFβ1-Fc compared with CD4-Fc administration (Fig. 6c–f). TGFβ1 signaling is known to promote differentiation of TH17 cells, a type of osteoclastogenic T cells implicated in bone destruction41,42. Indeed, in an LPS-induced model of bone destruction, we found that TH17 cell frequency significantly increased in mice treated with LPS together with latent-TGFβ1-Fc compared with control mice treated with PBS plus latent-TGFβ1-Fc (Fig. 6g). The fact that bone destruction was inhibited by latent-TGFβ1-Fc, even under elevated TH17 cell conditions, suggests that latent-TGFβ1-Fc could antagonize bone destruction in osteoclast-activating conditions.

Latent TGFβ1 is converted to an active form by osteoclastic activity.
(a) Osteoclast progenitors from wild-type mice were cultured in the presence of M-CSF and RANKL with or without either active- or latent-TGFβ1 (10 ng/ml each) for primary culture. Quantitative real-time PCR analysis of osteoclastic mRNAs was then undertaken. Data represent mean Ctsk or NFATc1 expression relative to β-actin ± SD (n = 3). (b) Primary culture supernatants were collected from wild-type cells and transferred to secondary cultures of wild-type osteoclast progenitors, which were then treated with M-CSF and RANKL, and Ctsk and NFATc1 expression was analyzed by realtime PCR. Data represent mean Ctsk or NFATc1 expression relative to β-actin ± SD (n = 3). (c–g) LPS (50 mg/kg) was administered subcutaneously onto the skull of living 8-week-old female wild-type mice with or without 16 mg of latent-TGFβ1. Five days later, osteolysis in calvariae was analyzed by μCT (c, low magnification; d, high magnification). PBS injection served as a negative control. The number of resorption pits per calvariae was scored. (e). Data represent mean resorption pit number per calvariae ± SD (n = 5). Sections were stained with mouse anti-Ctsk and rabbit anti-NFATc1 antibodies, followed by Alexa488-conjugated goat anti-mouse Ig’ antibody, Alexa488-conjugated goat anti-rabbit Ig’ antibody and DAPI. Sections were then observed by fluorescence microscopy (f). Spleen cells were stained with anti-CD4 and anti-IL-17 antibodies, and the frequency of TH17 cells (CD4+IL-17+ cells) was analyzed by flow cytometry (g). Data represent mean TH17 cell frequency ± SD (n = 5). Bar = 100 μm. *P < 0.05; NS, not significant. Representative data of at least two independent experiments are shown.
Discussion
Numerous bone-regulating factors maintain bone homeostasis1,43. Among them, factors activating signals via Smad4, including TGFβ and BMP, reportedly support osteoblastic cell migration, proliferation, differentiation and bone formation in vivo and in vitro (Supplementary Fig. 12a)19,44. This study demonstrates that Smad4 mediates osteoprotective signals that are coupled with osteoclastic bone resorption and acts as part of a negative feedback mechanism (Supplementary Fig. 12b,c). Our findings suggest overall that Smad4 plays a role in both inhibiting bone resorption and activating bone formation (Supplementary Fig. 12). Here, we show that latent-TGFβ is activated by osteoclasts, which inhibits their activity (Supplementary Fig. 12b).
The activity of TGFβ superfamily members in osteoclasts reportedly varies26,27,28,29,32, and we show that TGFβ1/β3 inhibits osteoclastogenesis, while TGFβ2/BMP2 stimulates it. However, the significant reduction in bone mass and elevated osteoclast formation we report here in Smad4 cKO mice suggests that in this system inhibitory signals via Smad4 are dominant over stimulators. Since Smad4 null mice exhibit embryonic lethality45, Smad4 function in osteoclasts and bones has not previously been characterized. The Cre/loxP system employed here did not completely abrogate Smad4 activity in osteoclasts, and some Smad4 function may remain. Nonetheless, it allowed us evaluate Smad4 function in osteoclastogenesis and bone at adult stages. Those signals via TGFβ result from conversion of latent-TGFβ to TGFβ1, which in turn blocks expression of Prdm1, a repressor of osteoclastogenesis. Loss of the repressor encoded by Prdm1 upregulates Bcl6 and Irf8, both of which repress osteoclast differentiation (Supplementary Fig. 12c). Although, at present, molecular mechanisms underlying are not clear, we found that Irf8 or Bcl6 expression was significantly inhibited in Bcl6- or Irf8-deficient osteoclasts, respectively (Fig. 4i), suggesting that these factors regulate each other in osteoclasts.
TGFβ and BMP signaling is regulated in a complex manner in osteoblasts44. Indeed, TGFβ1 is reportedly required for osteoblastogenesis19,44, while it is also reported to inhibit osteoblastogenesis induced by BMP246. However, there is net decrease in bone mass seen in osteoblast-specific Smad4-deficient mice19, suggesting that Smad4 signals in osteoblasts positively regulate bone formation. Thus overall, although why high concentration of BMP2 (1,000 ng/ml) inhibited osteoclast formation was not clear, Smad4 signaling in both osteoclasts and osteoblasts results in increases in bone mass.
Recent advances in developing anti-osteoporosis drugs have resulted in both anti-resorptive agents such as bisphosphonate or anti-RANKL antibodies, and bone-forming drugs, such as teriparatide47,48,49,50. Both types have significant therapeutic effects in increasing bone mass and preventing fractures in osteoporosis patients51. However, the broad effects of anti-resorptive or bone-forming agents in inhibiting or promoting osteoclast differentiation/function, respectively, can cause adverse side effects such as jaw osteonecrosis, super suppressive bone turnover or osteosarcoma formation4,52. As alternatives, investigators are currently seeking novel reagents targeting specific sites where bone formation is required following resorption. Our data strongly suggests that the TGFβ/Smad4 system is specifically activated at such sites. Our observations therefore provide a molecular basis for developing agents that both inhibit bone-resorption and activate bone-formation.
Methods
Mice
Wild-type mice were purchased from Sankyo Labo Service (Tokyo, Japan). Ctskcre/+, Smad4f/f, Prdm1f/f, Bcl6-deficient and Irf8-deficient mice were prepared as previously described39,40,53,54. Animals were maintained under specific pathogen-free conditions in animal facilities certified by the Keio University Institutional Animal Care and Use Committee, and animal protocols were approved by that committee. All animal studies were performed in accordance with the Guidelines of the Keio University animal care committee.
Analysis of skeletal morphology
Ctskcre/+Smad4f/f, Ctskcre/+Smad4f/fPrdm1f/f and control littermates were necropsied, and their hind limbs were removed, fixed in 70% ethanol, and subjected to DEXA analysis to measure bone mineral density, and analysis of bone histomorphometric parameters. Bones were collected from 8-week-old female mice.
In vitro osteoclast formation
For in vitro analysis, bone marrow cells isolated from femurs and tibias were cultured for 72 h in MEM (Sigma-Aldrich Co.) containing 10% (vol/vol) heat-inactivated FBS (JRH Biosciences) and GlutaMax (Invitrogen Corp.) supplemented with M-CSF (50 ng/mL, Kyowa Hakko Kirin Co.). Subsequently, adherent cells were collected and cultured in 96-well plates (1 × 105 cells per well) under indicated conditions containing M-CSF (50 ng/mL) and recombinant soluble RANKL (25 ng/mL, PeproTech Ltd.) with or without latent-TGFβ1 (10 ng/ml, R & D Systems), TGFβ1 (0.016–10 ng/ml, R & D Systems), TGFβ3 (0.016–10 ng/ml, R & D Systems) or BMP2 (40–1,000 ng/ml, Pepro Tech Ltd.). Medium was changed every 2 days. Osteoclastogenesis was evaluated by TRAP staining, and TRAP-positive multi-nuclear cells containing more than three nuclei were scored as osteoclasts.
For some experiments, supernatants from osteoclast culture for five days with or without latent-TGFβ1-Fc (10 μg/ml, R & D Systems) were added to secondary cultures, and osteoclastogenesis was evaluated by TRAP staining or expression of osteoclastic genes.
Quantitative PCR analysis
Total RNAs were isolated from bone marrow cultures using TRIzol reagent (Invitrogen Corp.), and cDNA synthesis was performed using oligo(dT) primers and reverse transcriptase (Wako Pure Chemicals Industries). Quantitative PCR was performed using SYBR Premix ExTaq II reagent and a DICE Thermal cycler (Takara Bio Inc.), according to the manufacturer’s instructions. β-actin (Actb) expression served as an internal control. Primers used for realtime PCR analysis were as follows.
β-actin-forward: 5′-TGAGAGGGAAATCGTGCGTGAC-3′
β-actin-reverse: 5′-AAGAAGGAAGGCTGGAAAAGAG-3′
Ctsk-forward: 5′-ACGGAGGCATTGACTCTGAAGATG-3′
Ctsk-reverse: 5′-GGAAGCACCAACGAGAGGAGAAAT-3′
NFATc1-forward: 5′-CAAGTCTCACCACAGGGCTCACTA-3′
NFATc1-reverse: 5′-GCGTGAGAGGTTCATTCTCCAAGT-3′
Smad4-forward: 5′-TATCACTATGAGCGGGTTGTCTCA-3′
Smad4-reverse: 5′-TCAAAATCTGGGCTCTTGTTCAG-3′
Prdm1-forward: 5′-TTCTTGTGTGGTATTGTCGGGACTT-3′
Prdm1-reverse: 5′-TTGGGGACACTCTTTGGGTAGAGTT-3′
Bcl6-forward: 5′-AGACGCACAGTGACAAACCATACAA-3′
Bcl6-reverse: 5′-GCTCCACAAATGTTACAGCGATAGG-3′
Irf8-forward: 5′-CAGGATTACAATCAGGAGGTGGA-3′
Irf8-reverse: 5′-AATCGAATGTCCTTCAGTGGGTAA-3′
BMP2-forward: 5′-CTAGATCTGTACCGCAGGCACT-3′
BMP2-reverse: 5′-TTTTCCCACTCATCTCTGGAAG-3′
TGFβ1-forward: 5′-GACCCTGGATACCAACTATTGC-3′
TGFβ1-reverse: 5′-CAGACAGAAGTTGCCATGGTAGC-3′
TGFβ2-forward: 5′-ATGAACCCAAAGGGTACAATGCT-3′
TGFβ2-reverse: 5′-AGCTTCGGGATTTATGGTGTTGT-3′
TGFβ3-forward: 5′-CCCTGGACACCAATTACTGCTTC-3′
TGFβ3-reverse: 5′-GCCTGAGCAGAAGTTGGCATAGT-3′
Western blot analysis
Whole cell lysates were prepared from 8-week-old Smad4f/f (control) or Ctskcre/+Smad4f/f mice bone marrow cultures using RIPA buffer (1% Tween 20, 0.1% SDS, 150 mM NaCl, 10 mM Tris-HCl (pH 7.4), 0.25 mM phenylmethylsulfonylfluoride, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mM Na3VO4, 5 mM NaF (Sigma-Aldrich Co.)). Equivalent amounts of protein were separated by SDS-PAGE and transferred to a PVDF membrane (EMD Millipore Corp.). Proteins were detected by using anti-Smad4 (9515, Cell Signaling) or anti-Actin (Sigma-Aldrich Co., St Louis, MO) antibody.
Chromatin immune precipitation sequence (ChIP seq) assay
Osteoclasts cultured with M-CSF + RANKL + TGFβ1 were harvested and ChIP-seq assay performed using anti-Smad2/3 antibody (BD biosciences, San Jose, CA, USA) as described37.
In vivo osteolysis model
100 μl of PBS containing LPS (50 mg/kg) was injected with or without latent-TGFβ1-Fc onto the periosteal surface of calvariae in living 8-week old wild-type mice. Five days later, mice were euthanized, and calvariae and spleen were harvested for micro-computed tomography (micro-CT) and flow cytometry, respectively. Micro-CT was performed using a (micro-CT) scan R_mCT2 system (Rigaku Corp., Tokyo, Japan). For flow cytometry, spleen cells were stained with anti-CD4 and anti-IL-17 antibodies, and analyzed by FACSCanto™ II (BD Biosciences, San Jose, CA, USA) as described55. Spleen cells were collected from each group.
Immunofluorescent staining
Surgical sections of calvaria were stained with mouse anti-Cathepsin K (Ctsk) (1:100 Daiichi Finechemical Co., Toyama, Japan) and anti-NFATc1 (NFATc1) (1:00 Santa Cruz Biotechnology) followed by Alexa488-conjugated goat anti-mouse Ig’ (1:200; Invitrogen, Carlsbad, CA). DAPI (1:750; Wako Pure Chemicals Industries, Osaka, Japan) was used for a nuclear stain.
Statistical analysis
Results are expressed as the mean ± s.d. Differences between groups were examined for statistical significance using Student t test.
Additional Information
How to cite this article: Morita, M. et al. Smad4 is required to inhibit osteoclastogenesis and maintain bone mass. Sci. Rep. 6, 35221; doi: 10.1038/srep35221 (2016).
References
Sims, N. A. & Martin, T. J. Coupling the activities of bone formation and resorption: a multitude of signals within the basic multicellular unit. Bonekey Rep 3, 481, doi: 10.1038/bonekey.2013.215 (2014).
Hattner, R., Epker, B. N. & Frost, H. M. Suggested sequential mode of control of changes in cell behaviour in adult bone remodelling. Nature 206, 489–490 (1965).
Zaidi, M. et al. Bone loss or lost bone: rationale and recommendations for the diagnosis and treatment of early postmenopausal bone loss. Curr Osteoporos Rep 7, 118–126 (2009).
Kuhl, S., Walter, C., Acham, S., Pfeffer, R. & Lambrecht, J. T. Bisphosphonate-related osteonecrosis of the jaws–a review. Oral Oncol 48, 938–947, doi: 10.1016/j.oraloncology.2012.03.028 (2012).
Matsuo, K. & Irie, N. Osteoclast-osteoblast communication. Arch Biochem Biophys 473, 201–209, doi: 10.1016/j.abb.2008.03.027 (2008).
Zhao, C. et al. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab 4, 111–121, doi: 10.1016/j.cmet.2006.05.012 (2006).
Irie, N. et al. Bidirectional signaling through ephrinA2-EphA2 enhances osteoclastogenesis and suppresses osteoblastogenesis. J Biol Chem 284, 14637–14644, doi: 10.1074/jbc.M807598200 (2009).
Negishi-Koga, T. et al. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat Med 17, 1473–1480, doi: 10.1038/nm.2489 (2011).
Hayashi, M. et al. Osteoprotection by semaphorin 3A. Nature 485, 69–74, doi: 10.1038/nature11000 (2012).
Lee, S. H. et al. v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat Med 12, 1403–1409, doi: 10.1038/nm1514 (2006).
Xu, J., Cheng, T., Feng, H. T., Pavlos, N. J. & Zheng, M. H. Structure and function of V-ATPases in osteoclasts: potential therapeutic targets for the treatment of osteolysis. Histol Histopathol 22, 443–454 (2007).
Takeshita, S. et al. Osteoclast-secreted CTHRC1 in the coupling of bone resorption to formation. J Clin Invest 123, 3914–3924, doi: 10.1172/JCI69493 (2013).
Assoian, R. K., Komoriya, A., Meyers, C. A., Miller, D. M. & Sporn, M. B. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J Biol Chem 258, 7155–7160 (1983).
Wrann, M. et al. T cell suppressor factor from human glioblastoma cells is a 12.5-kd protein closely related to transforming growth factor-beta. EMBO J 6, 1633–1636 (1987).
Derynck, R. et al. A new type of transforming growth factor-beta, TGF-beta 3. EMBO J 7, 3737–3743 (1988).
Jakowlew, S. B., Dillard, P. J., Sporn, M. B. & Roberts, A. B. Nucleotide sequence of chicken transforming growth factor-beta 1 (TGF-beta 1). Nucleic Acids Res 16, 8730 (1988).
Roberts, A. B. et al. Mesoderm induction in Xenopus laevis distinguishes between the various TGF-beta isoforms. Growth Factors 3, 277–286 (1990).
Derynck, R. & Zhang, Y. E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584, doi: 10.1038/nature02006 (2003).
Tan, X. et al. Smad4 is required for maintaining normal murine postnatal bone homeostasis. J Cell Sci 120, 2162–2170, doi: 10.1242/jcs.03466 (2007).
Zhang, J. et al. Smad4 is required for the normal organization of the cartilage growth plate. Dev Biol 284, 311–322, doi: 10.1016/j.ydbio.2005.05.036 (2005).
Canalis, E., McCarthy, T. & Centrella, M. Growth factors and the regulation of bone remodeling. J Clin Invest 81, 277–281, doi: 10.1172/JCI113318 (1988).
Urist, M. R. Bone: formation by autoinduction. Science 150, 893–899 (1965).
Hayden, J. M., Mohan, S. & Baylink, D. J. The insulin-like growth factor system and the coupling of formation to resorption. Bone 17, 93S–98S (1995).
Xian, L. et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat Med 18, 1095–1101, doi: 10.1038/nm.2793 (2012).
Tang, Y. et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med 15, 757–765, doi: 10.1038/nm.1979 (2009).
Takai, H. et al. Transforming growth factor-beta stimulates the production of osteoprotegerin/osteoclastogenesis inhibitory factor by bone marrow stromal cells. J Biol Chem 273, 27091–27096 (1998).
Fuller, K., Lean, J. M., Bayley, K. E., Wani, M. R. & Chambers, T. J. A role for TGFbeta(1) in osteoclast differentiation and survival. J Cell Sci 113 (Pt 13), 2445–2453 (2000).
Quinn, J. M. et al. Transforming growth factor beta affects osteoclast differentiation via direct and indirect actions. J Bone Miner Res 16, 1787–1794, doi: 10.1359/jbmr.2001.16.10.1787 (2001).
Weivoda, M. M. et al. Osteoclast TGF-beta Receptor Signaling Induces Wnt1 Secretion and Couples Bone Resorption to Bone Formation. J Bone Miner Res 31, 76–85, doi: 10.1002/jbmr.2586 (2016).
Itoh, K. et al. Bone morphogenetic protein 2 stimulates osteoclast differentiation and survival supported by receptor activator of nuclear factor-kappaB ligand. Endocrinology 142, 3656–3662, doi: 10.1210/endo.142.8.8300 (2001).
Sotillo Rodriguez, J. E. et al. Enhanced osteoclastogenesis causes osteopenia in twisted gastrulation-deficient mice through increased BMP signaling. J Bone Miner Res 24, 1917–1926, doi: 10.1359/jbmr.090507 (2009).
Okamoto, M. et al. Conditional deletion of Bmpr1a in differentiated osteoclasts increases osteoblastic bone formation, increasing volume of remodeling bone in mice. J Bone Miner Res 26, 2511–2522, doi: 10.1002/jbmr.477 (2011).
Kim, J. H. & Kim, N. Signaling Pathways in Osteoclast Differentiation. Chonnam Med J 52, 12–17, doi: 10.4068/cmj.2016.52.1.12 (2016).
Teitelbaum, S. L. & Ross, F. P. Genetic regulation of osteoclast development and function. Nat Rev Genet 4, 638–649, doi: 10.1038/nrg1122 (2003).
Takayanagi, H. et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 3, 889–901 (2002).
Ishida, N. et al. Large scale gene expression analysis of osteoclastogenesis in vitro and elucidation of NFAT2 as a key regulator. J Biol Chem 277, 41147–41156, doi: 10.1074/jbc.M205063200 (2002).
Omata, Y. et al. Genomewide comprehensive analysis reveals critical cooperation between Smad and c-Fos in RANKL-induced osteoclastogenesis. J Bone Miner Res 30, 869–877, doi: 10.1002/jbmr.2418 (2015).
Nishikawa, K. et al. Blimp1-mediated repression of negative regulators is required for osteoclast differentiation. Proc Natl Acad Sci USA 107, 3117–3122, doi: 10.1073/pnas.0912779107 (2010).
Miyauchi, Y. et al. The Blimp1-Bcl6 axis is critical to regulate osteoclast differentiation and bone homeostasis. J Exp Med 207, 751–762, doi: 10.1084/jem.20091957 (2010).
Zhao, B. et al. Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat Med 15, 1066–1071, doi: 10.1038/nm.2007 (2009).
Mangan, P. R. et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441, 231–234, doi: 10.1038/nature04754 (2006).
Veldhoen, M., Hocking, R. J., Atkins, C. J., Locksley, R. M. & Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189, doi: 10.1016/j.immuni.2006.01.001 (2006).
Canalis, E., McCarthy, T. L. & Centrella, M. Growth factors and cytokines in bone cell metabolism. Annu Rev Med 42, 17–24, doi: 10.1146/annurev.me.42.020191.000313 (1991).
Chen, G., Deng, C. & Li, Y. P. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci 8, 272–288, doi: 10.7150/ijbs.2929 (2012).
Sirard, C. et al. The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev 12, 107–119 (1998).
Yoshida, S. et al. PDGFBB promotes PDGFRalpha-positive cell migration into artificial bone in vivo. Biochem Biophys Res Commun 421, 785–789, doi: 10.1016/j.bbrc.2012.04.084 (2012).
Jilka, R. L. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone 40, 1434–1446, doi: 10.1016/j.bone.2007.03.017 (2007).
McClung, M. R. et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med 370, 412–420, doi: 10.1056/NEJMoa1305224 (2014).
Teitelbaum, S. L. Bone: the conundrum of glucocorticoid-induced osteoporosis. Nat Rev Endocrinol 8, 451–452, doi: 10.1038/nrendo.2012.89 (2012).
Kumagai, Y., Hasunuma, T. & Padhi, D. A randomized, double-blind, placebo-controlled, single-dose study to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of denosumab administered subcutaneously to postmenopausal Japanese women. Bone 49, 1101–1107, doi: 10.1016/j.bone.2011.08.007 (2011).
Das, S. & Crockett, J. C. Osteoporosis - a current view of pharmacological prevention and treatment. Drug Des Devel Ther 7, 435–448, doi: 10.2147/DDDT.S31504 (2013).
Watanabe, A. et al. Osteosarcoma in Sprague-Dawley rats after long-term treatment with teriparatide (human parathyroid hormone (1–34)). J Toxicol Sci 37, 617–629 (2012).
Yang, X., Li, C., Herrera, P. L. & Deng, C. X. Generation of Smad4/Dpc4 conditional knockout mice. Genesis 32, 80–81 (2002).
Nakamura, T. et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell 130, 811–823, doi: 10.1016/j.cell.2007.07.025 (2007).
Kashiwagi, I. et al. Smad2 and Smad3 Inversely Regulate TGF-beta Autoinduction in Clostridium butyricum-Activated Dendritic Cells. Immunity 43, 65–79, doi: 10.1016/j.immuni.2015.06.010 (2015).
Acknowledgements
Prdm1 flox mice were kindly provided by Dr. A. Sakamoto and Dr. T. Tokuhisa. CtskCre/+ mice were provided by Dr. S. Kato. T. Miyamoto was supported by a grant-in-aid for Scientific Research in Japan and a grant from Japan Agency for Medical Research and Development. Y. Sato and K. Miyamoto were supported by a grant-in-aid for Scientific Research in Japan. This study was supported in part by a Grant-in-aid for Scientific Research and a grant from the Translational Research Network Program.
Author information
Authors and Affiliations
Contributions
Mayu Morita, S.Y., R.I. and Y.S. performed culture and animal experiments. A.Y. performed FACS experiments. T.Y., S.T. and H.A. performed Chip sequence experiments. T.K., M.T., K.O. and C.-X.D. prepared animals for experiments. R.W., T.O. and K.M. analyzed data. Y.T., Morio Matsumoto, M.N., H.K., T.N. and T.M. designed the study. T.M. wrote the manuscript with input from all authors. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Morita, M., Yoshida, S., Iwasaki, R. et al. Smad4 is required to inhibit osteoclastogenesis and maintain bone mass. Sci Rep 6, 35221 (2016). https://doi.org/10.1038/srep35221
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35221
- Springer Nature Limited
This article is cited by
-
The roles and regulatory mechanisms of TGF-β and BMP signaling in bone and cartilage development, homeostasis and disease
Cell Research (2024)
-
TGFβ reprograms TNF stimulation of macrophages towards a non-canonical pathway driving inflammatory osteoclastogenesis
Nature Communications (2022)
-
Transcriptomic Data Identified Key Transcription Factors for Osteoporosis in Caucasian Women
Calcified Tissue International (2018)
-
The nicotinic acetylcholine receptor α7 subunit is an essential negative regulator of bone mass
Scientific Reports (2017)