
Abstract
Loss of airway epithelial integrity contributes significantly to asthma pathogenesis. Thymic stromal lymphopoietin (TSLP) may have dual immunoregulatory roles. In inflammatory disorders of the bowel, the long isoform of TSLP (lfTSLP) promotes inflammation while the short isoform (sfTSLP) inhibits inflammation. We hypothesize that lfTSLP contributes to house dust mite (HDM)-induced airway epithelial barrier dysfunction and that synthetic sfTSLP can prevent these effects. In vitro, airway epithelial barrier function was assessed by monitoring transepithelial electrical resistance, fluorescent-dextran permeability, and distribution of E-cadherin and β-catenin. In vivo, BALB/c mice were exposed to HDM by nasal inhalation for 5 consecutive days per week to establish an asthma model. sfTSLP and 1α,25-Dihydroxyvitamin D3 (1,25D3) were administered 1 h before HDM exposure. After 8 weeks, animal lung function tests and pathological staining were performed to evaluate asthma progression. We found that HDM and lfTSLP impaired barrier function. Treatment with sfTSLP and 1,25D3 prevented HDM-induced airway epithelial barrier disruption. Moreover, sfTSLP and 1,25D3 treatment ameliorated HDM-induced asthma in mice. Our data emphasize the importance of the different expression patterns and biological properties of sfTSLP and lfTSLP. Moreover, our results indicate that sfTSLP and 1,25D3 may serve as novel therapeutic agents for individualized treatment of asthma.
Similar content being viewed by others


Introduction
Asthma is a chronic inflammatory disease of the conducting airways in which many cells of the innate and adaptive immune systems act along with epithelial cells to cause airway inflammation, mucus overproduction, bronchial hyper-reactivity, and airway narrowing1,2. Airway epithelium has typically been thought to function mainly as the first defensive barrier by impeding the access of allergens3. The integrity of the epithelial barrier depends on apical tight junctions composed of zonula occludens 1-3, occludin, and claudin 1-5, and on adherens junctions, which consist of E-cadherin, β-catenin, and junctional adhesion molecule, that keep bronchial epithelial cells together and maintain their apicobasal polarity4. E-cadherin is regarded as the ‘gatekeeper’ in the airway mucosa and in allergic sensitization due to its key role in suppressing the production of allergenic mediators and promoting the establishment of tolerance5. Structural and functional abnormalities of E-cadherin may lead to enhanced signaling between the epithelium and the underlying immune and structural cells.
Thymic stromal lymphopoietin (TSLP) is a novel interleukin (IL)-7–like cytokine, initially isolated from a murine thymic stromal cell line Z210R.1 and described to be a lymphocyte growth factor6. During allergic inflammation, TSLP is produced by various cell types, including epithelial cells, epidermal keratinocytes, dendritic cells (DCs), and mast cells7. TSLP may promote immune responses that can be protective or detrimental to the host. On the one hand, TSLP has been connected with protective functions. For example, DCs produce TSLP that acts directly on T-cells by controlling Th17 cell differentiation, fosters regulatory T-cell development, and protects against colitis8. Nasal epithelial-derived TSLP can also preserve the mucosal barrier via the upregulation of tight-junction proteins9. The C-terminal antimicrobial region of TSLP exerts potent antimicrobial effects10. Furthermore, TSLP can regulate the capacity of tolerogenic DCs to drive the differentiation of natural regulatory T cells both in the intestine and thymus11,12. In contrast to these protective effects, TSLP overexpression can be detected in airway epithelia of asthmatic patients and in mice with asthma13,14,15 and is correlated with the severity of asthma16. In addition, TSLP-activated DCs can create a Th2-permissive microenvironment and induce inflammatory Th2 cells that produce the classical Th2 cytokines IL-4, IL-5, and IL-13, and large amounts of tumor necrosis factor (TNF)-α17,18.
TSLP is present in 2 distinct isoforms, short- and long-form TSLP (hereafter called sfTSLP and lfTSLP, respectively). LfTSLP has 159 amino acids and sfTSLP encompasses the last 63 residues of lfTSLP and is identical to its C-terminal portion19. In normal human bronchial epithelial cells, two TSLP splice variants have been reported and lfTSLP, but not sfTSLP, was highly induced after exposure to polyinosinic-polycytidylic acid (polyI:C)20. Moreover, in primary human keratinocytes, toll-like receptor ligands, a pro-inflammatory cytokine (TNF-α), and Th2 cytokines (IL-4 and IL-13) predominantly upregulate gene expression of lfTSLP, but not sfTSLP21. Another study found that synthetic sfTSLP exerts potent antimicrobial activity and that sfTSLP is the predominant form of TSLP, being the form widely and constitutively expressed in keratinocytes and saliva at steady state, both at the mRNA and protein levels22. In inflammatory disorders of the bowel, lfTSLP was shown to promote inflammation and is only expressed during inflammation, while sfTSLP is expressed under steady-state conditions and inhibits inflammation19.
In this study, we evaluated the expression of the two TSLP isoforms. Our data showed that lfTSLP is upregulated at the mRNA and protein levels, while sfTSLP is only slightly upregulated, in response to HDM in 16HBE cells. We also found that only lfTSLP contributes to HDM-induced airway epithelial barrier dysfunction and that synthetic sfTSLP prevents this HDM-induced disruption.
Results
Effects of HDM on TSLP expression and the barrier integrity of 16HBE cells
A previous study demonstrated that the expression of lfTSLP, which is a crucial cytokine for the induction of inflammatory Th2 responses, is highly induced by dsRNA in bronchial epithelial cells20. Initially, we found a significant upregulation of lfTSLP at the protein (Fig. 1b,c) and mRNA (Fig. 1d,e) levels. Unexpectedly, the mRNA expression of sfTSLP was scarcely induced (Fig. 1d,e) after stimulation with HDM in 16HBE cells. Next, we examined the effects of HDM on barrier permeability in monolayers of 16HBE cells. In the treated 16HBE cells, HDM exposure resulted in TER reduction (Fig. 1f), while the permeability increased (Fig. 1g) in a time-dependent manner. The expression of E-cadherin and β-catenin proteins was not altered (Fig. 1a,c).

Effects of HDM on the TSLP isoforms expression and the barrier integrity of 16HBE cells.
(a) The expression of E-cadherin and β-catenin was assessed using western blotting in 16HBE cells stimulated with HDM (400 U) for the indicated times. (b) Western blotting was used to detect lfTSLP protein in the cells and the culture medium of 16HBE cells stimulated with HDM for the indicated times. (c) Western blotting analysis of E-cadherin, β-catenin, and lfTSLP proteins in 16HBE cells stimulated with different concentrations of HDM for 6 h. (d) RT-PCR assay of the long and short splice forms of TSLP mRNA in 16HBE cells stimulated with HDM (400 U) for the indicated times. (e) RT-PCR assay of the long and short splice forms of TSLP mRNA in 16HBE cells stimulated with the indicated doses of HDM for 4 h. (f) Transepithelial electrical resistance (TER) was measured to determine epithelial barrier integrity after the cells were treated with HDM (400 U) for the indicated times. (g) The FITC-Dx permeability was measured to determine epithelial barrier integrity after the cells were treated with HDM (400 U) for the indicated times. Data are presented as the mean ± SD; n = 3–4.*P < 0.05 vs. the control group.
1,25D3 upregulates sfTSLP expression and inhibits HDM-induced barrier disruption in 16HBE cells
Recent studies show that 1,25D3 plays a positive role in the airway epithelia barrier function23,24. Therefore, we assessed the effects of 1,25D3 in HDM-treated 16HBE cells. The 16HBE cells were treated with culture medium (control group), 1,25D3 (10 nM)24, or HDM (400 U), with or without 1,25D3 for 1 h. We found that 1,25D3 upregulated sfTSLP expression and inhibited HDM-dependent upregulation of lfTSLP (Fig. 2a,b) and that 1,25D3 treatment significantly reversed the effects of HDM, including the decrease in TER (Fig. 2c), increase in fluorescein isothiocyanate (FITC) - dextran (Dx) permeability (Fig. 2d), and the delocalization of E-cadherin (Fig. 2e) and β-catenin (Fig. 2f).

Effect 1,25D3 on the HDM-induced TSLP isoforms expression and the HDM-induced barrier disruption.
16HBE cells were treated with culture medium (control group), 1,25D3 (10 nM), or HDM (400 U) with or without 1,25D3 for 1 h prior to stimulation with HDM. (a) RT-PCR assay of the long and short splice forms of TSLP mRNA in the treated cells. (b) The expression of E-cadherin, β-catenin, and lfTSLP proteins was assessed using western blotting. (c) TER and (d) FITC-Dx permeability was measured in the treated cells. (e) The distribution of E-cadherin was monitored using immunofluorescence. Green represents E-cadherin, blue represents the nucleus. (f) The distribution of β-catenin was monitored using immunofluorescence. Red represents β-catenin, blue represents the nucleus. Data are presented as the mean ± SD; n = 3–4. *P < 0.05 the HDM group vs. the control group; #P < 0.05 the 1,25D3+HDM group vs. the HDM group; **P < 0.05 the 1,25D3 group vs. the control group.
Mediation of HDM-induced lfTSLP upregulation by the mitogen-activated protein kinase (MAPK) signaling cascade
MAPKs are involved in diverse cell-mediated initiation by various stimuli, such as pro-inflammatory cytokines and growth factors25. In our study, HDM induced a rapid and marked phosphorylation of ERK1/2 and p38, but not c-Jun N-terminal kinase (JNK). Western blotting analysis showed that phosphorylation of ERK1/2 and p38 reached a maximum at 15–30 min after HDM exposure (Fig. 3a,b). These data indicate that HDM activates the ERK1/2 and p38 signaling pathways in 16HBE cells. To investigate the role of MAPK signaling pathway components in HDM-induced lfTSLP expression, we used SB203580 and U0126, specific and potent inhibitors of p38 and p42/p44 ERK, respectively. Treatment of 16HBE cells with SB203580 or U0126 1 h before stimulation with HDM led to a significant inhibition of lfTSLP expression (Fig. 3c,d). Taken together, these results indicate that activation of the MAPK pathway (ERK1/2 and p38) is essential for HDM-mediated lfTSLP expression in16HBE cells.

HDM-induced lfTSLP upregulation is mediated by the MAPK signaling cascade in 16HBE cells.
(a,b) Activation of three MAPK pathway proteins, (phosphorylated [p]-MAPK/total [t]-MAPK], p-ERK:t-ERK, p-p38:t-p38, and p-c-Jun N-terminal kinase (JNK):t-JNK was measured by western blotting and densitometric analyses in 16HBE cells stimulated with HDM (400 U) for the indicated times. (c) 16HBE cells were respectively treated with culture medium (control group), U0126 (10 μM), or HDM (400U) with or without U0126 (10 μM) for 1 h prior to stimulation with HDM. The expression of p-ERK, t-ERK, and lfTSLP proteins was evaluated using western blotting analysis in the treated cells. (d) 16HBE cells were respectively treated with culture medium (control group), SB203580 (10 μM), or HDM (400 U) with or without SB203580 (10 μM) for 1 h prior to stimulation with HDM. The expression of p-p38, t-P38, and lfTSLP protein was evaluated using western blotting analysis in the treated cells. Data are presented as the mean ± SD; n = 3–4. *P < 0.05 vs. the control group; #P < 0.05 vs. the HDM group.
Involvement of STAT5 phosphorylationin lfTSLP-induced barrier dysfunction
Previous studies have demonstrated that TSLP binds with TSLP receptor (TSLPR) to induce epithelial cell proliferation and wound healing through the phosphorylation of signal transducer and activator of transcription (STAT)5 in bronchial epithelial cells26. Firstly, our results show that treatment with lfTSLP (10 ng/ml) induces more marked abnormalities in TER (Fig. 4c), FITC-Dx permeability (Fig. 4d) and delocalization of E-cadherin (Fig. 4e) and β-catenin (Fig. 4f). As a mechanism responsible for the lfTSLP-induced barrier dysfunction, we proposed that the phosphorylation of STAT5 may contribute to this type of barrier dysfunction. We observed a significant and reproducible increase in STAT5 tyrosine phosphorylation when the 16HBE cells were stimulated with lfTSLP (Fig. 4a,b). To further establish the role of STAT5 activation, we assessed airway epithelial barrier function in the presence of a STAT5 inhibitor. Compared with the control groups, treatment with the STAT5 inhibitor partial reversed the lfTSLP-induced changes in TER and FITC-Dx permeability (Fig. 4c,d), and facilitated redistribution of E-cadherin (Fig. 4e) and β-catenin (Fig. 4f) and phosphorylation of STAT5 (Fig. 4a,b). These data suggest that STAT5 signaling contributes to the lfTSLP-induced dysfunction of the epithelial barrier.

Involvement of STAT5 phosphorylationin lfTSLP-induced barrier dysfunction in 16HBE cells.
16HBE cells were treated with culture medium (control group), STAT5 inhibitor (10 μM), or lfTSLP (10 ng/ml) with or without STAT5 inhibitor (10 μM) for 1 h prior to stimulation with lfTSLP. (a,b) p-STAT5, t-STAT5, E-cadherin, and β-catenin protein expression was evaluated by western blotting and densitometric analyses. (c) TER and (d) FITC-Dx permeability was measured in the treated cells. (e) The distribution of E-cadherin was monitored using immunofluorescence. Green represents E-cadherin, blue represents the nucleus. (f) The distribution of β-catenin was monitored using immunofluorescence. Red represents β-catenin, blue represents the nucleus. Data are presented as the mean ± SD; n = 3–4. *P < 0.05 vs. the control group; #P < 0.05 vs. the lfTSLP group.
Protective effect of sfTSLP on HDM- and lfTSLP-induced airway epithelial barrier disruption
A previous study showed that vitamin D3 induced increased transcription primarily of the sfTSLP isoform and that sfTSLP exerts anti-inflammatory effects in inflammatory disorders of the bowel19. In our previous results, we have found that 1,25D3 upregulated sfTSLP expression. Therefore, we postulated that sfTSLP might prevent HDM-induced airway epithelial barrier disruption. To test this hypothesis, we chemically synthesized sfTSLP and then treated 16HBE cells with culture medium (control group), sfTSLP (100 ng/ml), or HDM (400 U) with or without sfTSLP for 1 h prior to stimulation with HDM. Treatment with sfTSLP not only reduced the expression of HDM-induced phosphorylation of ERK1/2 and p38 (Fig. 5a), but also decreased the changes in TER (Fig. 5c), FITC-Dx permeability (Fig. 5d), and delocalization of E-cadherin (Fig. 5e) and β-catenin (Fig. 5f). Interestingly, in contrast to the effects of lfTSLP, sfTSLP also partially reversed lfTSLP-induced TER reduction (Fig. 5c), permeability increase (Fig. 5d), and E-cadherin (Fig. 5e) and β-catenin (Fig. 5f) redistribution. sfTSLP did not induce STAT5 phosphorylation (Fig. 5b). However, when 16HBE cells were stimulated with the two isoforms, either alone or together, sfTSLP inhibited lfTSLP-dependent STAT5 phosphorylation (Fig. 5b). These data indicate that sfTSLP plays a protective role on HDM-induced airway epithelial barrier disruption.

Protective effect of sfTSLP on HDM- and lfTSLP-induced airway epithelial barrier disruption in 16HBE cells.
(a) 16HBE cells were treated with culture medium (control group), sfTSLP (100 ng/ml), or HDM (400 U) with or without sfTSLP (100 ng/ml) for 1 h prior to stimulation with HDM. The expression of p-ERK, t-ERK, p-p38, t-P38, E-cadherin, and β-catenin proteins was assessed using western blotting analysis. (b) 16HBE cells were treated with culture medium (control group), sfTSLP (100 ng/ml), or lfTSLP (10 ng/ml) with or without sfTSLP (100 ng/ml) for 1 h prior to stimulation with lfTSLP. The expression of p-STAT5, t-STAT5, E-cadherin, and β-catenin proteins was assessed using western blotting. (c) TER and (d) FITC-Dx permeability was measured in the different six groups of treated cells. (e) The distribution of E-cadherin was monitored using immunofluorescence. Green represents E-cadherin, blue represents the nucleus. (f) The distribution of β-catenin was monitored using immunofluorescence. Red represents β-catenin, blue represents the nucleus. Data are presented as the mean ± SD; n = 3–4.*P < 0.05 the HDM group vs. the control group; #P < 0.05 the sfTSLP+HDM group vs. the HDM group; **P < 0.05 the lfTSLP group vs. the control group; ##P < 0.05 the sfTSLP+lfTSLP vs. the lfTSLP group.
SfTSLP and 1,25D3 decrease airway hyper-reactivity and inflammation in the mouse model of HDM-induced asthma
To assess the validity of the mouse model of HDM-induced, we monitored the enhanced pause (Penh) response to assess airway reactivity to methacholine. The Penh values were significantly increased in the HDM-treated mice, compared with the saline-treated control group following stimulation with 25, 50, and 100 mg/ml methacholine (Fig. 6a). Treatment with sfTSLP and 1,25D3 significantly inhibited the AHR to levels similar to that in mice that inhaled methacholine (Fig. 6a). Concomitantly, serum IgE levels were markedly raised by HDM, and this effect was inhibited by sfTSLP and 1,25D3 treatment (Fig. 6b). Next, we analyzed the levels of Th1/Th2-associated cytokines in the BALF of mice. The BALF IL-4, IL-5, IL-13, IL-33 (Th2-related; Fig. 6c), and lfTSLP (Fig. 6e) levels, but not IFN-γ (Th1-related; Fig. 6d) was increased in HDM-exposed mice, compared with PBS-treated control animals. These data support the model that asthmatic airway inflammation induced by HDM is a robust Th2 response in mice, and that sfTSLP and 1,25D3 markedly suppress the dominant production of Th2-associated cytokines and the upregulation of lfTSLP in BALF. Histological examination of lung sections from HDM-treated mice indicated markedly large numbers of infiltrating inflammatory cells in the peribronchial regions, as well as evident epithelial hyperplasia and a degree of epithelial shedding, compared to the control group, while treatment with sfTSLP and 1,25D3 mitigated this HDM-induced peribronchial inflammation (Fig. 6f).

sfTSLP and 1,25D3 attenuate airway hyper-reactivity and airway inflammation in the mouse model of HDM-induced asthma.
(a) Airway hyper-responsiveness was measured by whole body plethysmography. (b) IgE levels in serum were measured by ELISA. (c) IL-4, IL-5, IL-13, and IL-33 levels in BALF were measured by ELISA. (d) IFN-γ levels in BALF were measured by ELISA. (e) lfTSLP levels in BALF were measured by ELISA. (f) Representative hematoxylin/eosin-stained lung sections from the different groups. Magnification, 200× (upper panel) and 400× (lower panel). Data are presented as the mean ± SE; n = 5–8/group.*P < 0.05 vs. the control group; #P < 0.05 vs. the HDM group.
SfTSLP and 1,25D3 ameliorate HDM-induced redistributionof E-cadherin and β-catenin and upregulation of lfTSLP in airway epithelia
The lung sections analyzed using immunofluorescence staining presented a strong immunoreactivity of E-cadherin, β-catenin, and lfTSLP in airway epithelial cells. Exposure to HDM resulted in aberrant distribution of E-cadherin (Fig. 7a) and β-catenin (Fig. 7b) at the epithelial cell-cell contact sites and much stronger staining for lfTSLP (Fig. 7c) in airway epithelial cells. These effects were partially reversed by treatment with sfTSLP and 1,25D3 (Fig. 7a–c).

sfTSLP and 1,25D3 ameliorate the HDM-induced redistribution of E-cadherin and β-catenin and the upregulation of lfTSLP in bronchial tissue.
(a) Representative immunohistochemical staining images of E-cadherin in bronchial tissue. Magnification, 400×. (b) Representative immunohistochemical staining images of β-catenin in bronchial tissue. Magnification, 400×. (c) Representative immunohistochemical staining images of lfTSLP in bronchial tissue. Magnification, 400×. n = 5–8/group.
SfTSLP and 1,25D3 inhibit the HDM-induced downregulation in E-cadherin and β-catenin and phosphorylation of ERK1/2 and p38
We next investigated whether HDM affects the expression of E-cadherin and β-catenin proteins in lung. Western blotting analysis indicated a significant decrease in total expression of E-cadherin and β-catenin proteins in lungs of HDM-treated mice and partial reversal of this downregulation upon treatment with sfTSLP and 1,25D3 (Fig. 8A,B). Consistent with the HDM-induced changes seen in16HBE cells, there was a marked increase in the expression of phosphorylated (p)-ERK1/2 and p-p38 in the lungs of HDM-treated mice, which was significantly reversed by treatment with sfTSLP and 1,25D3 (Fig. 8A,B).

sfTSLP and 1,25D3 inhibit the HDM-induced downregulation in E-cadherin and β-catenin and phosphorylation of ERK1/2 and P38 in lungs.
(A) Western blotting analysis of p-ERK, t-ERK, p-p38, t-P38, E-cadherin, and β-catenin proteins in whole lung homogenates in the control, 1,25D3, HDM, and 1,25D3+HDM groups. (B) Western blotting analysis of p-ERK, t-ERK, p-p38, t-P38, E-cadherin, and β-catenin proteins in whole lung homogenates in the control, sfTSLP, HDM, and sfTSLP+HDM groups. Data are presented as the mean ± SD; n = 5/group. *P < 0.05 vs. the control group; #P < 0.05 vs. the HDM group.
Discussion
In this study, we have shown that the two isoforms of TSLP have different expression patterns and biological properties in HDM-induced asthmatic airway epithelial barrier disruption. To our knowledge, ours is the first study to demonstrate that lfTSLP damages the airway epithelial barrier, contributing to the pathogenesis of asthma, and that sfTSLP alleviates HDM-induced airway inflammation and prevents epithelial barrier disruption.
Asthma is one of the most common chronic inflammatory diseases of the airways2. Consistent with the findings of previous experimental models27,28, we successfully established a chronic asthmatic experimental mouse model using repeated, prolonged intranasal administration of HDM. The HDM-exposed mice developed typical asthmatic features, including airway hyper-reactivity, airway inflammation, increased serum IgE levels, as well as an imbalanced Th1/Th2 response.
The primary function of airway epithelium is to serve as a physical barrier for the underlying tissue that responds to the external environment29,30. The essential features that contribute to airway epithelial barrier dysfunction are loss of the cell-cell contact integrity, disruptions of the coordinate expression and interaction of proteins in cell-cell junctional complexes, especially adherens junctions. Adherens junctions, comprised of E-cadherin, β-catenin, and α-catenin, mechanically connect adjacent cells and initiate the formation and maturation of cell-cell contacts30,31,32. In our study, we selected the airway epithelial cell line 16HBE to assess the effects of HDM and lfTSLP on barrier properties in vitro. Compared with the effects of external stimuli on barrier function23,24,33, HDM caused an increase in both ionic and macromolecular permeability (TER reduction and permeability increase) in association with the E-cadherin and β-catenin cleavage. Subsequently, the in vivo experiments and in the mouse model showed an HDM-induced aberrant arrangement of E-cadherin and β-catenin at epithelial cell-cell contact sites. However, the expression of E-cadherin and β-catenin proteins in HDM-treated 16HBE cells remained unchanged, while that in lungs of HDM-treated mice was decreased. A reasonable explanation for the discrepancy in the cultured cells vs. the mouse model is that in the mice, the airway inflammation and the damage to epithelial barrier function influence each other. HDM-induced airway inflammation is persistent, which may cause greater damage to epithelial barrier function, causing liberation of larger amounts of adhesive proteins like E-cadherin and β-catenin from the membrane.
TSLP is produced by epithelial cells and has pleiotropic biological functions. Recent studies have addressed the role of the two isoforms of TSLP. The two isoforms of TSLP are expressed in many human tissues; while sfTSLP mRNA is constitutively expressed, lfTSLP mRNA is not20. Only lfTSLP was markedly induced by invasive bacteria or pro-inflammatory stimuli in normal human airway epithelial cells, primary human keratinocytes and intestinal epithelial cells19,20,21. The present study shows that 16HBE cells express lfTSLP only in response to HDM, whereas the expression of sfTSLP remains unaltered. We previously reported that 1,25D3 alleviates toluene diisocyanante- and cigarette smoke-induced airway inflammation and prevents epithelial barrier disruption23,24. In the current study, we also found that 1,25D3 inhibits HDM-induced asthmatic airway epithelial barrier disruption. Moreover, 1,25D3 upregulates sfTSLP expression and inhibits HDM-dependent upregulation of lfTSLP. These data are consistent with earlier studies that showed that the expression of sfTSLP is constitutive and can be upregulated by anti-inflammatory mediators (vitamin D3) in skin keratinocytes19 and that TSLP, predominantly sfTSLP, is upregulated by VDR agonists21. Hence, upregulation of sfTSLP may be one of the mechanisms of vitamin D-mediated protection against airway inflammation, supporting its advantages in the treatment of asthma34,35. sfTSLP has been confirmed to exert potent antimicrobial activity22 and have anti-inflammatory activities in inflammatory disorders of the bowel19. At the protein level, sfTSLP encompasses the last 63 residues of lfTSLP. We synthesized the sfTSLP peptide and found that treatment with sfTSLP prevents HDM-induced asthmatic airway epithelial barrier disruption and alleviates airway inflammation in vitro and in vivo. Nevertheless, the mechanisms by which sfTSLP enhances barrier function in HDM-induced airway epithelial barrier disruption remain to be elucidated.
MAPKs are well-known cell cycle regulators that convert extracellular stimuli into a wide range of cellular responses, and the ERK, JNK, and p38 MAPK are three well-characterized MAPK pathways25. Previous studies showed that IL-33-induced TSLP expression is mediated by MAPK signaling, via ERK, JNK, and p38 activation36 and that allergen-induced IL-25 and TSLP production were regulated via MAPK pathways37. In this study, HDM significantly induced the expression of p-ERK and p-p38, but not p-JNK. To further elucidate the relationship between the HDM-dependent upregulation of lfTSLP and the MAPK signaling pathway, we treated 16HBE cells with HDM in the presence of ERK and p38 inhibitors and found that both inhibitors reversed the HDM-mediated increase in lfTSLP. These data suggest that HDM induces lfTSLP expression via the ERK and p38 signaling pathways.
We also found that lfTSLP causes more severe damage to epithelial barrier function than HDM, suggesting that lfTSLP plays key roles in HDM-induced asthma. The mechanisms through which lfTSLP disrupt the airway epithelial barrier warrant further studies. Recent studies have demonstrated that TSLP promotes transcription of target genes via STAT5 activation26,38. Bronchial epithelial cells express TSLPR, which interacts with TSLP to induce cell proliferation and injury repair mediated by STAT5 phosphorylation26. In the present study, we showed that STAT5 inhibition prohibits lfTSLP-induced airway epithelial barrier disruption, indicating that lfTSLP-induced damage in asthma is STAT5-dependent. These data support the model that HDM exposure upregulates lfTSLP through the ERK and p38 signaling pathways and subsequently, lfTSLP contributes to airway epithelial barrier dysfunction via STAT5 phosphorylation.
Intriguingly, we found that sfTSLP can attenuate HDM- and lfTSLP-induced airway epithelial barrier disruption. In addition, sfTSLP significantly inhibited the activation of the ERK and p38 signaling pathways by HDM and the lfTSLP-induced phosphorylation of STAT5. However, a previous study that showed that sfTSLP does not block or inhibit lfTSLP-dependent STAT5 phosphorylation19. The different cell types utilized in these studies might explain the contradictory results. Our results demonstrate that sfTSLP prevents HDM-induced airway epithelial barrier disruption, and that the inhibition of the ERK/p38 pathway and the lfTSLP/TSLPR/STAT5 pathway may be involved in this process. However, the role of the interaction between sfTSLP and lfTSLP and the heterodimeric receptor IL-7Rα/TSLPR complex in this process needs to be investigated in future studies.
In conclusion, we demonstrated 2 distinct roles for the long and short TSLP isoforms; lfTSLP contributes to HDM-induced airway epithelial dysfunction, whereas synthetic sfTSLP exerts protective effects in HDM-induced airway inflammation and in the asthmatic mouse model. Moreover, our findings suggest that sfTSLP is a candidate for further development as a personalized therapeutic agent for the prevention and treatment of patients with asthma. Finally, our data emphasize the need for analyzing the roles of the two TSLP isoforms separately in future studies.
Methods
Preparation of TSLP peptides
Synthetic sfTSLP peptides (63aa: MFAMKTKAALAIWCPGYSETQINATQAMKKRRKRKVTTNKCLEQVSQLQGLWRRFNRPLLKQQ) were prepared by China Peptides (Shanghai, China). Recombinant Human TSLP (lfTSLP) was obtained from R&D systems.
Animals and experimental protocol
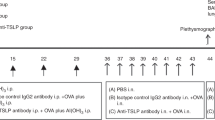
Specific-pathogen-free BALB/c mice (male, 6–8 weeks old, 20–24 g) were purchased from Southern Medical University (Guangzhou, China). The mice were housed in a specific pathogen-free environment (room temperature 24 °C, humidity range 40–70%, and a 12-h light/dark cycle). Sterilized water and food were provided ad libitum. The mice used in our study were treated according to an experimental plan approved by the committee of Southern Medical University on the use and care of animals. All procedures complied with the guidelines of the Institutional Animal Ethics Committee for the care and use of laboratory animals. Purified HDM extract was purchased from ALK-Abello A/S (Denmark) and 1α,25-dihydroxyvitamin D3 (1,25D3; molecular weight, 416.64) was obtained from Sigma (USA). Mice were randomly placed into one of the following six groups: (1) control group, in which the mice received phosphate-buffered saline (PBS; Gibco, Life Technology); (2) sfTSLP group; (3) 1,25D3 group; (4) HDM group; (5) sfTSLP+HDM group, in which mice were pretreated with sfTSLP, followed by HDM; and (6) 1,25D3+HDM group, in which the mice were pretreated with 1,25D3, followed by HDM. All treatments (except 1,25D3) were administered via intranasal inhalation. 1,25D3 was administered to the mice by intraperitoneal injection.
Briefly, mice were exposed to intranasal sevoflurane-anesthesia, then treated with 10 μL PBS, HDM (400 U/mouse each day, dissolved in de-ionized water), sfTSLP (0.1 μg/μL, dissolved in deionized water) and 300 μL 1,25D3 (100 ng/mouse, dissolved in 300 μL PBS containing 0.9% ethanol). In the sfTSLP+HDM and 1,25D3+HDM groups, the anaesthetized mice were pretreated with sfTSLP and 1,25D3 60 min prior to the administration of HDM. These treatment procedures were carried out daily for 5 consecutive days, followed by two days of rest, for 8 consecutive weeks.
Assessment of airway hyper-responsiveness (AHR) to methacholine
Airway parameters were measured 24 h after the last challenge. Mice were placed in a barometric plethysmographic chamber (Buxco Electronics, Troy, NY). The mice were first placed in a chamber for acclimatization, the baseline response was determined, and then normal saline was nebulized into the mice, followed by the administration of increasing concentrations (6.25, 12.5, 25, 50, and 100 mg/ml, respectively) of nebulized methacholine (Sigma Aldrich) in tandem. The reading interval was set to 5 min following each nebulization. The bronchopulmonary resistance was expressed as enhanced pause (Penh).
Enzyme-linked immunosorbent assay (ELISA) for serum IgE and analysis of bronchoalveolar lavage fluid (BALF)
As previously described39, mice were sacrificed with pentobarbital (100 mg/kg, i.p.) one day after the last airway challenge. Blood samples were allowed to rest for 2 h at room temperature, then centrifuged (3000 × g, 20 min), and supernatants were harvested and stored at −80 °C. Total serum IgE was measured by ELISA (eBioscience, San Diego, US) according to the manufacturer’s instructions. Next, the lungs were lavaged twice in situ with 0.8 ml sterile saline (0.9% NaCl, pre-warmed), and the recovered fluid was pooled. Then, the fluids were centrifuged (1000 × g, 10 min) and supernatants were stored for further analysis of IFN-γ, IL-4, IL-5, IL-13, IL-33, and TSLP(lfTSLP) by ELISA (eBioscience) according to the manufacturer’s instructions.
Pulmonary histologic examination
Left lungs were gently infused with 4% neutral formalin to fully inflate all lobes (inflation was judged visually) and immersed in formalin for at least 24 h, then fixed, paraffin-embedded, cut in 4-μm sections, and stained with hematoxylin and eosin (H&E) for blinded histopathologic assessment. For immunohistochemistry of lfTSLP, E-cadherin and β-catenin, lung sections (4 μm) were prepared with a Leica microtome 2030 (Leica Microsystems Nussloch GmbH, Nussloch, Germany), and then submerged in citrate buffer (pH 6.0) for antigen retrieval. Samples were treated with H2O2 for 15 min to block endogenous peroxidases, and then incubated overnight at 4 °C in recommended dilutions of anti-TSLP (Abcam), anti-E-cadherin (Santa Cruz), and anti-β-catenin (Santa Cruz) antibodies. After washing with PBS, slices were incubated with a secondary antibody for 30 min at room temperature. Signals were visualized with a DAB peroxidase substrate kit (ZhongShanJinQiao, Beijing).
Cell culture and treatment
The human bronchial epithelial cell line, 16HBEo- (16HBE; Shanghai Fuxiang Biological Technology Co. Ltd., ATCC, USA) was grown in RPMI 1640 medium (Gibco) with 10% fetal bovine serum (Gibco) and placed in a humidified incubator at 37 °C with an atmosphere of 5% CO2. When the cells reached 80–90% confluence, the cells were treated with trypsin and seeded into culture plates at a density of 104–105 cells per cm2 for use in the experiments. The medium was changed to serum-free RPMI 1640 when the cells reached 85% confluence, and after 12 h, the cells were rinsed with PBS and then stimulated with HDM for the indicated times and doses. The cells were also treated with other mediators and inhibitors, namely lfTSLP, the extracellular signal-regulated kinase (ERK)1/2 tyrosine kinase inhibitor U0126 (10 μM; Cell Signaling Technology, USA), the p38 kinase inhibitor SB203580 (10 μM; Cell Signaling Technology), the STAT5 Inhibitor (10 μM; Santa Cruz, USA), sfTSLP, and 1,25D3 for 1 h prior to stimulation with HDM and lfTSLP in serum-free medium.
Evaluation of epithelial barrier function
Epithelial barrier function was assessed by measuring TER and FITC-Dx flux across the monolayers of cultured epithelial cells. TER and FITC-Dx flux was quantified as previously described23. Briefly, confluent monolayers of 16HBE cells, polarized at an air-liquid interface, were cultured in 12-well Transwell inserts (Corning Costar). TER was measured using a Millicell ERS-2 Epithelial Volt-Ohm meter with an STX01 electrode (Millipore Corp, Billerica, MA, USA). Then, the apical medium (luminal side) was replaced with 200 μL of phenol red-free RPMI 1640 containing 0.5 mg/ml FITC-Dx (Sigma Chemical Co, USA), and the basal medium (non-luminal side) was replaced with 800 μL of phenol red-free RPMI 1640 without FITC-Dx, and the cells were incubated at 37 °C for 90 min. Samples were analyzed by fluorimetry (excitation 492 nm; emission 530 nm). Epithelial permeability was expressed as the percent leakage of FITC-Dx from apical to basolateral compartments.
Immunofluorescence
At the end of the treatment period, the cells were fixed with 4% paraformaldehyde at room temperature for 10 min, washed with PBS for 30 min, incubated with 0.2% Triton X-100 in PBS for 10 min, and rinsed again with PBS. Cells were blocked with 3–5% BSA in PBS for 2 h. The cell monolayers were then incubated overnight with a primary antibody at 4 °C in PBS containing 3–5% BSA, primary rabbit anti-E-cadherin (Santa Cruz) and anti-β-catenin (Santa Cruz) antibodies. Following PBS washes, the cell monolayers were incubated with a secondary antibody, Alexa Fluor 488 (R37118) or Alexa Fluor 594 (R37119) (1:200 diluted in PBS) (Invitrogen, USA), for 1 h at room temperature in the dark. The cell nuclei were stained with 4′,6-diamidino-2-phenylindole dihydrochloride (Sigma Aldrich) for 10 min. The labeled sections were viewed and images captured using a confocal microscope (FV1000, Olympus) at 100× objective magnification.
Western blotting analysis
The treated cells and/or the completely homogenized right lung tissue samples were centrifuged and resuspended in lysis solution containing lysis buffer, the protease inhibitor phenylmethylsulfonyl fluoride, and calcineurin inhibitors for 30 min at 4 °C. The samples were centrifuged at 13000 rpm for 15 min, and the supernatants were collected and boiled with standard SDS sample buffer. The samples were resolved by SDS-PAGE and western blotting analysis was performed for detection of the following antigens: secreted TSLP (lfTSLP) (Abcam) in CM(condition medium). TSLP (lfTSLP) (Abcam), p-ERK1/2 (Cell signaling technology), ERK1/2 (Cell signaling technology), p-p38 (Cell signaling technology), p38 (Cell signaling technology), p-JNK (Cell signaling technology.), JNK (Cell signaling technology), p-STAT5 (Cell signaling technology), STAT5 (Cell signaling technology), E-cadherin (Santa Cruz), and β-catenin (Santa Cruz). After incubation with a secondary antibody, signal intensities were analyzed by using the Odyssey infrared Image System (LiCor, USA). The densitometry results were normalized with that of β-actin.
Quantitative real-time PCR (RT-PCR)
Total RNA was extracted from the treated cells using Trizol (Takara, Japan). RNA samples were then reverse transcribed into first-strand cDNA using the PrimeScriptTM RT reagent kit (Takara). Reverse transcription polymerase chain reaction (RT-PCR) was done following the manufacturer’s instructions by Mx3005 P RealTime PCR Detection System (Stratagene, Santa Clara, CA, USA). Primers for amplifying glyceraldehyde 3-phosphate dehydrogenase (GAPDH) cDNA were 5′-ATCAGCAATGCCTCCTGCAC-3′ (forward) and 5′-TGGCATGGACTGTGGTCATG-3′ (reverse); primers for lfTSLP cDNA were 5′-CACCGTCTCTTGTAGCAATCG-3′ (forward) and 5′-TAGCCTGGGCACCAGATAGC-3′ (reverse); primers for sfTSLP cDNA were 5′-cgtaaactttgccgcctatga-3′ (forward) and 5′-ttcttcattgcctgagtagcatttat-3′ (reverse). The amplification protocol was set as follows: denaturation at 95 °C for 10 min; 40 or 50 cycles denaturation at 95 °C for 15 s; followed by 1 min of annealing/extension at 60 °C. The levels of TSLP mRNA were normalized to those of GAPDH mRNA (the internal control; ΔCt method) and were calculated and displayed as 2−ΔCt values.
Statistical analysis
Statistical analysis was carried out using the SPSS (version 19.0) software package. The variables were expressed as the mean ± standard deviation (SD). One-way ANOVA accompanied by Bonferonni’s post hoc test for multiple comparisons were utilized to compare differences between groups. Values of P < 0.05 were considered to be statistically significant.
Additional Information
How to cite this article: Dong, H. et al. Distinct roles of short and long thymic stromal lymphopoietin isoforms in house dust mite-induced asthmatic airway epithelial barrier disruption. Sci. Rep. 6, 39559; doi: 10.1038/srep39559 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Lambrecht, B. N. & Hammad, H. Allergens and the Airway Epithelium Response: Gateway to Allergic Sensitization. J Allergy Clin Immun. 134, 499–507 (2014).
Lambrecht, B. N. & Hammad, H. The Immunology of Asthma. Nat Immunol. 16, 45–56 (2014).
Lambrecht, B. N. & Hammad, H. The Airway Epithelium in Asthma. Nat Med. 18, 684–692 (2012).
Xiao, C. et al. Defective Epithelial Barrier Function in Asthma. J Allergy Clin Immun. 128, 549–556 (2011).
Nawijn, M. C., Hackett, T. L., Postma, D. S., van Oosterhout, A. J. M. & Heijink, I. H. E-Cadherin: Gatekeeper of Airway Mucosa and Allergic Sensitization. Trends Immunol. 32, 248–255 (2011).
Liu, Y. Thymic Stromal Lymphopoietin: Master Switch for Allergic Inflammation. The Journal of Experimental Medicine. 203, 269–273 (2006).
Takai, T. TSLP Expression: Cellular Sources, Triggers, and Regulatory Mechanisms. Allergology International. 61, 3–17 (2012).
Spadoni, I., Iliev, I. D., Rossi, G. & Rescigno, M. Dendritic Cells Produce TSLP that Limits the Differentiation of Th17 Cells, Fosters Treg Development, and Protects Against Colitis. Mucosal Immunol. 5, 184–193 (2012).
Kamekura, R. et al. Thymic Stromal Lymphopoietin Enhances Tight-Junction Barrier Function of Human Nasal Epithelial Cells. Cell Tissue Res. 338, 283–293 (2009).
Sonesson, A. et al. Thymic Stromal Lymphopoietin Exerts Antimicrobial Activities. Exp Dermatol. 20, 1004–1010 (2011).
Watanabe, N. et al. Hassall’s Corpuscles Instruct Dendritic Cells to Induce CD4+CD25+ Regulatory T Cells in Human Thymus. Nature. 436, 1181–1185 (2005).
Iliev, I. D. et al. Human Intestinal Epithelial Cells Promote the Differentiation of Tolerogenic Dendritic Cells. Gut. 58, 1481–1489 (2009).
Al-Shami, A., Spolski, R., Kelly, J., Keane-Myers, A. & Leonard, W. J. A Role for TSLP in the Development of Inflammation in an Asthma Model. The Journal of Experimental Medicine. 202, 829–839 (2005).
Zhou, B. et al. Thymic Stromal Lymphopoietin as a Key Initiator of Allergic Airway Inflammation in Mice. Nat Immunol. 6, 1047–1053 (2005).
Ying, S. et al. Thymic Stromal Lymphopoietin Expression is Increased in Asthmatic Airways and Correlates with Expression of TH2-attracting Chemokines and Disease Severity. J Immunol. 174, 8183–8190 (2005).
Ying, S. et al. Expression and Cellular Provenance of Thymic Stromal Lymphopoietin and Chemokines in Patients with Severe Asthma and Chronic Obstructive Pulmonary Disease. J Immunol. 181, 2790–2798 (2008).
Ito, T. et al. TSLP-activated Dendritic Cells Induce an Inflammatory T Helper Type 2 Cell Response through OX40 Ligand. The Journal of Experimental Medicine. 202, 1213–1223 (2005).
Soumelis, V. et al. Human Epithelial Cells Trigger Dendritic Cell Mediated Allergic Inflammation by Producing TSLP. Nat Immunol. 3, 673–680 (2002).
Fornasa, G. et al. Dichotomy of Short and Long Thymic Stromal Lymphopoietin Isoforms in Inflammatory Disorders of the Bowel and Skin. J Allergy Clin Immun. 136, 413–422 (2015).
Harada, M. et al. Functional Analysis of the Thymic Stromal Lymphopoietin Variants in Human Bronchial Epithelial Cells. Am J Resp Cell Mol . 40, 368–374 (2009).
Xie, Y., Takai, T., Chen, X., Okumura, K. & Ogawa, H. Long TSLP Transcript Expression and Release of TSLP Induced by TLR Ligands and Cytokines in Human Keratinocytes. J Dermatol Sci. 66, 233–237 (2012).
Bjerkan, L. et al. The Short Form of TSLP is Constitutively Translated in Human Keratinocytes and Has Characteristics of an Antimicrobial Peptide. Mucosal Immunol. 8, 49–56 (2014).
Zhang, R., Zhao, H., Dong, H., Zou, F. & Cai, S. 1Α,25-Dihydroxyvitamin D3 Counteracts the Effects of Cigarette Smoke in Airway Epithelial Cells. Cell Immunol. 295, 137–143 (2015).
Li, W., Dong, H., Zhao, H., Song, J. & Tang, H. 1,25-Dihydroxyvitamin D3 Prevents Toluene Diisocyanate-Induced Airway Epithelial Barrier Disruption. Int J Mol Med. 36, 263–270 (2015).
Cargnello, M. & Roux, P. P. Activation and Function of the MAPKs and their Substrates, the MAPK-Activated Protein Kinases. Microbiol Mol Biol R. 75, 50–83 (2011).
Semlali, A., Jacques, E., Koussih, L., Gounni, A. S. & Chakir, J. Thymic Stromal Lymphopoietin–Induced Human Asthmatic Airway Epithelial Cell Proliferation through an IL-13–Dependent Pathway. J Allergy Clin Immun. 125, 844–850 (2010).
Johnson, J. R. et al. Continuous Exposure to House Dust Mite Elicits Chronic Airway Inflammation and Structural Remodeling. Am J Resp Crit Care. 169, 378–385 (2004).
Chen, Z. et al. Neutralization of TSLP Inhibits Airway Remodeling in a Murine Model of Allergic Asthma Induced by Chronic Exposure to House Dust Mite. Plos One. 8, e51268 (2013).
Schleimer, R. P., Kato, A., Kern, R., Kuperman, D. & Avila, P. C. Epithelium: At the Interface of Innate and Adaptive Immune Responses. J Allergy Clin Immun. 120, 1279–1284 (2007).
Holgate, S. T. The Sentinel Role of the Airway Epithelium in Asthma Pathogenesis. Immunol Rev. 242, 205–219 (2011).
Ganesan, S., Comstock, A. T. & Sajjan, U. S. Barrier Function of Airway Tract Epithelium. Tissue Barriers. 1, e24997 (2013).
Huang, W. et al. High-Mobility Group Box 1 Impairs Airway Epithelial Barrier Function through the Activation of the RAGE/ERK Pathway. Int J Mol Med. 37, 1189–1198 (2016).
Hardyman, M. A. et al. TNF-α–Mediated Bronchial Barrier Disruption and Regulation by Src-Family Kinase Activation. J Allergy Clin Immun. 132, 665–675 (2013).
Tian, W. M., Yang, Y. G., Shang, Y. X., Cai, X. X. & Chen, W. W. Role of 1,25-Dihydroxyvitamin D3 in the Treatment of Asthma. European Review for Medical & Pharmacological. 18, 1762–1769. (2014).
Agrawal, T., Gupta, G. K. & Agrawal, D. K. Vitamin D Supplementation Reduces Airway Hyperresponsiveness and Allergic Airway Inflammation in a Murine Model. Clinical & Experimental Allergy. 43, 672–683. (2013).
Ryu, W. et al. IL-33 Induces Egr-1-dependent TSLP Expression Via the MAPK Pathways in Human Keratinocytes. Exp Dermatol. 24, 857–863 (2015).
Yu, H. S., Angkasekwinai, P., Chang, S. H., Chung, Y. & Dong, C. Protease Allergens Induce the Expression of IL-25 Via Erk and P38 MAPK Pathway. J Korean Med Sci. 25, 829 (2010).
Isaksen, D. E. et al. Requirement for Stat5 in Thymic Stromal Lymphopoietin-Mediated Signal Transduction. Journal of immunology (Baltimore, Md. 1950). 163, 5971–5977 (1999).
Yao, L. et al. The Receptor for Advanced Glycation End Products is Required for Β-Catenin Stabilization in a Chemical-Induced Asthma Model. Brit J Pharmacol. 173, 2600–2613 (2016).
Acknowledgements
This study was funded by the National Natural Science Foundation of China (81470228, 81270087, 81500023, 81300029, and 81270089), the Precision Medicine Research of The National Key Research and Development Plan of China (2016YFC0905800), the National Program on Key Basic Research Project of China (973 Program, 2012CB518203), and Natural Science Foundation of Guangdong Province (S2013040013505, 2014A030310325, and 2015A030313236) and Scientific and Technological Project of Guangdong Province (2016A020215117).
Author information
Authors and Affiliations
Contributions
C.S., D.H., and H.Y. designed the experiment and wrote the manuscript. H.Y., D.H. and L.L. performed the experiments and analyzed the data. Z.M., Z.F., H.C., Y.C., W.X., L.L., Z.H., C.J. and X.Z. helped with data analysis and revised the manuscript. All authors reviewed and approved the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Dong, H., Hu, Y., Liu, L. et al. Distinct roles of short and long thymic stromal lymphopoietin isoforms in house dust mite-induced asthmatic airway epithelial barrier disruption. Sci Rep 6, 39559 (2016). https://doi.org/10.1038/srep39559
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep39559
- Springer Nature Limited
This article is cited by
-
Role of thymic stromal lymphopoietin in allergy and beyond
Nature Reviews Immunology (2023)
-
Short isoform thymic stromal lymphopoietin reduces inflammation and aerobic glycolysis of asthmatic airway epithelium by antagonizing long isoform thymic stromal lymphopoietin
Respiratory Research (2022)
-
Thymic stromal lymphopoietin protects in a model of airway damage and inflammation via regulation of caspase-1 activity and apoptosis inhibition
Mucosal Immunology (2020)
-
TSLP Inhibitors for Asthma: Current Status and Future Prospects
Drugs (2020)
-
Association between methylation in nasal epithelial TSLP gene and chronic rhinosinusitis with nasal polyps
Allergy, Asthma & Clinical Immunology (2019)