
Abstract
The mosquito Aedes aegypti (Ae. aegypti) is the most notorious vector of illness-causing viruses such as Dengue, Chikugunya, and Zika. Although numerous genetic expression studies utilizing quantitative real-time PCR (qPCR) have been conducted with regards to Ae. aegypti, a panel of genes to be used suitably as references for the purpose of expression-level normalization within this epidemiologically important insect is presently lacking. Here, the usability of seven widely-utilized reference genes i.e. actin (ACT), eukaryotic elongation factor 1 alpha (eEF1α), alpha tubulin (α-tubulin), ribosomal proteins L8, L32 and S17 (RPL8, RPL32 and RPS17), and glyceraldeyde 3-phosphate dehydrogenase (GAPDH) were investigated. Expression patterns of the reference genes were observed in sixteen pre-determined developmental stages and in cell culture. Gene stability was inferred from qPCR data through three freely available algorithms i.e. BestKeeper, geNorm, and NormFinder. The consensus rankings generated from stability values provided by these programs suggest a combination of at least two genes for normalization. ACT and RPS17 are the most dependably expressed reference genes and therefore, we propose an ACT/RPS17 combination for normalization in all Ae. aegypti derived samples. GAPDH performed least desirably, and is thus not a recommended reference gene. This study emphasizes the importance of validating reference genes in Ae. aegypti for qPCR based research.
Similar content being viewed by others

Introduction
Aedes aegypti(Ae. aegypti) is widely regarded as the primary vector of arthropod-borne viruses (arboviruses) such as Dengue, Zika and Chikugunya. The past year has seen the mosquito garnering much international attention due to the roles it may have played in the widespread propagation of Zika, although historically the first-reported association of the virus to Ae. aegypti dates as far back as 50 years ago1. This reputation as a host to such organisms has created an Ae. aegypti research base more revolved around the viruses and parasites it carries, rather than the insect itself. However, its relatively recent re-emergence in parts of Europe and North America as well as associations with an ever-growing list of zoonotic diseases have renewed interests in the species as its own organism2,3. Studies involving gene expression and regulation are therefore imminent, as they will not only elucidate the biological significance of any particular gene within Ae. aegypti, but additionally provide a clearer understanding of the complexities behind the networks within which host-virus interaction occurs.
Presently, quantitative real-time polymerase chain reaction (qPCR) remains the most accessible and widely-applied technique for such purposes. The method not only requires minimal nucleic acid quantities when compared to more traditional quantification assays such as northern-blot, but is faster and more reproducible4. The ready availability of reagents and automated platforms has also added to its burgeoning popularity5. Despite its advantages, interpretation of qPCR data is made difficult due to inconsistencies in protocol as well as in template quality and enzymatic efficiencies6,7. Normalization of data against a ‘housekeeping’ or reference gene is therefore critical, as it compensates for differences in starting cDNA quantities amongst samples caused by variations encountered along the RNA extraction and subsequent reverse transcription steps8,9. It involves comparing the ratios of expression levels of the target gene against that of the selected reference gene(s)10. ACT and GAPDH are examples of genes utilized heavily for this purpose. However, it has quickly emerged that no one gene is stably expressed under all developmental and experimental conditions11,12,13,14. Algorithms such as BestKeeper, geNorm, and NormFinder have been developed to identify the best-fit reference gene to use with consideration to one’s protocol and biological samples10,15. Nonetheless, choosing a set of genes as reference rather than just one may in fact be necessary to normalize gene expression experiments16,17,18.
Up to date, the most commonly used gene for normalization in RNA quantification methods in Ae. aegypti is the RibosomalProteinS17 (RPS17)19,20,21,22,23. However, application of a singular reference gene for all tissue and morphological types of the developing mosquitos an arguably flawed scientific approach. This is especially true for Ae. aegypti as in addition to undergoing complete metamorphosis, the insect spends half its life-cycle as an aquatic organism. This indicates that it is exposed to many variables throughout its lifetime; not only in terms of natural progression, but from additional environmental influences. Growth and development patterns of larvae and adult mosquitoes have indeed been shown to be greatly affected by several factors such as diet and temperature24,25,26. There is thus a need for the stability of reference genes at different points of development to be validated to ensure robustness in gene expression normalization where samples are from individuals of a specific developmental stage.
Here, we comprehensively evaluated seven candidate reference genes i.e. ACT, eEF1α, GAPDH, α-tubulin, RPL8, RPL32 and RPS17 at nine points of development as well as inAag2cells. Three programs, namely geNorm, BestKeeper, and NormFinder, were used to analyse their stability and to rank the reference genes for usability at any particular point of development. We also applied our findings to the normalization of a chosen target gene i.e. CTP synthase (CTPsyn) to validate the consensus rankings generated. The suitable reference gene(s) can be applied for normalization of qPCR data for whole organism Ae. aegypti tissue at multiple developmental stages as well as cell culture.
Results and Discussion
Primer evaluation and amplification efficiency of candidate reference genes
Seven candidate genes from three functional classes were investigated: (i) structure-related genes: ACT and α-tubulin, (ii) ribosomal and protein-production genes: eEF1a, RPS17, RPL8 and RPL32, and (iii) metabolism-related gene: GAPDH. Primer pairs were evaluated through standard curve generation with serially diluted pooled cDNA. Efficiency(E) and linear regression coefficient (R2) values are observed to determine the performance of the designed primers in detecting and amplifying cDNA at very high to very low concentrations. All recorded acceptable E values between 92.5 and 100.8% with R2 values ranging from 0.979 to 0.997 (Table 1). Amplification specificity was displayed through the production of a singular peak in melt-curve analysis. Purified qPCR products were sequenced to show specificity and accuracy whereby (a) each primer produced a singular sequence output, and (b) the sequence aligns with the cDNA of the expected gene through BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Sequences are available on GenBank’s BankIt depository with the accession numbers KY000701 to KY000707. Post-run 2% agarose gel further confirmed single amplicon production (see Supplementary Figure S1B).
Expression levels and sample integrity
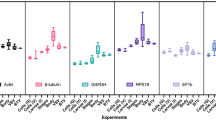
Expression levels were quantified and individual candidate variability in any developmental stage, cell culture, and throughout all assays are summarized as Box-Whisker plots in Fig. 1. The highest recorded Ct value amongst the seven genes was by RPS17 at 15.02, whereas the lowest was by α-tubulin at 33.55. RPS17 and α-tubulin each produced the highest and lowest mean Ct values of 17.86 and 23.83, respectively, when all sample types (n = 17) were taken into account. The other candidate genes were also expressed at high moderate to low moderate levels, with mean Ct values of 18.94, 19.61, 19.71, 20.12, 21.32, and 23.65 each to RPL32, RPL8, eEF1α, ACT, and GAPDH. The target gene CTPsyn had a mean Ct of 22.50, with values ranging from 18.99 to 26.32. This means that comparatively, CTPsyn displayed a narrower Ct variation than six out of the seven candidate genes i.e. ACT, eEF1α, α-tubulin, GAPDH, RPL8 and RPL32 (Table 1). Sample integrity is inferred from the intrinsic variation (InVar) score as generated by the BestKeeper algorithm. Removal of samples with scores in excess of ±3.0 is recommended27. In Var scores for most of the individual developmental stages were low and did not exceed the proposed exclusion value. Triplicate variability was acceptable for all samples. As expected, InVar scores were found to be excessively high when all Ct values were pooled. This strengthens the opinion that no single gene would satisfy all stages, and that each would likely have a unique set of reference genes most suitable for normalization.

Boxes encompass 25th to 75th percentiles. Whisker caps denote maximum and minimum Ct values.
BestKeeper analysis
BestKeeper estimates the standard deviation (SD) value of each candidate gene from raw Ct numbers. An SD > 1 signifies that the variations in expression of a gene within a sample of the same origin are high, and thus indicating its instability. Our data demonstrated that not all candidates were stable across all samples (see Supplementary Table S2). Expression appears to vary most within adult tissues. All barring RPS17 were unstably expressed in adult male samples. RPL8, α-tubulin, eEF1α and RPL32 showed high SDs in non-blood fed female adults. Additionally, ACT was shown to be unstable in 48 to 72 hour embryos; α-tubulin in 6 to 9 hour embryos; eEF1α in 48 to 72 hour embryos and fourth instar larvae; RPL8 throughout the 24 to 72 hour embryonic periods as well as cell samples; RPL32 in 24 to 48 hour embryos and both first instar and third instar larvae; and GAPDH in 48 to 72 hour embryos, all larval stages, and cell culture. The target gene CTPsyn gave relatively low SD values, only exceeding 1 in 48 to 72 hour embryos and Aag2 cells. Ranking of genes is based on the value given as BestKeeper vs Pearson correlation of coefficient. The closer this value is to 1, the greater the reliability of the gene. A third component of the program’s statistical analysis is a P-value, where P < 0.05 indicates the correlation of a candidate gene to the BestKeeper index calculated as the geometric mean of the Ct values. Our data showed that for all instances where the BestKeeper vs Pearson correlation of coefficient value is above 0.67, the gene would be significantly correlated to the BestKeeper index. With genes carrying SD values of above 1 excluded, RPS17 is most reliable for the 48 to 72 hour embryos, first instar larvae and adult male stages; α-tubulin for 0 to 3 hour and 24 to 48 h embryos as well as fourth instar larvae; eEF1α for both 6 to 9 hour and 18 to 24 hour embryos, along with third instar larvae; ACT for the 9 to 12 hour embryonic stage as well as both non-blood fed and blood-fed adult female samples; RPL8 for 3 to 6 hour and 12 to 18 hour embryos, second instar larvae, and pupae; and RPL32 for Aag2 cell culture samples. However, for comparison purposes, all genes are included regardless of SD values. Rankings are shown in Table 2.
geNorm analysis
geNorm determines the expression stability of selected candidate genes based on a data comprised of relative values, i.e. the degree of fold differences observed between Ct values of a sample set in relation to the lowest recorded value. Two assessment outcomes are provided by the software. The first is an average expression stability score as symbolized by M. The higher the M-value of a gene, the less stable it is perceived to be. This value should fall below 1.5. Rankings based on the M-value are in Table 3. Single-normalizer strategies can reliably apply RPL8 or eEF1α for embryo-derived samples of any time point within the first 24 hours, and either ACT or RPS17 for samples from 24 through to 72 hours. RPS17, α-tubulin or RPL8 appear suitable for larval stages; RPS17 or eEF1α for pupae samples; ACT or RPL32 for adult stages, and eEF1α or RPL32 for cell culture samples. A summarization of rankings in charts as provided by the software is as shown in Fig. 2A. The second outcome from geNorm is a pairwise variation or V value which estimates the effect of a gene addition event10. The proposed cut-off value is 0.15. Our data showed that for ten out of the seventeen sample types, two reference genes may be enough for normalization of target gene expression (Fig. 2B). In 0 to 3 hour embryos, the addition of a third gene is recommended for proper normalization. For the other developmental stages, none of the gene inclusion events resulted in satisfactory V values.

(A) Average stability values (M) of genes in individual developmental stages and cell culture. (B) Pairwise variation (V) analysis of candidate reference genes. For each Figure the graphs represent (A) 0 to 3 hour embryos (B) 3 to 6 hour embryos (C) 6 to 9 hour embryos (D) 9 to 12 hour embryos (E) 12 to 18 hour embryos (F) 18 to 24 hour embryos (G) 24 to 48 hour embryos (H) 48 to 72 hour embryos (I) First instar larvae (J) Second instar larvae (K) Third instar larvae (L) Fourth instar larvae (M) Pupae (N) Adult Male (O) Adult Female (P) Adult Female, 6 hours Post-Blood Meal (Q) Aag2 cells.
NormFinder analysis
Similar to geNorm, the data utilized by this program is based on relative values, and not raw Ct data. The algorithm produces a stability value for each gene where a lower value indicates greater stability. The program does not make suggestions for a cut-off value28. Rankings formed by NormFinder are summarized in Table 4. ACT and eEF1α are interchangeable in terms of usability as the reference gene for embryos aged between 0 to 48 hours and all adult samples, whereas RPL32 is best for embryos aged between 48 and 72 hours. RPS17 is the best performing gene in both first and second instar larvae as well as pupae; α-tubulin for the third and fourth larval stages, and eEF1α for cell culture samples. GAPDH and α-tubulin are not recommended for embryonic, larval nor cell samples, and RPS17 should not be the normalizer for adults. In the absence of group identifiers, it is presumed that the two genes with the lowest stability value within a sample set would provide the best combination for two-reference gene normalization strategies29.
Consensus list of reference genes
Consensus rankings are obtained through geometrically averaging the weights assigned to each gene (in the form of stability values from geNorm and NormFinder, and a function of 1-((BestKeeper vs. Pearson correlation coefficient value) from BestKeeper) as generated by the three programs. All genes are included regardless of BestKeeper SD values. Results are summarized in Table 5. The three top-ranked genes in the consensus list for any developmental stage are typically considered to be most reliable e.g. eEF1α, RPS17, GAPDH for pupal samples; α-tubulin, RPL8 and RPS17 for both the third and fourth instar stages; and RPL32 alongsideeEF1α andRPS17 in Aag2 cell culture samples. However, as these vary greatly from one developmental stage to the next, reliability across sample types is also assessed on the basis of overall frequency at which a gene appears amongst the top-three. ACT and RPS17 are the most reliable, with a frequency of 0.216(11/51) and 0.196 (10/51), respectively.
Validation of consensus rankings using CTPsyn target gene
To evaluate consensus ranking outcomes, an assumptive analysis was undertaken. Relative expression of the target gene CTPsyn was investigated ineach sample type. This gene was chosen due to its expected expression stability. Therefore, it is assumed that the ‘true’ fold change value for any developmental stage is derived solely from the △Ct of CTPsyn (2ΔCtCTPsyn). Fold-change as predicted by a normalizing gene(s) is calculated with the Livak method i.e. 2−ΔΔCt (see Supplementary Table S3). Outcomes of normalization against single, top-two consensus-ranked, top-three consensus-ranked, as well as ACT/RPS17 gene combinations are as shown in Table 6. For thirteen out of the sixteen developmental stages as well as cell samples, a single-normalizer strategy estimated CTPsyn fold-change most effectively. RPS17 was the best performing gene for six points of development including in blood-fed females, despite being ranked last within its consensus. RPL8, RPL32 and α-tubulin are each the best normalizer for two, whereas eEF1α and GAPDH each normalized best for one developmental stage. This suggests that if chosen carefully, application of one reference gene may be sufficiently robust for gene expression evaluation. Our analysis also demonstrated that the usage of sequentially ranked genes in combinations of three is preferable to two in estimating fold-change values for most sample types. However, an ACT/RPS17 pairing outperformed both two and three-gene combinations in eleven developmental stages as well as cell culture, suggesting that these two genes together could provide proper normalization regardless of sample origin.
Discussion
As qPCR increasingly becomes the method of choice in gene expression-focused studies, the need for reliable reference genes grows ever more urgent6,8. Misinformed selection of reference genes could lead to false positives or false negatives, effectively masking the true nature of a gene’s expression patterns30. Though Ae. aegypti has been recognized as an important vector of viral diseases for years, it is only recently that research involving the insect has shifted focus to the host back from the viruses it transmits. The Ae. aegypti genome is now fully sequenced and annotated31, and its developmental transcriptome is described32. Nonetheless, studies with Ae. aegypti involving qPCR often adopt genes already exhaustingly utilized as the reference genes for insects of other genera for normalization purposes. In most, the lack of dependable information regarding reference genes for Ae. aegypti has limited normalization to be against a singular reference gene, a practice which could lead to inaccurate data interpretation33,34. Now that it is thrusted into the spotlight as a major proponent of global epidemic threats, in-depth molecular research into the mosquito’s life cycle is vigorously ongoing and thus, it is high time that a detailed panel of reference genes uniquely catering to Ae. aegypti is defined.
In this study, a total of sixteen candidate genes including 18S, ATP5C1, PGK1, TBP and RNAPII were initially identified as candidates. However, these were gradually eliminated due to several factors, including the lack of introns; primer design difficulties due to consecutive base runs spanning exon-exon boundaries; and low basal expression levels (Ct ≥ 30) even at a high starting cDNA concentration discommending the gene from usage as a reference. Remaining genes such as ribosomal proteins RPL8, RPS17 and RPL32 were chosen on the basis of frequency of appearance in Ae. aegypti literature19,21,35,36,37,38. GAPDH, ACT, α-tubulin and eEF1α were selected as available transcriptome data have demonstrated their level expression across the diverse developmental states of this insect32.
Our study sampled subgroups comprising of sixteen pre-determined stages of development in Ae. aegypti as well as Aag2cells. Seven candidate genes were selected and their stability within each developmental stage is evaluated. Our data resulted in three differing rankings from the three evaluation programs employed, i.e. BestKeeper, geNorm, and NormFinder. Nonetheless, some degree of congruence was clearly demonstrated. Results from geNorm and NormFinder were similar in seven developmental stages as well as cell culture. The rankings generated from BestKeeper and NormFinder were alike in 3 to 6 hour and 18 to 24 hour embryos, whereas overlaps were seen between BestKeeper and geNorm in the 12 to 18 hour embryonic samples and blood-fed female adults. Good overall congruency was observed for male and non-blood fed female adults across all three programs. Conversely, major disagreements appeared in candidate gene rankings in 48 to 72 hour embryos (see Supplementary Table S3). As a whole, results generated by BestKeeper tended to contradict those of geNorm and NormFinder both, as was especially noticeable for results in second and third instar larvae. Disparities are to be expected, as each program is based off its own unique algorithm39,40. Moreover, many of the candidate genes in this study fall within the same functional groups. Algorithmic dissimilarities and the resulting differences in sensitivity each program would have towards co-regulated reference genes may have led to this observation10,27,28,41. BestKeeper also has more considerations in the form of InVar, SD, and P-values, all of which contribute to the BestKeeper vs. Pearson correlation coefficient value. These compounding factors result in the obvious differences in final outcomes.
Findings additionally reiterate the notion that there is no universal reference gene stable enough to counterbalance all age and developmental-point imposed variations in gene expression42. The candidate genes investigated here showed considerable variation in expression across different samples. Four displayed a range exceeding 10 Ct, with total standard deviation values of above ±2.2. Most exhibited a larger Ct value range than the target gene used for validation i.e. CTPsyn. Although this could be due to the possibility that CTPsyn – as a synthetase enzyme producing the constantly in-demand CTP molecule – may on its own be a reference gene, the significantly high standard deviations as seen in several samples emphasizes the necessity for an assessment of reference genes in accordance to situational parameters. However, it is impractical to evaluate a large number of candidate genes for every minor qPCR procedure. With developmental stages grouped by tissue and morphological characteristics, the consensus ranking suggests certain two-gene combinations for normalization. Throughout the critical first 24 hours of embryonic development, a pairing between eEF1α and either ACT or RPL8 is optimum. A combination of RPS17 and ACT is best for embryos aged 24 to 72 hours. RPS17 could also provide ideal normalization when simultaneously applied with α-tubulin during the first, third and fourth instar larval stages; with RPL8 in second instar larvae; and with eEF1α with pupal tissue. ACT along with α-tubulin should normalize adult female expression levels regardless of blood-feeding status, whereas RPL32 and eEF1α are suggested for samples of male adults and Aag2 cells. The genes least recommended as reference for egg or embryo-derived tissue samples are α-tubulin and GAPDH. For any larval stage, the usage of RPL32, eEF1α or GAPDH is unadvisable. RPL32 should be avoided when evaluating gene expression in samples from pupal tissue; RPS17 in male and blood fed female adult tissue; and GAPDH in samples derived from non-blood fed female adults as well as Aag2 cells. The frequency with which GAPDH appeared at the lower end of consensus rankings was surprisingly high, as the reliability of the gene as a reference in arthropods has been shown time and time again43,44,45. Nevertheless, given the high standard deviation and overall instability of gene as displayed by our data, we are comfortable in suggesting caution when utilizing GAPDH in regards to Ae. aegypti.
When normalizing expression of a target gene against a reference(s), the objective is to minimalize the normalization factors (NF) value. In geNorm, stepwise inclusion of the next best gene is given as a score denoted as pairwise variation or V value. When this falls below a 0.15 threshold, it suggests that the addition event will only slightly contribute towards decreasing the NF value, and may thus be unnecessary. The V values as generated from our data indicate that in ten out of the seventeen sample subgroups, the combination of two genes should be sufficient for normalization. However, our analysis with the target gene CTPsyn demonstrated that a top-two gene combination was the best normalizer only for second instar larval tissue. Although this same strategy performed well in general for all developmental stages and cell culture, in fourteen of the seventeen subgroups, a singular gene adequately provided proper normalization, though the most effective gene in these instances is never the top-ranked gene. This pairwise variation analysis additionally displayed that no consecutive gene inclusion events satisfied the 0.15 threshold value in 6 to 9 hour embryos, 24 to 48 hour embryos, first, third and fourth instar larvae, as well as adult male tissue samples. Several studies have suggested that as the threshold cutoff point serves as a guide rather than a rule, observing the changing trends as gene inclusion proceeds is more indicative of the ideal number of normalizing genes than the actual value themselves46,47. Others propose that the utilization of the three genes with the lowest M values should most appropriately assist with normalization10,48,49,50. It is therefore of great interest to note that when validated against CTPsyn, triple-combos of top-ranked genes consistently gave more accurate estimations of fold-change than double-combos, and together with findings from previous studies41,51, indicate that applying the suggested minimum of three genes may simultaneously be the most practicable and useful strategy.
Although certain candidates may indeed be able to serve as the sole reference gene for certain developmental stages, our validation analysis also showed that normalization power across most samples did not adhere to its consensus ranking. Top genes often deviate quite significantly from the ‘true’ fold-change value on their own. Selecting the most preferable reference gene may thus devolve into a matter of guesswork and luck. Furthermore, as the data set becomes vulnerable to the variables encountered along the qPCR process10,17, dependence on a singular reference gene for normalization remains an undesired practice. This is especially true when considering the complex nature of a sample set such as ours. The sixteen developmental stages in this study represent a series of transitions in the Ae. aegypti growth environment i.e. from terrestrial (egg or embryo), to aquatic (larval and pupal stages), and back to terrestrial (adult). Though in theory a ‘reference’ gene should not be influenced by such circumstantial stress, this is often not the case. Changes undergone by the organism throughout these periods may exacerbate gene expression variability amongst the sample subgroups. This weakens the gene’s stability, thus impairing its ability to reduce the NF value. Such an observation supports the claim that the suitability of reference genes could be experimentally exclusive, and that a panel of candidates should be simultaneously assessed within the confines of the variables of an assay.
As aforementioned, this may not however be feasible in scientific practice. Therefore, we proposed ACT/RPS17 combination for general usage in normalization practices for Ae. aegypti, regardless of tissue sample origins. This pair of genes appeared with the highest frequencies within the consensus top-three ranks of this study, alluding to their overall stability and dependability in countering developmentally-influenced variation. ACT encodes for a component of the cytoskeleton. Its importance in upholding structural integrity ensures that the gene is expressed at moderately high levels within every cell type. Over the years, ACT has acquired a rather bad reputation due to several instances whereby its seemingly excessive usage as a reference gene was proven to be unjustified. Regardless, ACT has been ranked as the most stable reference gene in a number of validation studies43,52,53. The gene RPS17 produces S17, a protein component of the 40S ribosomal subunit. It has long been used as the reference gene in Ae. aegypti transcriptional profiling36,54,55,56, and is relied upon as the reference gene in numerous studies involving insects57,58,59. Readily available transcriptomics data exhibited minimal ACT expression variation throughout Ae. aegypti development32. However, their levels within the mosquito head appear to be moderately affected by rhythmic circadian cycle changes60. Conversely, the same studies reported rather significant RPS17 fluctuations during developmental progression, but minimal changes due to light-dark switches. These clashes in situations where ACT/RPS17 are more likely to vacillate perhaps allows the genes to counterbalance one another as they act together to normalize gene expression.
Nonetheless, improvements could be made in the future through increasing the number and characteristic variability of candidate genes in further reference gene validation studies. In Drosophila, it has been shown that a larger panel of reference genes is required as the sample size and inherent complexity grows61,62,63. Selection of gene types is also crucial. Here, the candidate genes can be separated into only three categories: (i) genes linked to ribosomal functions and/or protein production, (ii) structural genes producing components of the cellular protein scaffold, and (iii) metabolism-related genes. In the future, including other common reference genes for evaluation such as genes encoding ubiquitin proteins, phosphatases e.g. PP2A, and oxygen-radical metabolizing proteins e.g. SOD and CAT, could improve normalization and consequently the integrity of gene expression studies in Ae. aegypti64,65,66.
Conclusion
To our best knowledge, this is the first study of its kind in Ae. aegypti. Through the utilization of algorithms specifically conceptualized for reference gene validation, a suitable panel of genes most robust for normalization are identified for each developmental stage and Aag2 cell culture. Our results show that although a singular reference gene may suffice for interpretation of target gene expression in most stages, a combination of at least two genes is recommended to minimize the effects of variables upon the data set and for consistency of normalization. Application of three genes for normalization is optimum. Based on consensus rankings, the proposed combinations are RPL8, ACT and eEF1α for early embryos between 0 to 24 hours post-oviposition; ACT, RPS17 and eEF1α for embryos 24 to 48 hours of age; RPS17, RPL32 and ACT for late embryos (aged between 48 to 72 hours); RPS17, α-tubulin, and ACT are recommended for first instar larvae; RPS17, RPL8 and GAPDH for second instar larvae; α-tubulin, RPL8 and RPS17 for both third and fourth instar larval samples; eEF1α, RPS17 and GAPDH for pupal samples; RPL32, eEF1α and ACT for adult male tissue; ACT, α-tubulin, and RPS17 for non-blood fed female adults; ACT, α-tubulin and RPS17 for blood-fed female adults; and finally, RPL32 along with eEF1α and RPS17 for Aag2 cell samples. GAPDH is ranked lowest for most developmental stages, and is thus not to be used as a reference gene. Overall, inferred stability suggests ACT and RPS17 as the most dependably expressed reference genes and therefore, an ACT/RPS17 combination is expected to provide robust normalization for genetic expression studies in all Ae. aegypti derived samples. These findings will benefit normalization practices in Ae. aegypti, and may additionally serve as a resource for screening reference genes in closely-related arthropods.
Materials and Methods
Rearing and sample collection
About 500 dried viable eggs of VCRU-lab strain Ae. aegypti were obtained from Vector Control Research Unit, Universiti Sains Malaysia and hatched in dechlorinated water. Larvae were maintained in relative humidity and natural light conditions at 28 °C in plastic containers. Rearing water is changed every other day. They were fed daily with crushed baby biscuits (Milna™ Rusks, Kalbe®). Larval samples collected were of first (1 L; 80 individuals), second (2 L; 60 individuals), third (3 L; 40 individuals), and fourth (4 L; 20 individuals) instar stages. Pupal samples were a mixture of an equal number of individuals at first, second, and third day of pupation (5 of each day; total of 15 per bioreplicate). Newly-eclosed adults were maintained in cages and fed on 10% sucrose solution. Adult samples collected comprised of equal numbers of males and non-blood fed females aged 1 to 10 days after eclosion (DAE) (two of each gender at each DAE; 20 total per bioreplicate). Food was removed from 5 to 7 DAE adults a full day before being blood-fed using artificial membrane blood feeding system. 20 females were collected 6 hours after the blood meal as post-blood meal samples. The remaining mosquitoes were returned to normal conditions for females to lay eggs. Around 250 embryos were collected at each of the following time points post-oviposition: 0 to 3 hours (0–3 h), 3 to 6 hours (3–6 h), 6 to 9 hours (6–9 h), 9 to 12 hours (9–12 h), 12 to 18 hours (12–18 h), 18 to 24 hours (18–24 h), 24 to 48 hours (24–48 h), and 48 to 72 hours (48–72 h). Aag2 cells cultured in Gibco® L-15 media supplemented with 10% FBS, 1% Pen-Strep and 10% Tryptose Phosphate Broth (all manufactured by ThermoFisher Scientific, USA) in a non-CO2 incubator at 28 °C were harvested at maximum confluency. Three biological replicates were collected for each developmental stage as well as cell culture.
RNA extraction and quality assurance
This study attempts to adhere to the Minimum Information for Publication of Quantitative Real-Time PCR guidelines or MIQE17. Samples collected throughout rearing were immediately stored in TRIzol® reagent (Invitrogen™, Ambion™, Life Technologies) at −20 °C. Total RNA extraction was done within two days of collection with a protocol previously described for mosquito tissue samples67. As much of the culturing media was aspirated away from Aag2 samples, prior to RNA extraction as described by Abcam®68. Extracts were quantified on the Hellma® Analytics TrayCell system in the SmartSpec Plus Spectrophotometer (Bio-Rad Laboratories, California). Those with an A260:A280 value between 1.75 and 2.05 were used immediately for downstream procedures. All showed clear 18S banding and minimal smearing in 1.0% agarose gel. An RNA gel with randomly chosen first bioreplicate extracts of differing degrees of freshness is shown as Supplementary Figure S1A. Extracts were kept at −20 °C for the duration of the experiment.
Reference gene selection, primer design, and primer validation
All genes are also commonly utilized reference genes in qPCR protocols. Primers for RPS17 were previously described19. Others were designed on the Primer 3 software (bioinfo.ut.ee/primer3–0.4.0/). Restrictive parameters for primer selection were: melting temperatures between 59.0 °C and 61.0 °C, GC content between 40 and 60%, nucleotide length between 18 and 24, and amplicon length of between 150 to 225 bases. Regions spanning exon-exon boundaries were specified for each primer pair. Other settings were kept at default. PCR product was confirmed in silico on the Sequence Manipulation Suite website (http://www.bioinformatics.org/sms2/) to be a singular amplicon from only the mature mRNA, and not the genomic DNA sequence. All genes, accession numbers, primer sequences and amplicon size used for this study is listed in Table 1.
Reverse transcription and qPCR
Reverse transcription with 1 μg of total RNA was carried out in 20 μl reactions using the iScript Reverse Transcription Supermix (Bio-Rad Laboratories, California; cat. no. 1708840) according to manufacturer’s protocol. Undiluted cDNA from all nine-developmental stages were pooled and serially diluted to the factor of 5 (1:1, 1:5, 1:25, 1: 125, 1:625 and 1:1875) for standard curve generation on the BioRad CFX96 qPCR platform. Optimum qPCR reactions were carried out in 10 μl reactions using iTaq™ Universal SYBR® Green Supermix (Bio-Rad Laboratories, California; cat. no. 1725120), ~1 ng total cDNA, and 500 nM each of forward and reverse primers. The standard run protocol is initial denaturation at 95 °C for 2.30 mins, followed by 40 cycles of denaturation at 95 °C for 20 s, annealing at 59 °C for 20 s and extension at 72 °C for 15 s. After a final extension at 72 °C for 20 s, the machine would perform a melting-curve analysis. All samples were amplified in technical triplicates. Expression levels were recorded as cycle quantification (Cq). Efficiency values (E) were calculated according to the equation: E = (10[−1/slope]−1) × 10069.
Data mining and selection of reference gene candidates with algorithms: geNorm, BestKeeper, and NormFinder
To assess the stability of candidate reference genes, publicly available evaluation tools i.e. BestKeeper (http://www.gene-quantification.com/bestkeeper.html)27,70, geNorm10 and Normfinder28 were utilized. The BestKeeper algorithm has been adapted for usage in Excel. The program generates a ranking through repeated pairwise correlation and regression analysis of a gene against all the other tested candidates. Up to ten genes can be evaluated at any one time for expression variations in up to one hundred samples. Raw data of Ct values (annotated as CP) and PCR efficiency of the primers were used to determine the correlation between each candidate gene and the index, expressed in the form of a coefficient of determination70. For geNorm and NormFinder, raw data was converted into linear values relative to the lowest Ct recorded for each candidate gene. In geNorm, the stability of a gene is assessed through the consistency of its expression ratio across all samples. The software generates both a stability value i.e. M, and a pairwise variation value i.e. V. M represents the average variation in transcript levels of a gene in comparison to all other candidate genes, achieved through a repeated process of stepwise exclusion commencing from the least stable gene. Pairwise variation estimates the effect of including another gene10 sequentially as per the established M-value rankings through the formula of Vn/Vn + 1. A threshold of 0.15 is set; a V value below this would mean that an additional reference gene would not improve normalization. NormFinder is a mixed-effects model statistical analysis which estimates the stability value of a gene as a function of the approximate expression variation it would impose onto the target gene data during normalization28. The lower this value is, the less variation one would introduce to a normalization exercise should the candidate gene be used as a reference. It also estimates the variation between sample subgroups of the sample set. The BestKeeper vs. Pearson correlation coefficient value, geNorm M value, and NormFinder stability value are perceived as weightage. Geometric means i.e. central tendencies of these weightages for a candidate gene forms the basis for generation of a consensus ranking.
Evaluation of results through target gene normalization
CTPsyn of Ae. aegypti was utilized as the target gene for candidate reference gene evaluation. This gene encodes for the enzyme CTP Synthetase, which converts UTP, ATP and glutamine into cytidine triphosphate (CTP) molecules71. Human isoforms of CTPsyn have been identified as potential reference genes72. Although the expression levels of the gene in insects and Ae. aegypti in particular are undefined, as a housekeeping gene, it is expected to be expressed stably across all developmental stages. For this reason, CTPsyn is chosen as the ‘target’ gene for the purpose of this analysis. Primer sequences for CTPsyn were forward 5′TTCCCCATTGCTACCCGAAC and reverse 5′GAAAACCCTTCCCCAGCGTA. The expected product size is 180 bp. ‘True’ fold-change is based solely on CTPsyn, in the function of 2−ΔCtCTPsyn. Fold changes estimated from normalization with (a) different genes, and (b) differing combinations and number of genes was calculated according to the Livak method i.e. 2−ΔΔCt 73. The degree of difference between the value predicted by normalizer(s) against fold change of CTPsyn △Ct is the basis for evaluation of the effectiveness of the candidate reference gene(s) in normalization.
Additional Information
How to cite this article: Dzaki, N. et al. Evaluation of reference genes at different developmental stages for quantitative real-time PCR in Aedes aegypti. Sci. Rep. 7, 43618; doi: 10.1038/srep43618 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Marchette, N. J., Garcia, R. & Rudnick, A. Isolation of Zika virus from Aedes aegypti mosquitoes in Malaysia. Am J Trop Med Hyg 18, 411–415 (1969).
Kraemer, M. U. G. et al. The global compendium of Aedes aegypti and Ae. albopictus occurrence. Scientific Data 2, 150035, doi: 10.1038/sdata.2015.35 (2015).
Schaffner, F. & Mathis, A. Dengue and dengue vectors in the WHO European region: past, present, and scenarios for the future. The Lancet. Infectious diseases 14, 1271–1280, doi: 10.1016/s1473-3099(14)70834-5 (2014).
Heid, C. A., Stevens, J., Livak, K. J. & Williams, P. M. Real time quantitative PCR. Genome research 6, 986–994 (1996).
Kheirelseid, E. A. H., Chang, K. H., Newell, J., Kerin, M. J. & Miller, N. Vol. 2010 12 (2010).
Bustin, S. A. & Nolan, T. Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. Journal of biomolecular techniques: JBT 15, 155–166 (2004).
Yeung, A. T., Holloway, B. P., Adams, P. S. & Shipley, G. L. Evaluation of dual-labeled fluorescent DNA probe purity versus performance in real-time PCR. BioTechniques 36, 266–270, 272, 274–265 (2004).
Bustin, S. A. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. Journal of molecular endocrinology 29, 23–39 (2002).
Huggett, J., Dheda, K., Bustin, S. & Zumla, A. Real-time RT-PCR normalisation; strategies and considerations. Genes and immunity 6, 279–284, doi: 10.1038/sj.gene.6364190 (2005).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology 3, research0034.0031-research0034.0011 (2002).
Fischer, M., Skowron, M. & Berthold, F. Reliable transcript quantification by real-time reverse transcriptase-polymerase chain reaction in primary neuroblastoma using normalization to averaged expression levels of the control genes HPRT1 and SDHA. The Journal of molecular diagnostics: JMD 7, 89–96, doi: 10.1016/s1525-1578(10)60013-x (2005).
Goidin, D., Mamessier, A., Staquet, M. J., Schmitt, D. & Berthier-Vergnes, O. Ribosomal 18S RNA prevails over glyceraldehyde-3-phosphate dehydrogenase and beta-actin genes as internal standard for quantitative comparison of mRNA levels in invasive and noninvasive human melanoma cell subpopulations. Analytical biochemistry 295, 17–21, doi: 10.1006/abio.2001.5171 (2001).
Nicot, N., Hausman, J. F., Hoffmann, L. & Evers, D. Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. Journal of experimental botany 56, 2907–2914, doi: 10.1093/jxb/eri285 (2005).
Selvey, S. et al. Beta-actin–an unsuitable internal control for RT-PCR. Molecular and cellular probes 15, 307–311, doi: 10.1006/mcpr.2001.0376 (2001).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic acids research 29, e45 (2001).
Bogaert, L. et al. Selection of a set of reliable reference genes for quantitative real-time PCR in normal equine skin and in equine sarcoids. BMC biotechnology 6, 24, doi: 10.1186/1472-6750-6-24 (2006).
Bustin, S. A. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical chemistry 55, 611–622, doi: 10.1373/clinchem.2008.112797 (2009).
Hamalainen, H. K. et al. Identification and validation of endogenous reference genes for expression profiling of T helper cell differentiation by quantitative real-time RT-PCR. Analytical biochemistry 299, 63–70, doi: 10.1006/abio.2001.5369 (2001).
Anderson, J. R. & Rico-Hesse, R. Aedes aegypti vectorial capacity is determined by the infecting genotype of dengue virus. The American journal of tropical medicine and hygiene 75, 886–892 (2006).
Frentiu, F. D. et al. Limited dengue virus replication in field-collected Aedes aegypti mosquitoes infected with Wolbachia. PLoS neglected tropical diseases 8, e2688, doi: 10.1371/journal.pntd.0002688 (2014).
Joy, T. K. et al. Aging field collected Aedes aegypti to determine their capacity for dengue transmission in the southwestern United States. PLoS One 7, e46946, doi: 10.1371/journal.pone.0046946 (2012).
Morlais, I., Mori, A., Schneider, J. R. & Severson, D. W. A targeted approach to the identification of candidate genes determining susceptibility to Plasmodium gallinaceum in Aedes aegypti. Molecular genetics and genomics: MGG 269, 753–764, doi: 10.1007/s00438-003-0882-7 (2003).
Morlais, I. & Severson, D. W. Identification of a polymorphic mucin-like gene expressed in the midgut of the mosquito, Aedes aegypti, using an integrated bulked segregant and differential display analysis. Genetics 158, 1125–1136 (2001).
Padmanabha, H., Lord, C. C. & Lounibos, L. P. Temperature induces trade-offs between development and starvation resistance in Aedes aegypti (L.) larvae. Medical and Veterinary Entomology 25, 445–453, doi: 10.1111/j.1365-2915.2011.00950.x (2011).
Rey, J. R., Hargraves, P. E. & O’Connell, S. M. Effect of selected marine and freshwater microalgae on development and survival of the mosquito Aedes aegypti. Aquatic Ecology 43, 987–997, doi: 10.1007/s10452-009-9232-0 (2009).
Tun-Lin, W., Burkot, T. R. & Kay, B. H. Effects of temperature and larval diet on development rates and survival of the dengue vector Aedes aegypti in north Queensland, Australia. Medical and Veterinary Entomology 14, 31–37, doi: 10.1046/j.1365-2915.2000.00207.x (2000).
Pfaffl, M. W., Tichopad, A., Prgomet, C. & Neuvians, T. P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnology letters 26, 509–515 (2004).
Andersen, C. L., Jensen, J. L. & Orntoft, T. F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer research 64, 5245–5250, doi: 10.1158/0008-5472.can-04-0496 (2004).
Mehdi Khanlou, K. & Van Bockstaele, E. A critique of widely used normalization software tools and an alternative method to identify reliable reference genes in red clover (Trifolium pratense L.). Planta 236, 1381–1393, doi: 10.1007/s00425-012-1682-2 (2012).
VanGuilder, H. D., Vrana, K. E. & Freeman, W. M. Twenty-five years of quantitative PCR for gene expression analysis. BioTechniques 44, 619–626, doi: 10.2144/000112776 (2008).
Nene, V. et al. Genome Sequence of Aedes aegypti, a Major Arbovirus Vector. Science 316, 1718–1723, doi: 10.1126/science.1138878 (2007).
Akbari, O. S. et al. The developmental transcriptome of the mosquito Aedes aegypti, an invasive species and major arbovirus vector. G3 (Bethesda, Md.) 3, 1493–1509, doi: 10.1534/g3.113.006742 (2013).
Schmittgen, T. D. & Zakrajsek, B. A. Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. Journal of biochemical and biophysical methods 46, 69–81 (2000).
Ferguson, B. S., Nam, H., Hopkins, R. G. & Morrison, R. F. Impact of Reference Gene Selection for Target Gene Normalization on Experimental Outcome Using Real-Time qRT-PCR in Adipocytes. PLoS ONE 5, e15208, doi: 10.1371/journal.pone.0015208 (2010).
Clifton, M. E. & Noriega, F. G. The fate of follicles after a blood meal is dependent on previtellogenic nutrition and juvenile hormone in Aedes aegypti. Journal of Insect Physiology 58, 1007–1019, doi: 10.1016/j.jinsphys.2012.05.005 (2012).
Cook, P. E. et al. Predicting the age of mosquitoes using transcriptional profiles. Nat. Protocols 2, 2796–2806, doi: http://www.nature.com/nprot/journal/v2/n11/suppinfo/nprot.2007.396_S1.html (2007).
Perez, M. H. & Noriega, F. G. Aedes aegypti pharate 1(st) instar quiescence affects larval fitness and metal tolerance. Journal of Insect Physiology 58, 824–829, doi: 10.1016/j.jinsphys.2012.03.005 (2012).
Mazzacano, C. A. & Fallon, A. M. Changes in ribosomal protein rpL8 mRNA during the reproductive cycle of the mosquito, Aedes aegypti. Insect Biochem Mol Biol 26, 563–570 (1996).
Guo, J., Ling, H., Wu, Q., Xu, L. & Que, Y. The choice of reference genes for assessing gene expression in sugarcane under salinity and drought stresses. Scientific Reports 4, 7042, doi: 10.1038/srep07042 http://www.nature.com/articles/srep07042#supplementary-information (2014).
Robledo, D. et al. Analysis of qPCR reference gene stability determination methods and a practical approach for efficiency calculation on a turbot (Scophthalmus maximus) gonad dataset. BMC Genomics 15, 648, doi: 10.1186/1471-2164-15-648 (2014).
Ponton, F., Chapuis, M.-P., Pernice, M., Sword, G. A. & Simpson, S. J. Evaluation of potential reference genes for reverse transcription-qPCR studies of physiological responses in Drosophila melanogaster. Journal of Insect Physiology 57, 840–850, doi: http://dx.DOI.org/10.1016/j.jinsphys.2011.03.014 (2011).
Bustin, S. A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. Journal of molecular endocrinology 25, 169–193 (2000).
Cardoso, G. A., Matiolli, C. C., de Azeredo-Espin, A. M. & Torres, T. T. Selection and validation of reference genes for functional studies in the Calliphoridae family. Journal of insect science (Online) 14, 2, doi: 10.1093/jis/14.1.2 (2014).
Lu, Y. et al. Identification and validation of reference genes for gene expression analysis using quantitative PCR in Spodoptera litura (Lepidoptera: Noctuidae). PLoS One 8, e68059, doi: 10.1371/journal.pone.0068059 (2013).
Van Hiel, M. B. et al. Identification and validation of housekeeping genes in brains of the desert locust Schistocerca gregaria under different developmental conditions. BMC Molecular Biology 10, 56, doi: 10.1186/1471-2199-10-56 (2009).
Hu, R., Fan, C., Li, H., Zhang, Q. & Fu, Y.-F. Evaluation of putative reference genes for gene expression normalization in soybean by quantitative real-time RT-PCR. BMC Molecular Biology 10, 93, doi: 10.1186/1471-2199-10-93 (2009).
Ray, D. L. & Johnson, J. C. Validation of reference genes for gene expression analysis in olive (Olea europaea) mesocarp tissue by quantitative real-time RT-PCR. BMC Research Notes 7, 304–304, doi: 10.1186/1756-0500-7-304 (2014).
Gu, C. et al. Reference gene selection for quantitative real-time PCR in Chrysanthemum subjected to biotic and abiotic stress. Mol Biotechnol 49, doi: 10.1007/s12033-011-9394-6 (2011).
Sun, M., Lu, M.-X., Tang, X.-T. & Du, Y.-Z. Exploring Valid Reference Genes for Quantitative Real-Time PCR Analysis in Sesamia inferens (Lepidoptera: Noctuidae). PLoS One 10, e0115979, doi: 10.1371/journal.pone.0115979 (2015).
Fu, J. et al. Identification of genes for normalization of quantitative real-time PCR data in ovarian tissues. Acta Biochim Biophys Sin (Shanghai) 42, 568–574, doi: 10.1093/abbs/gmq062 (2010).
Teste, M.-A., Duquenne, M., François, J. M. & Parrou, J.-L. Validation of reference genes for quantitative expression analysis by real-time RT-PCR in Saccharomyces cerevisiae. BMC Molecular Biology 10, 99, doi: 10.1186/1471-2199-10-99 (2009).
Jiang, H. B., Liu, Y. H., Tang, P. A., Zhou, A. W. & Wang, J. J. Validation of endogenous reference genes for insecticide-induced and developmental expression profiling of Liposcelis bostsrychophila (Psocoptera: Liposcelididae). Molecular biology reports 37, 1019–1029, doi: 10.1007/s11033-009-9803-0 (2010).
Scharlaken, B., de Graaf, D. C., Goossens, K., Brunain, M., Peelman, L. J., & Jacobs, F. J. Reference gene selection for insect expression studies using quantitative real-time PCR: The head of the honeybee, Apis mellifera, after a bacterial challenge. Journal of Insect Science 8, 33, http://doi.org/10.1673/031.008.3301 (2008).
Cook, P. E. et al. The use of transcriptional profiles to predict adult mosquito age under field conditions. Proceedings of the National Academy of Sciences of the United States of America 103, 18060–18065, doi: 10.1073/pnas.0604875103 (2006).
Klasson, L., Kambris, Z., Cook, P. E., Walker, T. & Sinkins, S. P. Horizontal gene transfer between Wolbachia and the mosquito Aedes aegypti. BMC Genomics 10, 33, doi: 10.1186/1471-2164-10-33 (2009).
Hugo, L. E. et al. Adult Survivorship of the Dengue Mosquito Aedes aegypti Varies Seasonally in Central Vietnam. PLoS neglected tropical diseases 8, e2669, doi: 10.1371/journal.pntd.0002669 (2014).
Vogt, R. G. et al. The insect SNMP gene family. Insect biochemistry and molecular biology 39, 448–456 (2009).
Weinberg-Shukron, A. et al. A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis. The Journal of Clinical Investigation 125, 4295–4304, doi: 10.1172/JCI83553.
Ezzeddine, N. et al. A subset of Drosophila integrator proteins is essential for efficient U7 snRNA and spliceosomal snRNA 3′-end formation. Molecular and cellular biology 31, 328–341, doi: 10.1128/mcb.00943-10 (2011).
Leming, M. T., Rund, S. S. C., Behura, S. K., Duffield, G. E. & O’Tousa, J. E. A database of circadian and diel rhythmic gene expression in the yellow fever mosquito Aedes aegypti. BMC Genomics 15, 1128, doi: 10.1186/1471-2164-15-1128 (2014).
Chintapalli, V. R., Wang, J. & Dow, J. A. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nature genetics 39, 715–720, doi: 10.1038/ng2049 (2007).
Zhai, Y. et al. Identification and Validation of Reference Genes for Quantitative Real-Time PCR in Drosophila suzukii (Diptera: Drosophilidae). PLoS One 9, e106800, doi: 10.1371/journal.pone.0106800 (2014).
Ling, D. & Salvaterra, P. M. Robust RT-qPCR Data Normalization: Validation and Selection of Internal Reference Genes during Post-Experimental Data Analysis. PLoS One 6, e17762, doi: 10.1371/journal.pone.0017762 (2011).
Liu, M., Jiang, J., Han, X., Qiao, G. & Zhuo, R. Validation of Reference Genes Aiming Accurate Normalization of qRT-PCR Data in Dendrocalamus latiflorus Munro. PLoS One 9, e87417, doi: 10.1371/journal.pone.0087417 (2014).
Meng, Y., Li, N., Tian, J., Gao, J. & Zhang, C. Identification and validation of reference genes for gene expression studies in postharvest rose flower (Rosa hybrida). Scientia Horticulturae 158, 16–21, doi: http://dx.DOI.org/10.1016/j.scienta.2013.04.019 (2013).
Zhu, X. et al. Selection and Evaluation of Reference Genes for Expression Analysis Using qRT-PCR in the Beet Armyworm Spodoptera exigua (Hübner) (Lepidoptera: Noctuidae). PLoS One 9, e84730, doi: 10.1371/journal.pone.0084730 (2014).
Kafatos, F. C. RNA isolation from mosquito Aedes aegypti tissue samples, http://kafatos.openwetware.org/totalRNA_extraction_from_adult_mosquitoes.html (2009).
Pattern, R. RNA isolation and reverse transcription protocol: Cells in culture, http://docs.abcam.com/pdf/protocols/rna-isolation-protocol-cells-in-culture.pdf.
Radonić, A. et al. Guideline to reference gene selection for quantitative real-time PCR. Biochemical and Biophysical Research Communications 313, 856–862, doi: http://dx.DOI.org/10.1016/j.bbrc.2003.11.177 (2004).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic acids research 29, e45–e45 (2001).
Long, C. W., Levitzki, A. & Koshland, D. E. Jr. The subunit structure and subunit interactions of cytidine triphosphate synthetase. The Journal of biological chemistry 245, 80–87 (1970).
Eisenberg, E. & Levanon, E. Y. Human housekeeping genes, revisited. Trends in genetics: TIG 29, 569–574, doi: 10.1016/j.tig.2013.05.010 (2013).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif.) 25, 402–408, doi: 10.1006/meth.2001.1262 (2001).
Acknowledgements
We would like to thank all our collaborators and colleagues for the discussion and the work conducted in this lab. We would especially like to thank the Vector Control Research Unit, Universiti Sains Malaysia for providing the mosquito samples. This study was funded by the Fundamental Research Grant Scheme (203/PBIOLOGI/6711457), Universiti Sains Malaysia Research University Grant (1001/PBIOLOGI/811320), Short Term Grant (304/PBIOLOGI/6313195) and ScienceFund grant (305/PBIOLOGI/613238). N.D. is funded by Yayasan Khazanah.
Author information
Authors and Affiliations
Contributions
G.A. conceived and design the experiments. N.D., K.N.R. and A.A. performed the experiments. G.A., N.D. and I.H.I. analysed the data and interpreted the results. G.A. and N.D. wrote the manuscript and generated the figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Dzaki, N., Ramli, K., Azlan, A. et al. Evaluation of reference genes at different developmental stages for quantitative real-time PCR in Aedes aegypti. Sci Rep 7, 43618 (2017). https://doi.org/10.1038/srep43618
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep43618
- Springer Nature Limited
This article is cited by
-
RNA-seq validation: software for selection of reference and variable candidate genes for RT-qPCR
BMC Genomics (2024)
-
Determination of internal controls for quantitative gene expression of Spodoptera litura under microbial pesticide stress
Scientific Reports (2024)
-
Effect of marker-free transgenic Chlamydomonas on the control of Aedes mosquito population and on plankton
Parasites & Vectors (2023)
-
Evaluation of potential reference genes in the biting midge Culicoides sonorensis for real-time quantitative PCR analyses
Scientific Reports (2023)
-
Ad libitum consumption of protein- or peptide-sucrose solutions stimulates egg formation by prolonging the vitellogenic phase of oogenesis in anautogenous mosquitoes
Parasites & Vectors (2022)